

Abstract
Extracellular signal-regulated kinase (ERK) activation has been shown to promote neuronal death in various paradigms. We demonstrated previously that the late and sustained ERK activation in cerebellar granule neurons (CGNs) cultured in low potassium predominantly promotes plasma membrane (PM) damage. Here, we examined the effects of a well established neuronal survival factor, insulin-like growth factor 1 (IGF-1), on the ERK cell death pathway. Stimulation of CGNs with IGF-1 induced an early and transient ERK activation but abrogated the appearance of late and sustained ERK. Withdrawal or readdition of IGF-1 after 4 h in low potassium failed to prevent sustained ERK activation and cell death. IGF-1 activated the protein kinase A (PKA) to mediate ERK inhibition via c-Raf phosphorylation at an inhibitory site (Ser259). Phosphatidylinositol 3-kinase (PI3K) or PKA inhibitors, but not a specific Akt inhibitor, abrogated PKA signaling. This suggests that the PI3K/PKA/c-Raf-Ser259 pathway mediates ERK inhibition by IGF-1 independent of Akt. In addition, adenoviral-mediated expression of constitutively active MEK (mitogen-activated protein kinase kinase) or Sindbis viral-mediated expression of mutant Raf Ser259Ala both attenuated IGF-1-mediated prevention of PM damage. Activation of caspase-3 promoted DNA damage. Its inhibition by IGF-1 was both PI3K and Akt dependent but PKA independent. 8-Br-cAMP, an activator of PKA, induced phosphorylation of c-Raf-Ser259 and inhibited ERK activation without affecting caspase-3. This indicates a selective role for PKA in ERK inhibition through c-Raf-Ser259 phosphorylation. Together, these data demonstrate that IGF-1 can positively and negatively regulate the ERK pathway in the same neuronal cell, and provide new insights into the PI3K/Akt/PKA signaling pathways in IGF-1-mediated neuronal survival.
Introduction
Intracellular signaling pathways, including phosphatidylinositol 3-kinase (PI3K), extracellular signal-regulated kinase (ERK), and protein kinase A (PKA) pathways have been shown to be important for regulating neuronal survival. In several neuronal cell types in vitro, the PI3K pathway appears to mediate the protective effects of growth factors. Activation of PI3K is involved in insulin-like growth factor 1 (IGF-1) and NGF promoted survival of cerebellar granule neurons (CGNs) and sympathetic neurons, respectively (Vemuri and McMorris, 1996; D'Mello et al., 1997; Parrizas et al., 1997; Crowder and Freeman, 1998). ERK activation has been implicated in the survival-promoting effects of BDNF on retinal ganglion cells (Meyer-Franke et al., 1998). The cAMP-PKA pathway can protect hippocampal neurons from oxide toxicity and anoxia (Maiese et al., 1993). Elevation of cAMP prevents death of spiral ganglion neurons, sympathetic neurons, and CGNs (D'Mello et al., 1993; Crowder and Freeman, 1999; Bok et al., 2003). Although the PI3K pathway has been exclusively implicated in cell survival, various neuronal injury models suggest that activation of ERK and PKA can also promote cell death (Ueda et al., 1996; Cha et al., 2000; Colucci-D'Amato et al., 2003; Cheung and Slack, 2004). Thus, identical intracellular signaling pathways can promote neuronal survival or death. Mechanisms underlying this ambiguity are poorly understood.
The Akt protein kinase has been implicated to be the transducer of PI3K-dependent survival signals (Jones et al., 1991; Burgering and Coffer, 1995; Franke et al., 1995). In response to PI3K activation, Akt binds to phosphorylated membrane lipids and is phosphorylated at Thr308 and Ser473, which propagates survival signal (Downward, 1998).
An elevation of the intracellular cAMP level is an important mediator of various biological responses. It is well established that elevated cAMP directly results in the activation of PKA (Taylor et al., 1990). PKA has been shown to promote neuronal survival independent of PI3K/Akt, and by converging at one or more downstream targets of PI3K/Akt (Miller et al., 1997; Crowder and Freeman, 1999; Li et al., 2000; Bok et al., 2003).
Putative cross talks among different signaling pathways resulting in neuronal survival are only beginning to emerge. Studies on non-neural cells have significantly advanced our understanding of cross talks among PI3K/PKA/ERK pathways. cAMP was shown to activate ERK or inhibit PI3K with distinct biological outcome (Englaro et al., 1995; Busca et al., 1996). Similarly, the PI3K target, Akt, was shown to inhibit the ERK pathway, and prevent proliferation via c-Raf phosphorylation at the inhibitory site Ser259 (Rommel et al., 1999; Zimmermann and Moelling, 1999; Moelling et al., 2002).
Cultured CGNs from postnatal rat require high potassium for survival. Changing the medium from high to low potassium induces death (Gallo et al., 1987; D'Mello et al., 1993; Miller and Johnson, 1996). We report here a novel cross talk between PI3K/PKA/ERK signaling in IGF-1-mediated neuronal survival. Our previous study has demonstrated that late and sustained ERK activation promotes plasma membrane (PM) damage, and that caspase-3 activation promotes DNA damage in CGNs (Subramaniam et al., 2004). Here, we demonstrate that IGF-1 prevents CGN death via two distinct pathways: the PI3K/PKA/c-Raf pathway, which inhibits ERK activation and PM damage, and the PI3K/Akt pathway, which prevents caspase-3 activation and DNA damage.
Materials and Methods
Reagents. IGF-1 was purchased from Ciba Geigy (Basel, Switzerland). Eagle's basal medium with Earle's salts, glutamine, penicillin, streptomycin, trypsin, PBS, HBSS, and TRIzol reagents were from Invitrogen (Karlsruhe, Germany). Fetal calf serum was obtained from Seromed (Berlin, Germany). 2-(4-Morpholinyl)-8-phenyl-1(4 H)-benzopyran-4-one (LY294002) (LY), rabbit polyclonal antibodies [anti-phospho-p44/42 (ERK1/2) mitogen-activated protein kinase (Thr202/Tyr204), anti-ERK1/2, anti-phospho c-Raf (Ser259), anti-caspase-3, anti-total Akt, anti-phospho Akt (Ser473), anti-IGF-1Rα], and anti-rabbit IgG HRP-conjugated antibody were from New England Biolabs (Frankfurt, Germany). Anti-total c-Raf was from BD Biosciences (San Diego, CA). 1,4-Diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene (U0126), Akt-I, Akt-IV, protein kinase inhibitor (PKI), N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl (H89), and wortmannin (W) were from Calbiochem (La Jolla, CA), and LY294002 and 2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one (PD98059) were from Promega (Madison, WI). Polyvinyldifluoride membrane and the ECL chemiluminescence kit were from Amersham Biosciences (Freiburg, Germany). All of the other chemicals including 8-Br-cAMP and anti-FLAG antibody (M5) were purchased from Sigma (St. Louis, MO). Antibody against hemagglutinin (HA) epitope was from Babco (Richmond, CA). All of the fluoroprobes were from Dianova (Hamburg, Germany). All of the reverse transcriptase (RT)-PCR reagents were purchased from Promega (Madison WI).
Cell culture. CGNs were isolated and cultured from 8-d-old Wistar rats as described previously (Subramaniam et al., 2003). Cells were resuspended in high K+ [Eagle's basal medium containing 10% fetal calf serum, 25 mm KCl, 2 mm glutamine, and 0.5% (v/v) penicillin/streptomycin], switched to low K+ (Eagle's basal medium, 5 mm KCl, 2 mm glutamine, and 0.5% penicillin/streptomycin) at day 4, treated with additives for the indicated time points, and processed for the specific assays described below. More than 98% of CGNs were positive for neuron-specific enolase, indicating that CGNs comprise a highly homogenous neuronal population (data not shown).
Cell death analysis. Cells were washed and fixed with 4% paraformaldehyde, and triple staining was performed for propidium iodide (PI) uptake, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), and 4′,6′-diamidino-2-phenylindole (DAPI) exactly as described previously (Subramaniam et al., 2004). Typically, ∼500 cells were counted for the number of total PI-stained (PM damage) or total TUNEL-positive (DNA-damaged) cells or total condensed nuclei. Cell numbers were presented as percentage of degenerated cells in relation to total cell numbers.
Preparation and infection of recombinant adeno and Sindbis viruses. The adenovirus expressing constitutively active (CA) mitogen-activated protein kinase kinase (MEK) (CA MEK-1) encoding an HA epitope tag was cloned, expressed, and infected as described previously (Subramaniam et al., 2004). N-terminally FLAG-tagged human full-length c-Rat S259A mutant (Raf-S259A) construct (Dhillon et al., 2002) was a generous gift from Amardeep Dhillon and Walter Kolch (The Beatson Institute for Cancer Research, Bearsden, Glasgow, UK). The Raf-S259A was sub-cloned into the pcDNA3.1 vector at KpnI-XhoI sites. For recloning into the pSinRep5 vector (a generous gift from Sondra Schlesinger, Washington University School of Medicine, St. Louis, MO), Raf-S259A was excised from the pcDNA3.1 vector using NheI and ApaI, and cloned into pSinRep5 vector, which was previously cut with XbaI and ApaI (pSinRep5-FLAG-c-Raf). As a control, a Sindbis expression vector was constructed with enhanced green fluorescent protein (EGFP) alone. EGFP was excised from pEGFP-C1 (Clontech, Heidelberg, Germany) with BglII, blunt-ended with Klenow fragment, cut with NheI, and cloned into pSinRep5 plasmid, which was previously cut with XbaI and ApaI (pSinRep5-EGFP).
pSinRep5-FLAG-c-Raf, pSinRep5-EGFP, and helper DH (26S) DNA (which encodes the structural proteins of the Sindbis virus) were transcribed in vitro (mMessage Machine SP6 kit; Ambion, Huntingdon, UK). The recombinant RNA and helper DH (26S) RNA were electroporated into baby hamster kidney cells using the Gene Pulser II electroporation system (Bio-Rad, München, Germany). The recombinant RNA encoding FLAG-c-Raf or EGFP alone was packaged and enveloped by the structural proteins produced by the helper RNA. The replication-incompetent pseudovirus, thus produced and released into the extracellular medium, was harvested after 24 h, filtered, and stored at -80°C. CGNs were infected with 20-30 μl of recombinant Sindbis viruses expressing FLAG-c-Raf-S259A or EGFP on day in vitro (DIV)3 in high-potassium culture medium. On DIV4, the medium was changed to low potassium. Depending on the experiments, CGNs infected with Sindbis viruses were processed for analysis of PM damage or Western blotting. By 24 h after the infection, 80-90% of the neurons appeared positive for EGFP. Sindbis virus expression does not cause apparent toxicity for CGN culture even up to 4 d after the infection.
Immunocytochemistry. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% normal horse serum (1 h) followed by incubation with a monoclonal antibody for activated ERK [phosphorylated ERK (pERK); 1:200; overnight]. Cells were then incubated with anti-mouse IgG conjugated with cyanin 3 (1: 1000; 1 h). DAPI (5 μg/ml) was added to visualize nuclei. After pERK/DAPI staining, the nuclear and cytoplasmic pERK was determined by counting 500 cells positive for nuclear pERK or cytoplasmic pERK per field. Cell numbers were presented as percentages of pERK-positive cells in relation to the total cell number.
RT-PCR. RNA was isolated before and 6, 12, and 24 h after the potassium change using TRIzol reagent (Invitrogen) as indicated in the manufacturer's protocol. Three to 5 μg of total RNA in a 11 μl volume was incubated with 0.5 μl of RNase inhibitor and 0.5 μl of RQ1 DNase for 15 min at 37°C. The mixture was heated to 70°C for 5 min and then slowly cooled down to room temperature. Reverse transcription was performed with reaction mixture (12 μl of RNA, 4 μl of 10× transcription buffer, 2 μl of 0.1 m DTT, 1 μl of 10 mm 2′-deoxynucleoside 5′-triphosphates, 1 μl of Moloney murine leukemia virus reverse transcriptase) in a total volume of 20 μl. The reaction mixture was incubated for 2 h at 37°C, heated to 90°C for 5 min, and stored at -20°C for additional use. Two to 4 μl was used for PCR amplification purposes. The PCR was performed for 22 cycles for IGF-1 receptor (IGF-1R) with forward (5′-CAA CGA CTA TCA GCA GCT GA-3′) and reverse (5′-TGG TGG AGA GGT AAC AGA GG-3′) primers (expected band, 343 bp). As a control, β-actin with forward (5′-TCA TGA AGT GTA ACG TTG ACA TCC GT-3′) and reverse (5′-CCT AGA ATT TGC GGT GCA CGA 3′) primers was used (expected band, 284 bp).
Western blot analysis. Western blot analysis was performed as described previously (Subramaniam et al., 2003). Densitometric quantification of Western blots was done using NIH Image (version 1.61) software. Isolation of nuclear and cytoplasmic cell fractions to detect ERK activation was performed essentially as described previously (Chen et al., 1992).
Protein kinase A activity assay. Determination of PKA kinase activity was performed using a solid-phase ELISA that uses a specific synthetic peptide (Arg-Arg-Xaa-Ser/Thr or Lys-Arg-Xaa-Xaa-Ser/Thr) as a substrate for PKA and a polyclonal antibody that recognizes the phosphorylated form of the substrate. The assay was designed for the analysis of PKA activity in the solution phase (Stressgen Bioreagents, Victoria, British Columbia, Canada). CGNs treated with specific agents were lysed after the indicated time points. Five micrograms of total protein extract were used for the assay, which was performed as described in the manufacturer's manual. Results were expressed as relative units of absorbance (450 nm).
Statistical analysis. Data were expressed as means ± SE. All of the experiments were performed in triplicate or duplicate and repeated at least twice. Statistical analysis among groups was performed using Student's t test (Origin 6.1). Values of p are as follows: */0p < 0.05, **/00p < 0.01, ***/000p < 0.001.
Results
IGF-1-stimulated ERK activation is not involved in neuronal survival
CGNs undergo neuronal death when medium is changed from high (25 mm) to low (5 mm) potassium (D'Mello et al., 1993). IGF-1 (100 ng/ml) was added to CGN cultures immediately after the potassium change. We used three markers for monitoring cellular damage, PI to monitor PM damage, enzymatic TUNEL staining to monitor DNA damage, and DAPI staining to monitor morphological changes of nuclei (Subramaniam et al., 2004). The number of degenerated neurons was counted after 24 h. IGF-1 markedly inhibited the cellular damage induced by low potassium (Fig. 1A,B).

IGF-1-stimulated ERK activation is not involved in neuronal survival. A, IGF-1 prevents low-K+-induced CGN degeneration. After the switch from high to low potassium, IGF-1 (100 ng/ml) was added, and the total numbers of cells stained with PI (PM damage), TUNEL (DNA damage), and condensed nuclei were determined after 24 h (see Materials and Methods). B, Quantification of the different forms of cellular damages. C, IGF-1-induced ERK activation is blocked by MEK inhibitors PD98059 (PD) and U0126. PD (50 μm) and U0126 (10 μm) were preadministered for 30 min before IGF-1 addition for 10 min. Cell extracts were processed for activated ERK (pERK) and total ERK (ERK). D, PD or U0126 do not affect IGF-1-promoted neuronal survival. PD (50 μm) and U0126 (10 μm) were coapplied with IGF-1, and cell death was determined after 24 h. Error bars indicate SE. ***p < 0.001 compared with low-potassium cultures.
Next, we tested the possible effect of IGF-1 on ERK activation and its involvement in IGF-1-promoted survival. IGF-1 (100 ng/ml) was added to CGN cultures in low potassium for 10 min, and cell extracts were processed for Western blotting to detect phosphorylated ERK (active, pERK). As shown in Figure 1C, IGF-1 induced a robust ERK activation in low potassium. Pretreatment with the MEK/ERK pathway inhibitors PD98059 (50 μm) or U0126 (10 μm) abrogated ERK activation by IGF-1 (Fig. 1C). To examine the possible involvement of ERK in IGF-1-promoted CGN survival, ERK activation was blocked with MEK/ERK inhibitors, and cell death was analyzed after 24 h. Treatment with PD98057 or U0126 did not interfere with IGF-1-mediated neuronal survival (Fig. 1D). As previously demonstrated (Subramaniam et al., 2003), MEK inhibitors alone prevented cell death induced by low potassium (Fig. 1D). Together, these data suggest that (1) IGF-1 induces an early activation of ERK, and (2) ERK is unlikely to be involved in IGF-1-dependent survival, because both MEK inhibitors failed to interfere with the IGF-1-mediated survival effect.
IGF-1 induces rapid and transient ERK but prevents slow and sustained ERK activation observed in low-potassium culture
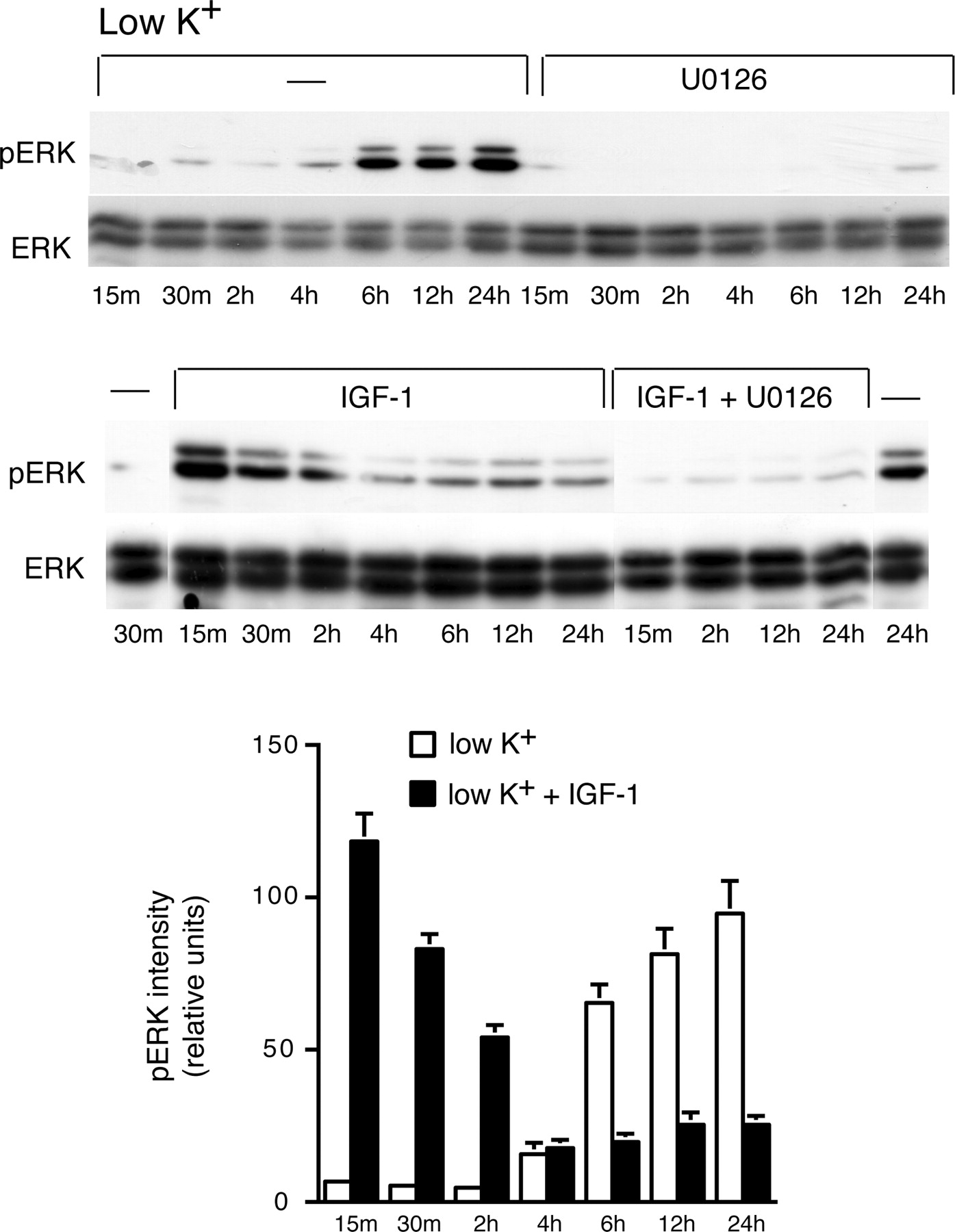
The above finding that MEK inhibitors failed to antagonize IGF-1-promoted survival prompted us to investigate whether IGF-1-induced survival may depend on the inhibition of sustained ERK activation that promoted CGN death (Subramaniam et al., 2003). To examine this, IGF-1 (100 ng/ml) was applied to CGNs, and cell extracts prepared at various time points were processed for the detection of ERK activation by Western blotting. As shown in Figure 2, ERK activation (pERK) was observed 6 h after the potassium change (low K+) and was sustained up to 24 h. Activation was attenuated by the MEK inhibitor U0126 (10 μm). IGF-1 induced a rapid activation of ERK when added immediately after the potassium change; this lasted for up to 2 h. At this time point, untreated low-K+ cultures revealed almost no activation of ERK (Fig. 2). However, the sustained ERK activation induced after 6 h in low potassium was dramatically inhibited by IGF-1 treatment (Fig. 2). U0126 attenuated both the low-potassium- and the IGF-1-induced activation of ERK (Fig. 2). Next, we investigated whether IGF-1-mediated inhibition of low-potassium-induced sustained ERK is required for IGF-1-mediated prevention of PM damage. IGF-1-treated CGNs were infected with recombinant adenovirus expressing CA MEK (Ad-CA MEK) and EGFP (Ad-GFP), each at 350 multiplicity of infection. As shown in Figure 3, A and B, a dramatic increase in PM-damaged neurons was observed in Ad-CA MEK-infected neurons compared with EGFP-infected neurons. This effect was associated with an increased activation of ERK in CA MEK-infected neurons in IGF-1-treated cultures (Fig. 3C). Together, these data suggest that IGF-1 has a dual effect on ERK: it induces an early and transient ERK activation, and downregulates a second induction of slow and sustained ERK observed in low-potassium CGN cultures, which is essential for IGF-1-mediated prevention of PM damage.

Role of IGF-1 in low-potassium-induced sustained ERK activation. IGF-1 induces a rapid and transient rise in ERK but prevents sustained ERK in low potassium. After the potassium change (low K+), CGN extracts were isolated from cultures treated with U0126 (10 μm), IGF-1 (100 ng/ml), and IGF-1 plus U0126. Western blotting was done to detect activated ERK (pERK) and total ERK (ERK). Error bars indicate SE.

Adenoviral-mediated CA MEK (Ad-CA MEK) overexpression attenuates IGF-1-inhibited PM damage. A, Fluorescence microscopy of CGNs infected with Ad-GFP, Ad-CA MEK infected CGNs. An antibody against HA epitope was used to detect CA MEK. B, Quantification of the percentage of total PI-positive cells in Ad-GFP- and in Ad-CA MEK-infected cultures. **p < 0.01 compared with Ad-GFP+PI alone. 000p < 0.001 compared with Ad-GFP+PI in IGF-1-treated cultures. Error bars indicate SE. C, Western blot analysis for pERK in Ad-GFP- and Ad-CA MEK-infected cultures.
Withdrawal or readdition of IGF-1 fails to interfere with the ERK-cell death pathway
We next investigated whether ERK inhibition is directly dependent on the continuous presence of IGF-1. If so, withdrawal of IGF-1 after 4 h should reinitiate the ERK activation. To examine this, IGF-1 was added to CGN cultures immediately after the potassium change. After 4 h, culture plates were washed three times and replaced with a medium without IGF-1. Cell extracts isolated from growth factor treated, and deprived, and control cultures were subjected to detection of ERK activation by Western blotting. IGF-1 withdrawal at 4 h inhibited the sustained ERK activation in low-potassium cultures; however, this inhibition did not last for >10 h (Fig. 4A). The shift in the induction of sustained ERK activation (pERK) was observed at ∼10 h rather than the usual 6 h (Fig. 4A). To investigate whether reactivation of ERK is followed by cell death, we quantified cell death by counting the total number of condensed nuclei. In these cultures, cell death was similar to control cultures at 24 h (Fig. 4B). Thus, constant inhibition of the ERK-cell death pathway requires the continued presence of IGF-1.

A, IGF-1 withdrawal reactivates ERK. IGF-1 (100 ng/ml) was administered for 4 h after the potassium change (low K+). Cultures were washed three times, and culture medium was replaced without IGF-1. Extracts cultures plus or minus IGF-1 were processed for Western blotting of active ERK (pERK) and total ERK (ERK). B, IGF-1 withdrawal induces cell death identical with that seen in untreated low-K+ cultures. After 4 h of IGF-1 treatment in low potassium, IGF-1 was withdrawn. Cell death was determined (number of condensed nuclei) after 24 h of potassium change (low K+). C, Right panel, Addition of growth factor at 6 h after the potassium change fails to prevent low-potassium-induced ERK. Six hours after the potassium change (low K+), CGN cultures were stimulated with IGF-1 for 1, 7, and 13 h (corresponding to 7, 13, and 19 h after potassium change), and cell extracts were processed for activated ERK (pERK) and total ERK (ERK). Left panel, The relative intensity level of pERK. **p < 0.01 was compared with low K+ alone. D, Addition of IGF-1 at 6 h after the potassium change fails to prevent low-potassium-induced cell death. IGF-1 was added at 6 h after the potassium change, and numbers of condensed nuclei were counted after 24 h. Error bars indicate SE. E, IGF-1R, a receptor for IGF-1, is not altered after potassium change. RT-PCR was performed to amplify IGF-1R and β-actin (see Materials and Methods). F, Western blot analysis to detect IGF-1Rα before potassium switch (0 h) and after potassium switch (6, 9, 12, 24 h).
Because ERK activation is observed at ∼6 h after the potassium change, we tested whether treatment with IGF-1, started at 6 h after the potassium change, inhibits ERK activation. IGF-1 was added at 6 h after the potassium change, and cell extracts were prepared after 1, 7, and 13 h of IGF-1 treatment, respectively (these time intervals correspond to 7, 13, and 19 h after potassium change). ERK activation (pERK) levels in IGF-1-treated cultures at 7 h after potassium change was significantly higher (p < 0.01) compared with untreated low-K+ cultures (Fig. 4C). However, at 13 and 19 h, ERK activation levels were similar to those of untreated low-K+ cultures. This suggests that IGF-1 retains the ability to induce ERK activation even if added at 6 h after the potassium change but failed to inhibit the slow and sustained ERK activation. IGF-1 applied at 6 h after the potassium change also failed to prevent cell death, determined at 24 h (Fig. 4D).
We hypothesized that the inability of IGF-1 to prevent ERK activation after 13 h after the potassium change is attributable to the downregulation of its receptor mRNA and protein. To test this hypothesis, we performed RT-PCR for IGF-1R (a receptor for IGF-1) from RNA isolated from CGNs, cultured in high- and low-potassium medium. Expression of IGF-1R was not altered after the potassium change. The level of IGF-1R at 6, 12, and 24 h is apparently comparable with that of high-potassium cultures (Fig. 4E). Western blot analysis to detect IGF-1Rα, a receptor subunit of IGF-1, revealed that IGF-1Rα expression was not altered after the potassium change (Fig. 4F). The level of IGF-1Rα at 6, 9, 12, and 24 h is apparently comparable with that of high-potassium cultures (0 h). This suggests that the failure of IGF-1 to prevent ERK activation or prevent cell death, if added after 6 h, is probably not attributable to downregulation of its receptor expression. After 6 h in low potassium, CGNs are irreversibly committed to die (Miller and Johnson, 1996); therefore, it is conceivable that the readdition of IGF-1 no longer could act on the cell death pathways. Together, these data suggest that the IGF-1-dependent inhibition of ERK activation is time dependent. IGF-1 no longer inhibits ERK or rescues neurons after a certain time point in low potassium. Addition of IGF-1 at 4 h after the potassium change (i.e., before the low-potassium-induced ERK activation) also failed to prevent ERK activation and cell death (data not shown).
PI3K and Akt inhibitors differentially interfere with ERK inhibition and IGF-1-mediated neuronal survival
The PI3K/Akt pathway mediates IGF-1-induced neuronal survival in various neuronal populations, including CGNs (Linseman et al., 2002; Zheng et al., 2002; Fukunaga and Kawano, 2003). We tested whether the PI3K/Akt pathway plays a role in IGF-1-mediated ERK inhibition to promote CGN survival. As shown in Figure 5A, IGF-1 induced the phosphorylation (activation) of Akt, which was abrogated by two structurally unrelated but specific PI3K inhibitors, LY (10 μm) and W (100 nm). IGF-1-mediated survival was quantified in the presence of LY or W after 24 h. Percentages of neurons with PM damage (PI), DNA damage (TUNEL), and condensed nuclei were significantly increased in the presence of either LY or W in IGF-1-treated cultures (Fig. 5B). Furthermore, we tested whether the specific Akt inhibitors (Akt-I and Akt-IV) (Hu et al., 2000; Kau et al., 2003) can interfere with IGF-1-mediated survival. Akt-I (10 μm) and Akt-IV (10 μm) completely abrogated the IGF-1-mediated inhibition of DNA damage without affecting the PM damage (Fig. 5B). Together, these data suggest that IGF-1-induced activation of the PI3K pathway regulates both PM damage and DNA damage, whereas its downstream target Akt regulates DNA damage only. Note that nuclear condensation, the step preceding PM or DNA damage (Subramaniam et al., 2004), was also increased by PI3K and Akt inhibitors. None of these inhibitors had any apparent effect on neuronal degeneration in untreated low-K+ cultures (Fig. 5B).

Role of PI3K on IGF-1-promoted neuronal survival. A, IGF-1 induces Akt phosphorylation. After the potassium change, CGNs were stimulated with IGF-1, alone or in combination with the PI3K inhibitor LY (10 μm) or W (100 nm). B, The PI3K inhibitor and the Akt inhibitor (Akt-I or Akt-IV) differentially affect IGF-1-promoted CGN survival. LY (10 μm) or W (100 nm) or Akt-I (10 μm) or Akt-IV (10 μm) were coapplied with IGF-1 (100 ng/ml), immediately after the potassium change (low K+), and cell death was analyzed after 24 h as mentioned in Materials and Methods. PI, PM damage; TUNEL, DNA damage. ***p < 0.001 was compared with IGF-1 alone. Error bars indicate SE. C, Top panel, Role of PI3K and Akt on ERK and caspase-3 activation. W, LY, and Akt-IV were added to CGNs; IGF-1 was administered for 10 min, 12 h, and 24 h to detect pERK and caspase-3. Bottom panel, The density determination for pERK and caspase-3 levels. Error bars indicate SE.
The apparent difference in the effect of PI3K and Akt inhibitors on neuronal degeneration prompted us to investigate their role on the activation of ERK (which promotes PM damage) and caspase-3 (which promotes DNA damage) (Subramaniam et al., 2004). PI3K or Akt inhibitors were applied for 30 min, 12 h, and 24 h followed by stimulation with IGF-1 for 10 min, 12 h, and 24 h. Cell extracts were subjected to the detection of ERK and caspase-3 activation by Western blotting. Treatment with IGF-1 induced an early and transient induction of ERK (Fig. 5C). PI3K inhibitors, LY or W, abrogated IGF-1-induced transient ERK activation. In contrast, the Akt inhibitor Akt-IV did not affect the transient IGF-1-induced ERK activation. Caspase-3 activation (cleavage) was observed after 6 h after the potassium switch (Subramaniam et al., 2004). IGF-1 inhibited activation of the caspase-3 observed in low-K+ cultures at 12 and 24 h tested (Fig. 5C). The PI3K inhibitors LY or W reversed the inhibitory effect of IGF-1 on sustained ERK and caspase-3 activation (Fig. 5C). Interestingly, a specific Akt inhibitor, Akt-IV, had no significant effect on IGF-1-mediated sustained ERK inhibition (Fig. 5C). However, Akt-IV almost completely abrogated IGF-1-mediated caspase-3 inhibition, to an extent similar to LY or W (Fig. 5C). Together, these data suggest that IGF-1 inhibits caspase-3 via PI3K in an Akt-dependent manner, and inhibits slow and sustained ERK activation via PI3K in an Akt-independent manner. In addition, PI3K but not Akt, regulates early and transient ERK activation induced by IGF-1.
IGF-1 induces c-Raf phosphorylation at the inhibitory site Ser259
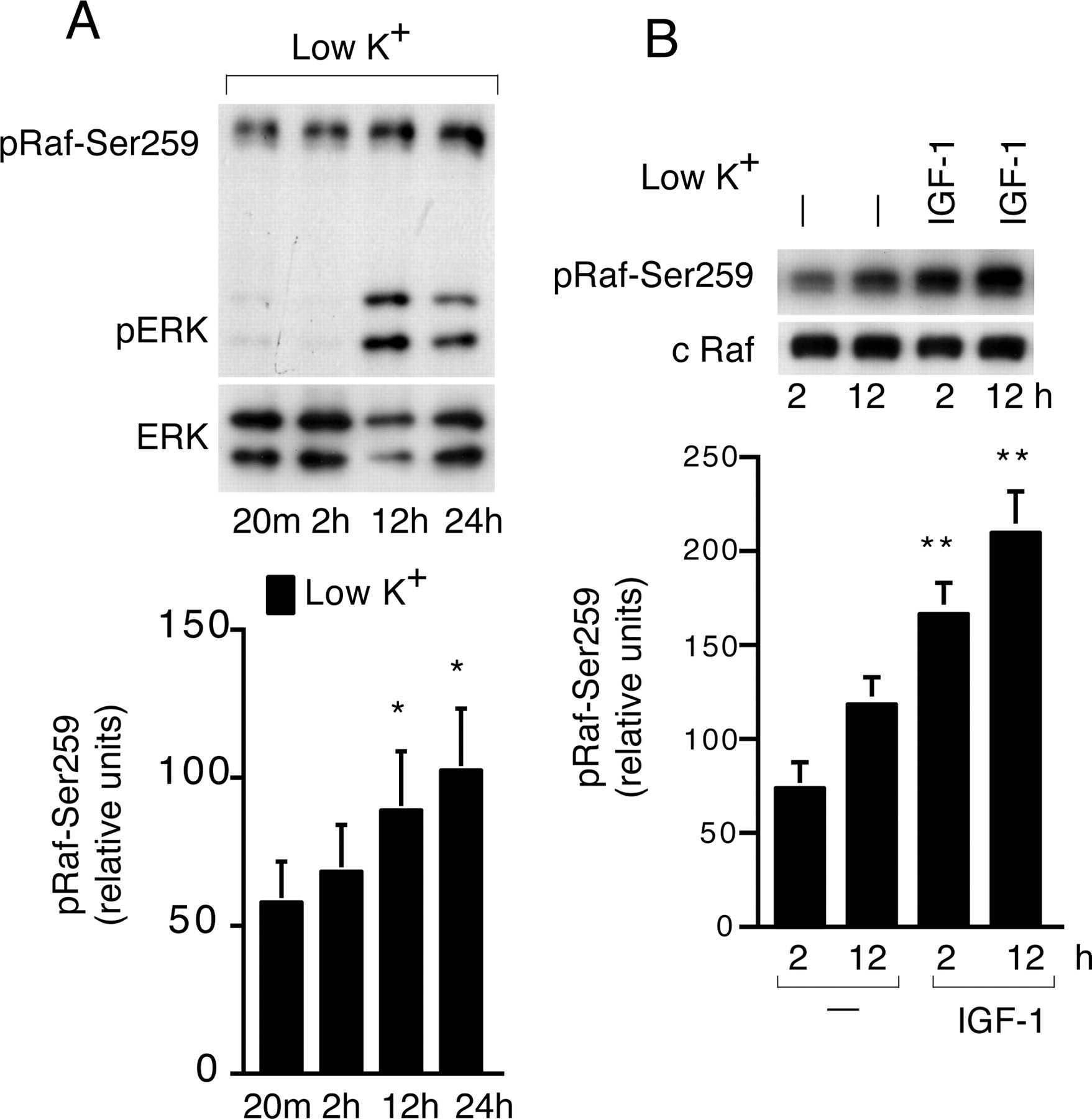
c-Raf, an upstream activator of the MEK/ERK pathway, has been shown to downregulate the MEK/ERK pathway when it is phosphorylated at an inhibitory site Ser259 in non-neuronal cells. Phosphorylation of c-Raf-Ser259 was shown to be mediated either by Akt or PKA (Zimmermann and Moelling, 1999; Dhillon et al., 2002). First, we tested the status of c-Raf-Ser259 phosphorylation in low-potassium CGN cultures in relation to sustained ERK activation (pERK). Cell extracts were isolated at various time points and subjected to Western blotting using phospho-c-Raf-Ser259 followed by pERK antibodies. c-Raf-Ser259 phosphorylation (pRaf-Ser259) was not altered in low potassium up to 2 h (at that time point, no ERK activation was detected) but was significantly increased (p < 0.05) at 12 and 24 h (at these time points, high ERK activation was detected) (Fig. 6A). Whether this increase in pRaf-Ser259 in low-potassium cultures is causally linked to the sustained ERK activation was addressed in the next series of experiments.

Regulation of pRaf-Ser259 in CGNs. A, Low potassium induces c-Raf-Ser259 phosphorylation (pRaf-Ser259). After the change to low potassium, CGN extracts were isolated at different time points and processed for the detection of pRaf-Ser259 and ERK activation (pERK), and total ERK (ERK) by Western blotting (top panel). The relative intensity of pRaf-Ser259 level (bottom panel). *p < 0.05 compared with 2 h. B, IGF-1 induces pRaf-Ser259. IGF-1 (100 ng/ml) was added, and the extracts were prepared after 2 and 12 h to detect pRaf-Ser259 and total c-Raf (c-Raf) by Western blotting (top panel). The relative intensity of c-Raf-Ser259 phosphorylation level (bottom panel). **p < 0.01 compared with untreated cultures (-). Error bars indicate SE.
First, we tested the effect of IGF-1 on pRaf-Ser259. IGF-1 induced a robust pRaf-Ser259 compared with low potassium, both at 2 and 12 h (Fig. 6B). To examine the signaling pathways involved in IGF-1-mediated pRaf-Ser259, we tested the effect of the PI3K (LY, 50 μm), Akt (Akt-IV, 10 μm), and PKA inhibitor (PKI, 20 μm) (Graves et al., 1993). Each inhibitor was coapplied with IGF-1, and cell extracts were isolated and subjected to Western blotting to detect pRaf-Ser259 and ERK activation. As shown in Figure 7A, LY had no apparent effect on pRaf-Ser259 induced by IGF-1 (at 2 h); however, LY inhibited IGF-1-induced activation of ERK (at 2 h). PKI prevented IGF-1-induced pRaf-Ser259 at 2 h but did not affect activation of ERK. Akt-IV did not affect either IGF-1-induced pRaf-Ser259 or ERK at 2 h (Fig. 7A). On the contrary, both LY and PKI markedly decreased pRaf-Ser259 induced by IGF-1 (Fig. 7A) and reversed the IGF-1-induced sustained ERK inhibition at 12 and 24 h. Akt-IV did not affect either IGF-1-induced pRaf-Ser259 or IGF-1-induced sustained ERK inhibition at 12 and 24 h (Fig. 7A). Different inhibitors of PI3K (wortmannin, 100 nm), Akt (Akt-I, 10 μm), and PKA (H89; 5 μm) showed a similar effect on IGF-1-regulated pRaf-Ser259 and ERK activation (Fig. 7B). None of the PI3K inhibitors or the Akt inhibitor affected the low-potassium-induced pRaf-Ser259 or ERK (data not shown). However, the PKA inhibitor (PKI; 20 μm), despite reducing pRaf-Ser259 at both early (2 h) and late time points (12 h), potentiated only the late and sustained ERK activation observed in low potassium (Fig. 7C). The basal ERK level was not affected by PKI at 2 h (Fig. 7C).

PI3K inhibitors (LY and W), Akt inhibitor (Akt-IV), and PKA inhibitor (PKI) differentially affect IGF-1-induced pRaf-Ser259 and ERK inhibition. A, Top panel, After the potassium change (low K+), CGNs were treated with IGF-1, alone or in combination with LY (50 μm), Akt-IV, (10 μm), or PKI (20 μm). CGN extracts were prepared after 2, 12, and 24 h and processed for detection of activated pRaf-Ser259 and ERK (pERK), and total ERK (ERK) by Western blotting. Bottom panel, Relative intensity of pERK and pRaf-Ser259. Error bars indicate SE. B, Role of different inhibitor of PI3K (Wort; 100 nm), PKA (H89; 5 μm), and Akt (Akt-I; 10 μm) on IGF-1-induced pRaf-Ser259 and ERK. C, The effect of PKA inhibitor PKI (20 μm), on pRaf-Ser259 and pERK at 2 and 12 h in low-potassium culture.
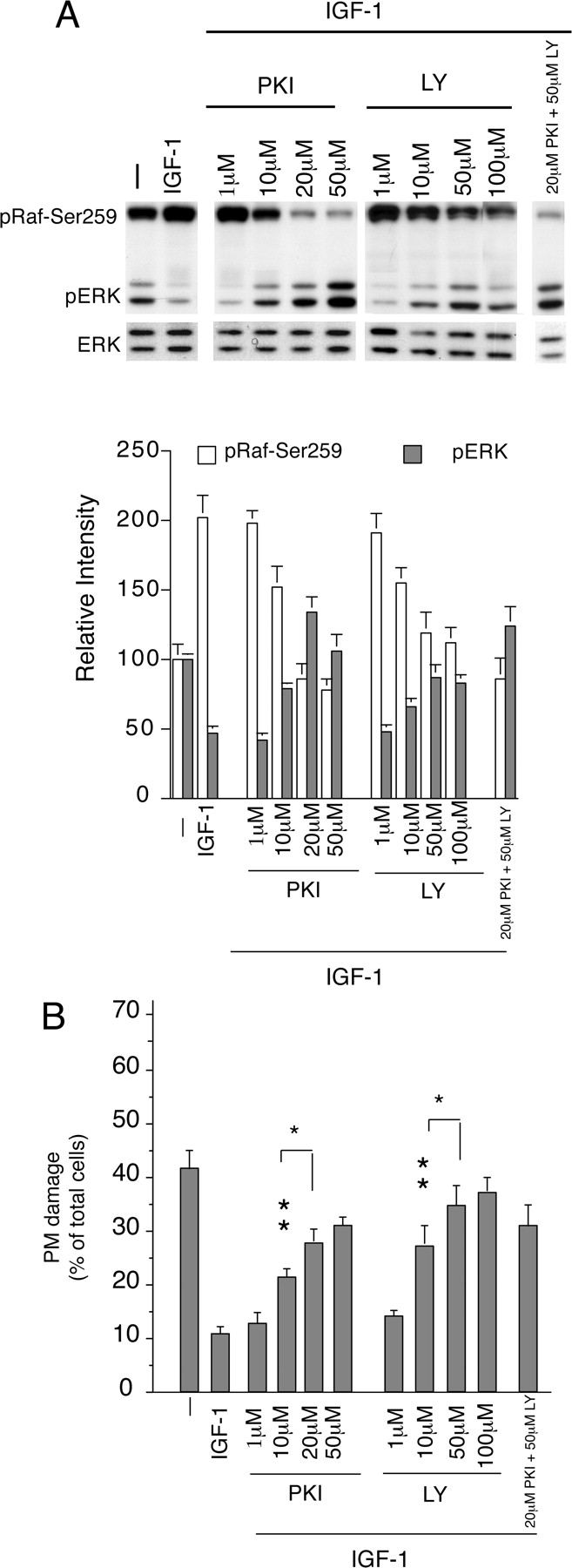
In addition, we investigated the concentration-dependent effect of PKI and LY on IGF-1-induced pRaf-Ser259, pERK inhibition, and PM damage. As shown in Figure 8 A, PKI and LY decreased pRaf-Ser259 and increased pERK in a concentration-dependent manner. PKI maximally inhibited IGF-1-induced pRaf-Ser259 between 20 and 50 μm, and LY between 50 and 100 μm. PKI and LY both attenuated the IGF-1 effect on PM damage in a concentration-dependent manner (Fig. 8B). We tested the specificity of inhibitors that were used in our experiments by checking their effect on pRaf-Ser259, phospho-Akt, phospho-glycogen synthase kinase (GSK), and phospho-ERK. As shown in Figure 9, the PI3K inhibitors (W and LY) specifically inhibit pRaf-Ser259, phospho-Akt, and phospho-GSK but induce the activation of ERK in IGF-1-treated CGN cultures. The PKA inhibitors (H89 and PKI) decreased IGF-1-induced pRaf-Ser259 and increased ERK activation, but apparently did not affect GSK and Akt. The Akt inhibitors (Akt-IV and Akt-I) did not affect IGF-1-induced pRaf-Ser259 and ERK inhibition; however, both inhibitors attenuated the activation of GSK, suggesting that Akt inhibitors act by suppressing downstream targets of Akt. Note that both Akt-IV and Akt-I inhibited Akt phosphorylation, although the magnitude of inhibition was somewhat stronger with Akt-IV compared with Akt-I.

A, Top panel, Concentration-dependent effect of PKA inhibitor (PKI) and PI3K inhibitor (LY) on IGF-1-induced activation of pRaf-Ser259 and inhibition of pERK. Bottom panel, The density determination for pRaf-Ser259 and pERK levels. B, Concentration-dependent effect of PKI and LY on IGF-1-mediated inhibition of PM damage. Error bars indicate SE. *p < 0.05 versus IGF-1 plus (10 μm) inhibitor; **p < 0.01 versus IGF-1 alone.

Specificity of W (100 nm), LY (50 μm), H89 (5 μm), PKI (20 μm), Akt-IV (10 μm), and Akt-I (10 μm) on IGF-1-induced pRaf-Ser259, Akt, and GSK, and on ERK inhibition. Western blotting was performed after 9 h of administration of IGF-1 together with inhibitors. The blot was probed with antibodies to pRaf-Ser259 and pERK followed by antibodies to pAkt and pGSK. The bottom panel indicates the relative intensity of the bands.
Together, these data imply that (1) PI3K and PKA play a crucial role in IGF-1-induced pRaf-Ser259 and IGF-1-mediated inhibition of ERK, (2) Akt does not affect either IGF-1-induced pRaf-Ser259 or IGF-1-mediated inhibition of ERK, (3) the PKA and PI3K inhibitors affected the effect of IGF-1 on CGNs in a concentration-dependent manner, and (4) the inhibitors used exhibited specificity with respect to their inhibitory effect.
IGF-1-induced pRaf-Ser259 is essential for the prevention of PM damage
To study the significance of IGF-1-induced pRaf-Ser259 in preventing PM damage, we expressed the mutant Raf-S259A using a Sindbis viral-mediated gene expression system. The Sindbis virus expression system expressing mutant c-Raf-S259A was generated as described in Materials and Methods. More than 80% of CGNs were infected with Sindbis virus expressing c-Raf-S259A (Sin-Raf-S259A) or EGFP (Sin-EGFP) (Fig. 10). As shown in Figure 10, A and B, coexpression of Sin-Raf-S259A in IGF-1-treated cultures completely abrogated the effect of IGF-1 on PM damage. More than 50% of Sin-Raf-S259A-infected CGNs were positive for PM damage compared with Sin-EGFP-expressing CGNs (∼10%) (Fig. 10A,B). Sin-Raf-S259A alone potentiated low-potassium-induced PM damage compared with Sin-EGFP (∼65% vs ∼50%). Sin-Raf-S259A expression increased ERK activation both in low potassium alone and IGF-1-treated cultures (Fig. 10C). Note that Sin-Raf-S259A expression attenuated phosphorylation of endogenous c-Raf-Ser259 (Fig. 10C). Together, these data suggest that phosphorylation of c-Raf on S259 is an essential event in the prevention of neuronal PM damage.

Sindbis viral-mediated Raf Ser259Ala (Sin-Raf-S259A) overexpression attenuates IGF-1-mediated inhibition of PM damage. A, Fluorescence microscopy of CGNs infected with Sin-EGFP or Sin-Raf-S259A. An antibody (M5) against the FLAG epitope was used to detect Raf-S259A. B, Quantification of the percentage of total PI-positive cells in Sin-EGFP- and in Sin-Raf-S259A-infected neurons. **p < 0.01 compared with Sin-EGFP+PI alone. 000p < 0.001 compared with Sin-EGFP+PI in IGF-1-treated cultures. Error bars indicate SE. C, Western blot analysis for the detection of pERK in Sin-EGFP- and Sin-Raf-S259A-infected cultures.
Low K+ induces PKA activity and is enhanced by IGF-1 in a PI3K-dependent manner
The ability of the PKA inhibitor to inhibit c-Raf-Ser259 phosphorylation and induce ERK activation both in untreated and in IGF-1-treated cultures suggests the presence of PKA activity in low-K+ cultures. Therefore, we next investigated the temporal induction of PKA activity in these cultures. Low-K+ cultures showed a slow increase in PKA activity that was observed after 4 h, and remained high until 24 h (Fig. 11A). Addition of IGF-1 further enhanced PKA activity compared with untreated low-K+ cultures, up to 24 h tested (Fig. 11A). Next, we determined the effect of the PI3K inhibitor (LY; 10 μm), the Akt inhibitor (Akt-I; 10 μm), and the PKA inhibitor (H89; 5 μm) on PKA activity at 12 h. Cotreatment with H89 abolished PKA activity both in untreated and IGF-1-treated cultures (Fig. 11B). LY had no effect on PKA activity in untreated cultures. However, it did diminish PKA activity in IGF-1-treated cultures (Fig. 11B). Akt-I did not affect PKA activity in both untreated and IGF-1-treated cultures (Fig. 11B). Another inhibitor of Akt, Akt-IV, also did not affect PKA activity (data not shown). The attenuation of PKA activity by the PKA inhibitor H89 suggests that the activity observed was mostly contributed by PKA. To further confirm this, we used another PKA inhibitor, PKI. PKI (20 μm) was added to the cultures, and the PKA activity was measured in the presence and absence of PKI. The presence of PKI strongly reduced (∼80%) the activity observed in low-potassium- and in IGF-1-treated cultures, suggesting that the activity observed is mostly contributed by PKA (Fig. 11C). Together, these data suggest that low potassium induces a slow increase in PKA activity, which was further enhanced by IGF-1 in a PI3K-dependent and Akt-independent manner.

A, Low-potassium-induced PKA activity is further enhanced by IGF-1. After the potassium change (low K+), CGN extracts were isolated in the presence and in the absence of IGF-1. PKA activity was measured as indicated in Materials and Methods. **p < 0.01, ***p < 0.001 compared with low K+ at 2 h. 00p < 0.01, 000p < 0.001 compared at respective time points in low K+. B, The PI3K inhibitor (LY) and the PKA inhibitor (H89), but not Akt inhibitor (Akt-I), abolish PKA activity. After the potassium change (low K+), CGNs were treated with IGF-1 and LY (50 μm), Akt-I (10 μm), or H89 (5 μm). CGN extracts were isolated at 12 h and processed for PKA activity as mentioned in Materials and Methods. **p < 0.01 compared with respective untreated cultures (-); ***p < 0.001. C, Role of the PKA inhibitor (PKI) on PKA activity. Error bars indicate SE.
PKA does not interfere with DNA damage in CGNs
Because treatment with the PKA inhibitors (H89; PKI) resulted in a decreased c-Raf-Ser259 phosphorylation in association with increased ERK, we tested the effect of H89 (5 μm) and PKI (20 μm) on neuronal degeneration. H89 and PKI potentiated PM damage and nuclear condensation without affecting DNA damage in untreated low-K+ cultures (Fig. 12A). When H89 and PKI were applied to IGF-1-treated cultures, they abolished the ability of IGF-1 to prevent PM damage and nuclear condensation but did not interfere with DNA damage. The inability of the PKA inhibitors to affect DNA damage, in light of our previous finding that DNA damage is regulated by caspase-3 (Subramaniam et al., 2004), may suggest that PKA might not interfere with caspase-3 activation. To test this, cell extracts of H89- and PKI-treated cultures maintained in the presence or in the absence of IGF-1 were subjected to Western blotting to detect caspase-3. H89 or PKI did not affect IGF-1-mediated inhibition of caspase-3 (Fig. 12B). Note that the coapplied Akt inhibitor (Akt-I) abolished the effect of IGF-1 on caspase-3, loaded as a control.

A, The PKA inhibitors (H89, PKI) potentiate PM damage. Cell death was analyzed in IGF-1-treated and in untreated low-K+ cultures in the presence of H89 (5 μm) and PKI (20 μm) after 24 h, as mentioned in Materials and Methods. PI, PM damage; TUNEL, DNA damage. Significance, 000p < 0.001 compared with IGF-1 alone. *p < 0.05, **p < 0.01 compared with untreated cultures (-). Error bars indicate SE. B, H89 does not affect caspase-3 activation. Detection of caspase-3 by Western blotting in IGF-1-treated and in untreated cultures in the presence of H89, 12 h after the potassium change. The Akt inhibitor (Akt-I; 10 μm) reversed the effect of IGF-1 on caspase-3 (cas-3). C, 8-Br-cAMP, an activator of PKA, prevents PM damage. 8-Br-cAMP (500 μm) was added at the time of the potassium change (low K+), and degeneration was determined as described in Materials and Methods. ***p < 0.001 compared with untreated cultures (-). D, 8-Br-cAMP induces c-Raf-Ser259 phosphorylation and inhibits ERK without affecting caspase-3. After the potassium change, CGNs were treated with 8-Br-cAMP (500 μm), and cell extracts were prepared after 2 and 12 h to detect activated ERK (pERK) and caspase-3 (cas-3).
Because inhibition of PKA decreased c-Raf-Ser259 phosphorylation (Fig. 11B,C), and potentiated ERK activation and its associated PM damage (Fig. 8A,B), we hypothesized that activation of PKA itself should be able to induce c-Raf-Ser259 phosphorylation and prevent activation of ERK and PM damage. Western blotting and cell death analysis were performed to detect phospho-c-Raf-Ser259, pERK, and caspase-3 in cultures treated with 8-Br-cAMP (500 μm), an activator of PKA. As expected, 8-Br-cAMP increased the phosphorylation of c-Raf-Ser259, and decreased ERK activation and its associated PM damage (Fig. 12C,D). Note that caspase-3 or DNA damage was not affected by 8-Br-cAMP. Together, this suggests that activation of PKA in low potassium may involve negative regulation of ERK and its associated PM damage but has no role in caspase-3 activation or caspase-3-associated DNA damage.
8-Br-cAMP compensates for the role of PI3K in PKA induction in IGF-1-plus-LY-treated cultures
The above experiments had provided evidence that blocking PI3K (using the inhibitor LY) also blocked IGF-1-induced PKA activity (Fig. 11B). Furthermore, the PKA inhibitors blocked IGF-1-induced c-Raf-Ser259 phosphorylation (Fig. 8A). Together, these data only indirectly suggested that IGF-1-induced PI3K stimulates phospho-c-Raf-Ser259 via PKA, to inhibit ERK activation. To establish a direct link between PI3K and PKA, we blocked the IGF-1-mediated PI3K effect on PKA by LY, and stimulated PKA by 8-Br-cAMP. LY abolished c-Raf-Ser259 phosphorylation and ERK inhibition induced by IGF-1. However, combined addition of LY and 8-Br-cAMP failed to interfere with IGF-1-stimulated c-Raf-Ser259 phosphorylation and ERK inhibition (Fig. 13A). This suggests that 8-Br-cAMP acts independent of PI3K. Note that cotreatment with LY and 8-Br-cAMP prevented IGF-1-mediated caspase-3 inhibition and Akt activation (Fig. 13A). Coapplication of LY and 8-Br-cAMP also abolished the effect of IGF-1 on DNA damage, without affecting PM damage (Fig. 13B), suggesting that PKA activity alone was sufficient to control PM damage. We also confirmed that the 8-Br-cAMP concentration used was sufficient to induce PKA activity, which was comparable with that induced by IGF-1 (Fig. 13C).

8-Br-cAMP compensates the effect of PI3K on PKA. A, After the potassium change (low K+), CGNs were treated with 8-Br-cAMP (500 μm), IGF-1 (100 ng/ml) alone, or IGF-1 plus LY (10 μm), 8-Br-cAMP plus IGF-1, or 8-Br-cAMP plus IGF-1 plus LY. CGN extracts were isolated after 9 h and processed for Western blot analysis for phospho c-Raf-Ser259, activated ERK (pERK), activated Akt (pAkt), and caspase-3 (cas-3). B, Quantification of CGN death in the above-mentioned additives. **p < 0.001 compared with IGF-1 plus LY. C, 8-Br-cAMP induces PKA. After the potassium change, CGNs were treated with 8-Br-cAMP (500 μm) or with IGF-1 (100 ng/ml). After 9 h, extracts were assayed for PKA activity as mentioned in Materials and Methods. Error bars indicate SE. ***p < 0.001 versus untreated (-).
ERK is differentially regulated in response to IGF-1 and low potassium alone
Our observation that there is a marked difference in temporal and duration patterns of ERK activation induced by IGF-1 and by low potassium prompted us to investigate additional putative cellular and molecular differences. We therefore tested the cellular distribution of activated ERK in the CGN model system. After IGF-1 stimulation for 5 min, 30 min, 2 h, and 24 h, CGN cultures were processed for immunocytochemistry for activated ERK, using monoclonal antibodies against pERK. Total numbers of neurons positive for pERK and numbers of neurons with nuclear translocated ERK were counted. In IGF-1-treated cultures, >80% of neurons at 2 h displayed pERK immunoreactivity (transient ERK), localized to the cytoplasm (Fig. 14A,B). At 24 h, pERK immunoreactivity was dramatically reduced compared with untreated, low-K+ cultures. However, in low-K+ cultures, high pERK immunoreactivity (sustained ERK) was observed at 24 h, and ∼35% of neurons showed nuclear pERK (Fig. 14A,B). In addition, we performed Western blot analysis for the detection of pERK in the cytoplasmic and nuclear fractions of CGNs treated with or without IGF-1. As shown in Figure 14C, a high nuclear expression of activated ERK was observed in low-potassium cultures. In contrast, in IGF-1-treated cultures, high levels of activated ERK were found in the cytoplasmic fraction.

IGF-1-induced (early) transient ERK and low-K+-induced (late) sustained ERK are differentially regulated. A, IGF-1-induced ERK remains in the cytoplasm. IGF-1 (100 ng/ml) was added to CGN cultures after the potassium switch (low K+). Cultures were processed to detect activated ERK (pERK) by immunocytochemistry after 5 min, 30 min, 2 h, and 24 h. Similarly, untreated cultures at 2 and 24 h were processed for pERK detection. DAPI was added for nuclear staining. The arrowhead indicates pERK immunoreactivity in the cytoplasm. Arrow indicates pERK immunoreactivity in the nucleus. B, Shown are the percentage of total cells that are positive for pERK (total pERK) and percentage of cells that are positive for nuclear pERK. C, Western blot to detect cytoplasmic pERK and nuclear pERK. D, Akt-I (5 μm), H89 (10 μm), CHX (10 μg/ml), 8-Br-cAMP (500 μm), U0126 (10 μm), and LY (10 μm) were applied 30 min before stimulation with IGF-1 (30 min), and pERK was detected by Western blotting. Similarly, untreated cultures were processed for pERK detection in the presence of additives after 12 h.
Furthermore, hints concerning differences and similarities between IGF-1-induced ERK and low-K+-induced ERK were obtained from experiments using various inhibitors [LY, Akt-I, cyclohexamide (CHX), and U0126] and the activator 8-Br-cAMP (Fig. 14D). Together, our data suggest that IGF-1-induced ERK can be distinguished from low-potassium-induced ERK by the differential cellular distribution and differential regulation.
In summary, our data demonstrate that IGF-1 not only induces ERK but also inhibits ERK in the same neuronal cell. Although the role of the early ERK induction remains to be investigated, the inhibition of ERK appears to be an important regulatory mechanism to promote survival. Although ERK is induced by IGF-1 as well as in low potassium alone, the two distinct modes of induction are subjected to complex regulations of cell signaling players. In addition, we demonstrated a novel role of the PI3K/Akt/PKA/c-Raf pathways in IGF-1-mediated neuronal survival. Supplemental Figure 15 (available at www.jneurosci.org as supplemental material) summarizes the mechanism of IGF-1-mediated CGN survival. IGF-1 stimulates PI3K, which activates both Akt and PKA. Akt controls caspase-3 and DNA damage, whereas PKA controls ERK and PM damage via c-Raf phosphorylation at the inhibitory site Ser259.
Discussion
Interaction between various signaling pathways ultimately determines the outcome of cellular functions. The observation that ERK and caspase-3 promote distinct cellular demise in CGNs (Subramaniam et al., 2004) has fostered efforts directed at understanding how the survival factor IGF-1 regulates ERK and caspase-3 in CGNs. IGF-1 inhibited both ERK and caspase-3 through PI3K, because both PI3K inhibitors abolished the effect of IGF-1. PI3K is an established important mediator of IGF-1-promoted survival in various neuronal systems, including CGNs (D'Mello et al., 1997; Miller et al., 1997; Kermer et al., 2000; Zheng et al., 2002). However, a different role of PI3K in controlling a distinct form of cellular damage in neurons has not been addressed. This report provides evidence that PI3K induces PKB/Akt and PKA as its downstream targets, both being involved in regulating two distinct forms of neuronal damage. Akt regulates DNA damage, and PKA controls PM damage. Akt is a well established target for PI3K (Burgering and Coffer, 1995). PKA has not been recognized as a PI3K target in neurons before. However, in non-neuronal cells, PI3K can induce activation of PKA, and PI3K-mediated induction of adenylate cyclase and PKA has been demonstrated previously (Edinger et al., 1999; Shpakov et al., 2002; Nguyen et al., 2003). However, PKA can apparently also act upstream to PI3K, and can inhibit PI3K in both neuronal and non-neuronal cells. Thus, cAMP-PKA activation was shown to inhibit PI3K and induce cortical neuronal injury (Poser et al., 2003). Inhibitory cross talk between the cAMP and PI3K signal transduction cascades differentially affecting proliferation and differentiation has been described in different non-neuronal cell lines (Ahmed et al., 1995; Kim et al., 2001; Wang et al., 2001; Mei et al., 2002). Interestingly, in the rescue of sympathetic neurons, NGF induces both PI3K and PKC pathways; however, inhibition of either one of the pathways failed to completely abrogate NGF-dependent rescue, suggesting that both PI3K and PKC synergize to promote NGF-mediated sympathetic neuron survival (Pierchala et al., 2004). Thus, growth factors promote neuronal survival by inducing varied intracellular signaling repertoires.
Previous studies have established that cAMP can promote the survival of different neuronal populations, including CGNs (Rydel and Greene, 1988; D'Mello et al., 1993; Hanson et al., 1998). However, the intracellular network that regulates cAMP-dependent pathways is not completely understood. In most cell types, PKA downregulates Ras/ERK signaling by interfering with Ras-mediated activation of Raf-1 (Cook and McCormick, 1993; Erhardt et al., 1995). However, in melanocytes and PC12 cells, an increase in the cAMP concentration activates the Ras/ERK pathway through the GTP-binding protein Rap-1 and/or B-Raf (Englaro et al., 1995; Vossler et al., 1997; Yao et al., 1998). We show here that the cAMP-PKA pathway elicits an inhibitory effect on ERK activation through c-Raf-Ser259 phosphorylation to promote neuronal survival, specifically to prevent PM damage. Previously, Akt was shown to mediate inhibition of c-Raf by phosphorylation at Ser259 (Rommel et al., 1999), yet this cross talk appears to occur only in certain cell types (Zimmermann and Moelling, 1999). Notably, we observed that PI3K, rather than Akt, is involved in c-Raf inhibition, because PI3K inhibitors, but not the specific Akt inhibitors, prevented IGF-1-mediated c-Raf-Ser259 phosphorylation. Although LY (PI3K inhibitor) selectively inhibited PI3K/Akt, it did not interfere with PKA/c-Raf-Ser259 phosphorylation, when it was coapplied with 8-Br-cAMP (activator of PKA). This suggests that PKA activation mediated by PI3K is involved in c-Raf-Ser259 phosphorylation. One study in COS-1 cells has shown that Ser259 is a target for phosphorylation by PKA, and that it is the main site mediating PKA-induced c-Raf inhibition, which negatively controls ERK activity (Dhillon et al., 2002). This is consistent with our present study, suggesting that PI3K regulation of PKA and c-Raf-Ser259 phosphorylation also operate in neurons.
As physiological regulators, phosphatases play a crucial role in inactivating ERK (Camps et al., 2000; Zhou et al., 2002). Although we did not address the issue of an involvement of phosphatases in our model, okadaic acid (OA), a serine/threonine phosphatase inhibitor has been demonstrated to induce death of CGNs and hippocampal neurons (Fernandez-Sanchez et al., 1996; Runden et al., 1998). In hippocampal neurons, OA-induced cell death is accompanied by sustained activation of ERK. Inhibition of the ERK pathway prevented OA-induced cell death in this model, suggesting that cell death, followed by phosphatase inhibition, involves an upregulation of ERK activity (Runden et al., 1998). It is therefore conceivable that low potassium in CGNs may decrease the phosphatase activity, thereby releasing its constraints on ERK activation. IGF-1 may increase phosphatase activity and thereby downregulate sustained ERK. In support of this notion, we observed that IGF-1 induces MKP3 (mitogen-activating protein kinase phosphatase 3), activation in CGNs (our unpublished observation). In addition, protein phosphatase 2A (PP2A), a stress-responsive phosphatase, has been shown to be induced after a death stimulus, and has been shown to be critically involved in the dephosphorylation of S259, a step required for Raf activation (Abraham et al., 2000). Although the status of PP2A activation in the CGN low-potassium model is not clear, the possibility that IGF-1 can inhibit PP2A activation, which in turn fails to dephosphorylate Raf at S259, cannot be ruled out. The present study shows an involvement of PKA in phosphorylating Raf S259, suggesting that ERK regulation via S259 Raf may involve direct activation by kinases rather than downregulation of phosphatases, in growth factor-mediated neuronal survival. Raf activation has shown to be regulated by multiple phosphorylation sites. Studies with Raf mutants have suggested that Ser338 and Thr341 are required and operate in Raf-1 activation (Mason et al., 1999). Ser338 phosphorylation has been shown to be induced by growth factors (Diaz et al., 1997; King et al., 1998). The effect of IGF-1 on Ser338 or Thr341 sites in our model is not known. In addition, it is also possible that sustained ERK activation in low-potassium CGN cultures is not achieved through Raf-1 (c-Raf) activation. B-Raf has been implicated in inducing sustained ERK activation in various non-neuronal models (York et al., 1998; Tsukamoto et al., 2004). It is therefore tempting to speculate that both positive and negative regulations of ERK in CGNs might be orchestrated by the members of the Raf family of proteins, B-Raf and c-Raf, respectively.
The dual regulation of ERK by IGF-1 to initially activate and subsequently inhibit ERK signaling within the same neuronal population has not been documented before. However, the importance of differential kinetics of ERK activation by epidermal growth factor (EGF) and NGF in rat PC12 cell has been described. EGF induces transient ERK phosphorylation, with no significant nuclear translocation, and thereby promotes proliferation. However, NGF induces sustained ERK phosphorylation and nuclear translocation that promotes PC12 cell differentiation (Qiu and Green, 1991; Traverse et al., 1992; Nguyen et al., 1993). Similarly, we observed that IGF-1-induced transient ERK remains in the cytoplasm, whereas low-potassium-induced sustained ERK translocates to the nucleus, which is protein synthesis dependent. This observation may suggest that sustained ERK activation has distinct consequences for gene expression compared with transient ERK activation, because nuclear translocation of ERK may result in the activation of transcription factors that promote cell death.
Furthermore, sensitivity of IGF-1-induced ERK and low-potassium-induced ERK to various inhibitors differed considerably. Although activation of ERK by IGF-1 is PI3K dependent and CHX independent, activation of ERK by low potassium is PI3K independent and CHX dependent. On the contrary, as described in Results, the suppression of low-potassium-induced sustained ERK by IGF-1 is PI3K dependent, suggesting that IGF-1-induced PI3K has a dual role: it inhibits slow and sustained ERK through the PKA/c-Raf pathway but also activates early ERK through a yet-unknown mechanism. The possibility that PAK (p21-activated kinase), a downstream target of PI3K, can induce MEK/ERK via activation of Rac, has been demonstrated for human kidney fibroblasts (Frost et al., 1996, 1997). Nevertheless, how PI3K establishes this dual function in CGNs requires additional investigations.
What is the role of the growth factor-mediated early rise in ERK? Is it essentially required for the prevention of cell death, or just a prelude to the inhibition of the late sustained ERK activation? This question is difficult to address. To our knowledge, there are no tools available that would specifically block the IGF-1-induced rise in ERK without interfering with the induction of ERK by low potassium. The PI3K inhibitor LY294002, which specifically blocked IGF-1-induced ERK, without affecting the low-potassium-induced ERK (Fig. 14), might be a candidate to test. However, it should be noted that the PI3K inhibitor also prevents CGN survival by suppressing Akt signaling. Thus, the precise role of the early transient induction of ERK is currently unclear. Nevertheless, it is worth noting that growth factor-induced ERK might activate signals that are involved in differentiation, synaptic plasticity, and long-term memory formation rather than neuronal survival (Lin and Bulleit, 1997; Impey et al., 1999; Bulleit and Hsieh, 2000; Schinder and Poo, 2000). Together, these findings shed new light on the mechanisms of IGF-1 signaling in mediating neuronal survival.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (STR 616/1-2) and by a fellowship to S.S. from DFG Graduiertenkolleg 791, University of Heidelberg, National Cancer Institute of Canada and Canadian Institutes for Health Research supported D.K. Ministry for Science and Culture of Lower Saxony supported N.S. and R.B. We thank Jutta Fey for technical assistance.
Correspondence should be addressed to Dr. Srinivasa Subramaniam, Neuroanatomy and Interdisciplinary Center for Neurosciences, University of Heidelberg, Im Neuenheimer Feld 307, D-69120 Heidelberg, Germany. E-mail: srinivasa.subramaniam{at}urz.uni-heidelberg.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252838-15$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}