

Abstract
It has been proposed that four members of the plexin A subfamily (plexin-As; plexin-A1, -A2, -A3, and -A4) and two neuropilins (neuropilin-1 and neuropilin-2) form complexes and serve as receptors for class 3 secreted semaphorins (Semas), potent neural chemorepellents. The roles of given plexin-As in semaphorin signaling and axon guidance, however, are mostly unknown. Here, to elucidate functions of plexin-A4 in semaphorin signaling and axon guidance events in vivo, we generated plexin-A4 null mutant mice by targeted disruption of the plexin-A4 gene. Plexin-A4 mutant mice were defective in the trajectory and projection of peripheral sensory axons and sympathetic ganglion (SG) axons and the formation of the anterior commissure and the barrels. The defects in peripheral sensory and SG axons were fundamentally related to those of neuropilin-1 or Sema3A mutant embryos reported but were more moderate than the phenotype in these mutants. The growth cone collapse assay showed that dorsal root ganglion axons and SG axons of plexin-A4 mutant embryos partially lost their responsiveness to Sema3A. These results suggest that plexin-A4 plays roles in the propagation of Sema3A activities and regulation of axon guidance and that other members of the plexin-A subfamily are also involved in the propagation of Sema3A activities. Plexin-A4-deficient SG axons did not lose their responsiveness to Sema3F, suggesting that plexin-A4 serves as a Sema3A-specific receptor, at least in SG axons. In addition, the present study showed that plexin-A4 bound class 6 transmembrane semaphorins, Sema6A and Sema6B, and mediated their axon-repulsive activities, independently of neuropilin-1. Our results imply that plexin-A4 mediates multiple semaphorin signals and regulates axon guidance in vivo.
Introduction
In the development of nervous systems, axons navigate particular paths toward their final targets after interacting with various axon guidance cues of either an attractive or repulsive nature that are provided by intermediate targets. Semaphorin (Sema) is a family of well known axon guidance molecules. To date, >20 semaphorins of secreted or membrane forms have been identified and divided into seven classes (Semaphorin Nomenclature Committee, 1999). Among them, class 3 secreted semaphorins, in particular Sema3A and Sema3F, are well characterized neural chemorepellents (Luo et al., 1993; Fan and Raper, 1995; Püschel et al., 1995, Chédotal et al., 1998; Chen et al., 1998) and regulate nerve fiber projection in vivo (Taniguchi et al., 1997; Sahay et al., 2003).
Two molecular families, the neuropilin and the plexin, serve as receptors for class 3 semaphorins. Two neuropilins, neuropilin-1 and neuropilin-2, have been identified in vertebrates (Kawakami et al., 1996; Chen et al., 1997; Kolodkin et al., 1997) and bind class 3 semaphorins except Sema3E (Chen et al., 1997; He and Tessier-Lavigne, 1997; Kolodkin et al., 1997; Gu et al., 2005). Dorsal root ganglion (DRG) axons of neuropilin-1 gene knock-out mouse embryos completely lose their responsiveness to Sema3A (Kitsukawa et al., 1997). Sympathetic ganglion (SG) axons of neuropilin-2 mutant embryos lose their responsiveness to Sema3F (Chen et al., 2000; Giger et al., 2000). Moreover, these neuropilin-deficient mice are defective in nerve fiber projections, suggesting a requirement of neuropilins in the signaling of class 3 semaphorins and axon guidance.
Neuropilins themselves, however, do not propagate activities of class 3 semaphorins into cells (Nakamura et al., 1998) and must form receptor complexes with plexins of subfamily A (plexin-A subfamily) (Takahashi et al., 1999; Tamagnone, 1999). In developing nervous tissues, two neuropilins and four members of the plexin-A subfamily (plexin-As; plexin-A1, -A2, -A3, and -A4) (Kameyama et al., 1996a,b; Maestrini et al., 1996; Suto et al., 2003) are expressed in various patterns (Murakami et al., 2001; Suto et al., 2003), raising a question, that is, what combinations of neuropilins and plexins are involved in the signaling of given class 3 semaphorins and guidance of given axons in vivo? Studies on plexin-A3 knock-out mice have revealed that the neuropilin-2/plexin-A3 complex serves as a receptor for Sema3F (Cheng et al., 2001) and plays a role in axon pruning in the hippocampus (Bagri et al., 2003). Roles of plexin-As and neuropilins in the signaling of class 3 semaphorin and axon guidance, however, are far from being understood.
In this study, we produced plexin-A4 mutant mice by targeted disruption of the plexin-A4 gene and elucidated functions of plexin-A4 in semaphorin signaling and axon guidance in vivo. Phenotypic analysis of plexin-A4 mutant mice reveals the requirement of plexin-A4 for the proper projection of peripheral sensory axons and SG axons and the formation of the anterior commissure and the barrels. In vitro analyses on the responsiveness of plexin-A4-deficient DRG and SG axons to class 3 semaphorins suggest that plexin-A4 is required for the propagation of repulsive activities of Sema3A but not Sema3F. In addition, we reveal that plexin-A4 serves as a direct receptor for two class 6 transmembrane semaphorins, Sema6A and Sema6B, both of which are repellents for SG axons. The present results indicate that plexin-A4 can propagate repulsive activities of both secreted and transmembrane semaphorins and plays roles in a wide variety of axon guidance events.
Materials and Methods
Histological procedures. Embryos and brains of postnatal mice were fixed with 4% paraformaldehyde (PFA) in PBS. Cryostat sections (20 μm thick) were thaw-mounted on poly-l-lysine (Sigma, St. Louis, MO)-coated glass slides. The anterior limb of the anterior commissure [pars anterior (acA)] (see Results) was detected with a monoclonal antibody against Tag-1 (4D7/TAG-1; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) and cyanine 3-labeled anti-mouse IgM (1:200 dilution; Jackson ImmunoResearch, West Grove, PA) as a secondary antibody. Detection of the barrels by cytochrome oxidase histochemistry in tangential and coronal sections (50 μm thick) of postnatal day 1 (P1) brains was performed following procedures reported previously (Ulupinar et al., 1999). Immunohistochemistry using anti-tyrosine hydroxylase (TH) antibodies (a gift from Dr. T. Nagatsu, Fujita Health University, Toyoake, Japan) and whole-mount immunostaining of embryos with an anti-neurofilament antibody 2H3 (Dodd et al., 1988) were performed as described previously (Kitsukawa et al., 1997).
Isolation of mouse Sema6A and Sema6B cDNAs and in situ hybridization. cDNA clones encoding mouse Sema6A and Sema6B were isolated by reverse transcription-PCR (RT-PCR) on embryonic brain mRNAs. Primers for the RT-PCR were constructed based on the sequences of mouse Sema6A cDNA (Xu et al., 2000) and rat Sema6B (r-Sema Z) cDNA (Kikuchi et al., 1997). Nonradioactive digoxygenin (DIG)-labeled cRNA probes for Sema6A (bases 502-1047 of the Sema6A cDNA) and Sema6B (bases 1-833 of Sema6B cDNA) were generated using DIG labeling mix (Roche, Mannheim, Germany) according to the manufacturer's recommendations. In situ hybridization (ISH) using the DIG-labeled probes was performed as described previously (Suto et al., 2003). The sense RNA probes gave no significant ISH signals. ISH for plexin-A4 has been reported previously (Suto et al., 2003).
Production of recombinant semaphorin proteins. The sequences corresponding to the ectodomain of mouse Sema6A (Sema6Aect; amino acids 18-648) and mouse Sema6B (Sema6Bect; amino acids 26-599) were amplified and inserted into the Aptag-4 vector (a gift from Dr. J. Flanagan, Harvard Medical School, Boston, MA) (AP-Sema6Aect; AP-Sema6Bect) (Flanagan et al., 2000). To dimerize recombinant proteins, fragments encoding AP-Sema6Aect and AP-Sema6Bect were inserted into pEF-Fc (AP-Sema6Aect-Fc; AP-Sema6Bect-Fc; the pEF-Fc expression vector was a gift from Dr. S. Nagata, Osaka University). Human embryonic kidney 293T (HEK293T) cells were transfected with pEF-AP-Sema6Aect-Fc, pEF-AP-Sema6Bect-Fc, pCAGGS-AP-Sema3A (Kitsukawa et al., 1997) (the pCAGGS expression vector was a gift from Dr. J. Miyazaki, Osaka University), or pSec Tag1-human Sema3F [human Sema3F-AP (Chen et al., 1998); a gift from Dr. M. Tessier-Lavigne, Stanford University, Stanford, CA] using Lipofectamine Plus (Invitrogen, Carlsbad, CA) and cultured in DMEM containing 10% fetal bovine serum (FBS) for 5-7 d in 5% CO2 at 37°C. Culture supernatants were collected and filtered using 0.22 μm filters.
Binding of Sema6A and Sema6B to plexin-A4. L-cells that stably express the full-length mouse plexin-A4 proteins (see below) were incubated with 250 μl of HBSS with 0.5 mg/ml BSA, 0.1% NaN3, and 20 mm HEPES, pH 7.0 (HBHA solution) (Flanagan and Leder, 1990) containing 1% FBS and the AP-Sema6Aect-Fc or AP-Sema6Bect-Fc recombinant proteins (the culture supernatant) for 1 h on ice. After removal of the HBHA solution, cells were treated with 250 μl of 10 mm Tris-HCl, pH 8.0, supplemented with 0.1% Triton X-100 to dissolve the recombinant proteins bound to the cell surface. The cell lysates were subjected to colorimetric analysis to measure AP activity as described previously (Flanagan and Leder, 1990).
Binding of Sema6A to cultured neurons. SG explant cultures (see below) were incubated with the AP-Sema6Aect-Fc recombinant proteins (the culture supernatant) for 1 h at 37°C, washed five times with HBHA solution, and then postfixed with PFA. The specimens were washed with PBS, kept at 65°C for 1 h to inactivate endogenous AP activity, washed with 10 mm Tris-HCl, pH 7.8, supplemented with 0.9% NaCl and 0.1% Triton X-100, and then developed with nitroblue-tetrazolium-chloride/5-bromo-4-chlor-indolyl-phosphate.
Growth cone collapse assay. DRGs of embryonic day 12.5 (E12.5) mouse embryos and SGs from E13.5 mouse embryos were cultured in DH10 medium (a mixture of 45% DMEM, 45% Ham's F-12, and 10% FBS) supplemented with 2.5S NGF (50 ng/ml). Culture dishes were pre-treated with 100 μg/ml poly-l-lysine overnight at 4°C and with 10 μg/ml mouse laminin for 2 h at room temperature. After 8 h (for DRGs) and 18 h (for SGs), the AP-Sema3A, Sema3F-AP, or AP-Sema6Aect-Fc recombinant proteins (culture supernatants) were added to the cultures for 1 h.
Axon avoidance assay. SGs of E15.5 embryos were cocultured with aggregates of Sema3F-expressing HEK293T cells (see above) in collagen gel for 2 d, following procedures reported previously (Chen et al., 1998). In this assay, 100 ng/ml 2.5S NGF was added to the cultures. Cultures were immunostained with an anti-β-tubulin antibody (a gift from Dr. Y. Arimatsu, Mitsubishi Kagaku Institute of Life Sciences, Machida, Japan).
Cell-contraction assay. L-cells, mouse fibroblastic line cells, were transfected with expression plasmids for the myc-tagged full-length plexin-A4 protein or the cytoplasmic segment-deleted plexin-A4 protein (Suto et al., 2003). L-cells were cultured with DH10 medium. Cell lines that stably expressed the plexin-A4 proteins were isolated by immunostaining with an anti-myc antibody, 9E10 (Evan et al., 1985). The plexin-A4-expressing cells were incubated with the AP-Sema6Aect-Fc recombinant proteins (15 nm; culture supernatant) for 1 h.
Neurite outgrowth assay. To isolate L-cells that stably express Sema6B (L6B), the cells were cotransfected with expression plasmids for the myc-tagged full-length Sema6B protein and pST-neoB (Katoh et al., 1987) using Effectene (Qiagen, Hilden, Germany), selected with Geneticin (Invitrogen), and stained with the anti-myc antibody. Small explants of SGs of E13.5 embryos were cultured on monolayer sheets of L-cells or L6B cells for 24 h.
Results
Trajectories and projections of peripheral sensory fibers are abnormal in plexin-A4 mutant mice
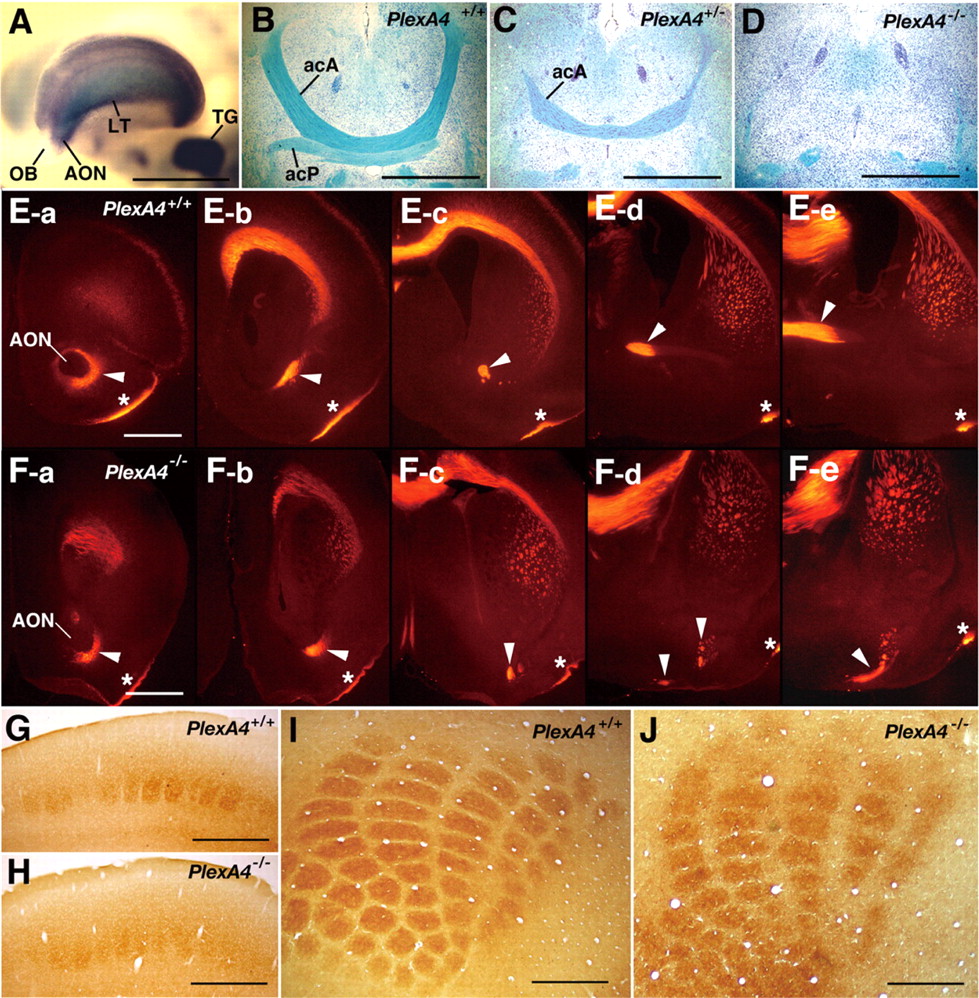
Plexin-A4 is expressed in embryonic cranial and spinal sensory ganglia and most motor nuclei of the brainstem (Suto et al., 2003) (also see Fig. 5A). Therefore, we generated plexin-A4 null mutant mice by targeted disruption of the plexin-A4 gene (supplemental Fig. 1 and Table 1, available at www.jneurosci.org as supplemental material) and examined the trajectories and projections of peripheral nervous system (PNS) fibers.

Expression of Sema6A, Sema6B, and plexin-A4 in mouse embryos and binding of Sema6A and Sema6B to plexin-A4. A, Expression of Sema6A, Sema6B, and plexin-A4 in E12.5 mouse embryos, detected by ISH on adjacent coronal sections. Skeletal muscles (arrows) including the anterior vertebrate muscle (AVM) show strong ISH signals for Sema6A and Sema6B but not plexin-A4. Sema6A transcripts are also detected in connective tissues surrounding the vertebrae (V) (arrowheads). In contrast, plexin-A4 ISH signals are detected solely in SGs and DRGs. SC, Spinal cord. B, Binding of the AP-tagged recombinant proteins for the ectodomain of Sema6A (AP-Sema6Aect-Fc) and Sema6B (AP-Sema6Bect-Fc) to plexin-A4-expressing L-cells, detected by AP histochemistry, is shown. C, Quantitative analysis on the binding of AP-Sema6Aect-Fc to plexin-A4. To obtain specific binding of the recombinant proteins to plexin-A4, the AP activities derived from the binding of AP-Sema6Aect-Fc to parental L-cells were subtracted. The protein concentration for AP-Sema6Aect-Fc was converted as a monomer. D, Scatchard plots derived from data in C. The estimated KD for the binding of AP-Sema6Aect-Fc to plexin-A4 is 3.56 nm. E, Binding of AP-Sema6Aect-Fc to cultured SG neurons of E13.5 wild-type (PlexA4+/+) and plexin-A4 homozygous (PlexA4-/-) embryos, detected by AP histochemistry. Scale bars: A, 500 μm; B, 100 μm; E, 200 μm.
Immunostaining of embryos with an anti-neurofilament antibody, 2H3, in whole-mount preparations showed that trajectories of the cranial and spinal nerves were abnormal in all plexin-A4 homozygous (PlexA4-/-) embryos examined (n = 8), although the position and configuration of the cranial and spinal ganglia were normal. In PlexA4-/- embryos, the ophthalmic nerve fibers overshot and fanned out in their peripheral target fields (Fig. 1B, E, O). The distal parts of the maxillary, mandibular, facial, glossopharyngeal, and vagus nerves were also disarrayed (Fig. 1B). No obvious defect of axon guidance was detected in the oculomotor or trochlear nerves (Fig. 1B, E). The pathways and projections of the spinal nerves in PlexA4-/- embryos were also disorganized: several 2H3-positive fibers grew into the skin surface in an irregular manner (Fig. 1H), although segmental patterns of skin-sensory innervation were mostly retained. Spinal nerve fibers within developing limbs in PlexA4-/- embryos were partially disarrayed and sometimes overshot (Fig. 1K). Labeling of DRGs of plexin-A4 mutant embryos at E15.5 with the lipophilic dye DiI revealed no apparent abnormality in the projection of DRG afferents within the spinal cord (data not shown).

Defects in pathway and projection of peripheral nerve fibers in plexin-A4 and neuropilin-1 mutant mouse embryos. Whole-mount immunostaining with anti-neurofilament antibody 2H3 of wild-type (PlexA4+/+), plexin-A4 heterozygous (Pl-exA4+/-), and homozygous (PlexA4-/-) embryos and neuropilin-1 homozygous (Nrp1-/-) embryos at E11.5 (A-C) and E12.5 (D-O) is shown. A-F, Pathways and projections of the cranial nerves. III, The oculomotor nerve; IV, the trochlear nerve; V, the trigeminal nerve and ganglia; VII, the facial nerve; IX, the glossopharyngeal nerve; X, the vagus nerve; op, the ophthalmic nerve; mx, the maxillary nerve; ma, the mandibular nerve. G-I, Pathways and projections of the spinal nerves at the trunk level. Arrowheads and arrows in G indicate penetration points of the lateral branches and the lateral cutaneous branches of skin-sensory fibers, respectively. df, Dorsal funiculus. J-L, Trajectory of the spinal nerves within the forelimbs is shown. The arrowhead in J indicates the penetration point of the skin-sensory rami. The arrow in K indicates overshooting fibers. M-O, The ophthalmic nerve in wild-type (M; a high-power picture of D), plexin-A4 heterozygous (N), and plexin-A4 homozygous (O; a high-power picture of E) embryos. Arrows in N and O indicate aberrant fibers. Scale bars: (in A-O) 1 mm.
The phenotype in the PNS of plexin-A4 mutant embryos, however, was moderate compared with that of neuropilin-1 mutant embryos. Overshooting of the ophthalmic nerve was less prominent in the PlexA4-/- embryos than neuropilin-1 homozygous (Nrp1-/-) embryos (Fig. 1, compare B, C, and E, F). Overshooting of spinal nerve fibers toward the opposite side of embryos after crossing the dorsal midline was a common phenotype in Nrp1-/- embryos (Kitsukawa et al., 1997) (Fig. 1I) but not in PlexA4-/- embryos (Figs. 1H). It is worth noting that abnormal trajectories and projections of the cranial and spinal nerves were also observed in all plexin-A4 heterozygous (Plex-A4+/-) embryos examined (n = 15) but were less severe than those in homozygous littermates (Fig. 1M-O).
Projection of sympathetic ganglion axons is abnormal in plexin-A4 mutant mouse embryos
Plexin-A4 was also expressed in the sympathetic ganglia (Suto et al., 2003) (also see Fig. 5A). Immunohistochemistry with anti-TH antibodies revealed an aberrant projection of SG axons in all PlexA4-/- embryos at E12.5 (n = 5) (Fig. 2B, C). The abnormal projection of SG axons was prominent in PlexA4-/- embryos at E15.5: several TH-positive axons invaded the anterior vertebrate muscles located just medial to SGs and connective tissues surrounding the vertebrae (Fig. 2F-H). We reported previously that more than one-half of SG neurons in neuropilin-1 and Sema3A mutant embryos failed to be accumulated into ganglia and were displaced instead (Kawasaki et al., 2002) (Fig. 2D). However, a few SG neurons were displaced in plexin-A4 mutant embryos. The sympathetic nervous system of PlexA4+/- embryos did not show any abnormality.

Defects in the sympathetic nervous system in plexin-A4 and neuropilin-1 mutant mice. Frontal sections of embryos at E12.5 (A-D) and at E15.5 (E-H) were immunostained with an anti-TH antibody and counterstained with toluidine blue. Arrowheads indicate aberrant TH-positive SG axons that grow into the anterior vertebral muscle (AVM) or connective tissues surrounding the vertebrae (V). Scale bars, 200 μm.
The anterior commissure fails to decussate normally in plexin-A4 mutant mice
The anterior commissure is comprised of an anterior limb, which is a tract connecting the left and right anterior olfactory nuclei, and a posterior limb [pars posterior (acP)], which is a tract connecting the temporal lobes of both sides. ISH analysis indicated a prominent expression of plexin-A4 in the anterior olfactory nucleus and lateral telencephalon of embryos at E13.5 when axons from these regions are actively growing to form the anterior commissure (Fig. 3A). Therefore, we analyzed the morphology of the anterior commissure in wild-type and plexin-A4 mutant mice. Luxol fast blue staining of horizontal brain sections showed that both the acA and acP tracts were absent in all adult PlexA4-/- mice examined (n = 7) (Fig. 3D). The anterior commissure was also abnormal in PlexA4+/- mice: the acA tract was thin in all mice examined (n = 5), and the acP tract was missing in two mice and extremely thin in three mice (Fig. 3C). To determine whether axons forming the anterior commissure are missing or displaced in plexin-A4 mutants, serial coronal sections of P1 mouse brains were immunostained with an anti-Tag-1 antibody, which is a marker for the acA tract (Cowan et al., 2004). In wild-type mice, the Tag-1-positive acA tract ran dorsomedially from the anterior olfactory nucleus (Fig. 3E-c, E-d) and then crossed the midline of the forebrain (Fig. 3E-e). In contrast, in PlexA4-/- mice, Tag-1-positive fibers from the anterior olfactory nucleus never ran dorsally but shifted ventrally (Fig. 3F-c,F-d) and then turned medially at the ventral surface of the forebrain (Fig. 3F-d, F-e). These results indicate that deficiency of plexin-A4 induces a pathfinding error for the axons of the anterior commissure.

Expression of plexin-A4 in the forebrain and defects in the anterior commissure and the barrel formation in plexin-A4 mutant mice. A, Expression of plexin-A4 in the forebrain of an embryo at E13.5, detected by whole-mount ISH. Strong ISH signals are detected in the anterior olfactory nucleus (AON), the lateral telencephalon (LT), and the trigeminal ganglion (TG) but not the olfactory bulb (OB). B-D, The anterior commissure in adult wild-type and plexin-A4 mutant mice. Horizontal sections were stained with Luxol fast blue to detect myelinated fibers and counterstained with cresyl violet. E, F, The acA tract of the anterior commissure in wild-type and plexin-A4 mutant mice at P1. Serial coronal sections were immunostained with an anti-Tag-1 antibody. The sections are aligned in the rostrocaudal order from a to d. Arrowheads indicate the acA tracts. Asterisks indicate the lateral olfactory tract. G-J, The barrels detected by histochemistry for cytochrome oxidase in coronal (G, H) and tangential (I, J) sections of adult brains of wild-type (G, I) and plexin-A4 mutant (H, J) mice. Scale bars: A-D, 1 mm; E-J, 500 μm.
Formation of the barrels is abnormal in plexin-A4 mutant mice
Trigeminal sensory inputs from the mystacial vibrissas are somatotopically represented as the barrels in layer 4 of the S1 facial area of the somatosensory cortex (Woolsey and Van der Loos, 1970). Because plexin-A4 is strongly expressed in neuronal components of the trigeminal sensory pathways, including the trigeminal ganglia (Fig. 3A), the trigeminal brainstem nuclei, and the ventroposteromedial nucleus (VPM) in the thalamus (Suto et al., 2003), we examined whether formation of the barrels is defective in plexin-A4 mutant mice. Cytochrome oxidase histochemistry in the adult brains showed that the boundary of each barrel was indistinct in all PlexA4-/- mice examined (n = 8), although the number and arrangement of the barrels were mostly normal (Fig. 3G-J). The whisker pattern was normal in plexin-A4 mutant mice (data not shown).
Plexin-A4 is involved in Sema3A signaling
The abnormal trajectories and projections of PNS efferent fibers and SG axons induced in plexin-A4 mutant embryos are closely related to the phenotype of neuropilin-1 and Sema3A mutant embryos (Kitsukawa et al., 1997; Taniguchi et al., 1997; Kawasaki et al., 2002) and suggest that plexin-A4 forms a receptor complex with neuropilin-1 to mediate the axon-repulsive activities of Sema3A. To test this possibility, we examined the responsiveness of plexin-A4-deficient and neuropilin-1-deficient DRG and SG axons to Sema3A with the growth cone collapse assay.
Sema3A induced growth cone collapse in both DRG and SG axons, depending on the dosage of the plexin-A4 gene: a significant deficit in the Sema3A response was observed for axons of PlexA4-/- and PlexA4+/- embryos, although the response was not completely missing in the axons of PlexA4-/- embryos (Fig. 4A, C). The results contrasted with the findings that growth cones of DRG and SG axons of Nrp1-/- embryos were never collapsed with Sema3A and that there was no clear difference in Sema3A-induced growth cone collapse between axons of wild-type and Nrp1+/- embryos (Fig. 4B, D).

Effects of Sema3A and Sema3F on DRG and SG axons of plexin-A4, neuropilin-1, or neuropilin-2 mutant embryos in vitro. A-D, Growth cone collapse induced by Sema3A in DRG axons of E12.5 plexin-A4 (A) and neuropilin-1 (B) mutant embryos and SG axons of E13.5 plexin-A4 (C) and neuropilin-1 (D) mutant embryos. In A-C, percentages of collapsed growth cones were plotted against different concentrations of the Sema3A recombinant proteins. The average percentage of growth cone collapse at each point was calculated for 30-40 DRG explants (from 4 plexin-A4 littermates and 3 neuropilin-1 littermates) and 15-20 SG explants (from 7 plexin-A4 littermates and 2 neuropilin-1 littermates). In D, average percentages of growth cone collapse for 5-10 SG explants (from 2 neuropilin-1 littermates) are given. E, F, Growth cone collapse induced by Sema3F in SG axons of E13.5 neuropilin-2 (E) and plexin-A4 (F) mutant embryos. The average percentage of growth cone collapse at each point was calculated for 15-20 SG explants (from 2 neuropilin-2 littermates and 6 plexin-A4 littermates). Vertical bars indicate SEM. *p < 0.001 (Student's t test). G, Repulsion of SG axons of E15.5 wild-type (PlexA4+/+) and plexin-A4 homozygous (PlexA4-/-) embryos by Sema3F in collagen gel cultures. SG axons were detected with the anti-β-tubulin antibody. Asterisks indicate aggregates of Sema3F-secreting HEK293T cells. Scale bars: (in G) 500 μm.
Taken collectively, the results obtained in the in vitro studies suggest that, in DRG and SG axons, plexin-A4 plays a role in the propagation of axon-repulsive activities of Sema3A.
Plexin-A4-deficient SG axons respond normally to Sema3F
Next, we tested whether plexin-A4 is also involved in Sema3F signaling. It has been shown that Sema3F repels SG axons in vitro (Chen et al., 1998) and that the Sema3F-induced response of SG axons required plexin-A3 as a receptor (Cheng et al., 2001), as well as neuropilin-2 (Chen et al., 1998; Giger et al., 2000). Therefore, we examined the response to Sema3F of SG axons of plexin-A4 and neuropilin-2 mutant embryos (Takashima et al., 2002).
Sema3F induced growth cone collapse in wild-type SG axons in a dose-dependent manner: 30 nm of AP-tagged human Sema3F recombinant protein (Sema3F-AP) induced the complete collapse of growth cones (Fig. 4E). The Sema3F-dependent growth cone collapse was completely abolished in SG axons of neuropilin-2 mutant embryos (Fig. 4E), confirming the requirement of neuropilin-2 in Sema3F signaling. In contrast, the response of plexin-A4-deficient SG axons to Sema3F was not abolished and actually increased (Fig. 4F). An increase in the response to Sema3F was observed even for SG axons of PlexA4+/- embryos. The axon avoidance assay in collagen gel showed that plexin-A4-deficient SG axons avoided Sema3F-secreting HEK293T cell aggregates, as wild-type axons did (Fig. 4G). These results suggest that, in SG axons, plexin-4 is not required for the propagation of Sema3F chemorepulsive activities, but rather that the deprivation of plexin-A4 results in the gain of Sema3F signaling.
Plexin-A4 interacts with class 6 transmembrane semaphorins, Sema6A and Sema6B
It has been shown that a class 6 transmembrane semaphorin, Sema6A, induced growth cone collapse in SG axons or repelled the axons in vitro (Xu et al., 2000). We examined the expression of Sema6A in mouse embryos by ISH analysis and found that Sema6A was present in skeletal muscles and connective tissues surrounding the vertebrae (Fig. 5A). It is of interest that Sema6A is expressed strongly in the anterior vertebrate muscles located just medial to the plexin-A4-expressing SGs. We also found that another class 6 semaphorin, Sema6B, was expressed in the anterior vertebrate muscles (Fig. 5A). As described in the previous part of this paper, several plexin-A4-deficient SG axons aberrantly projected into these Sema6A- and Sema6B-expressing tissues (Fig. 3F-H), implying that plexin-A4 is functionally correlated with these class 6 semaphorins.
To test this possibility, we first examined the physical interaction of plexin-A4 with Sema6A or Sema6B. Because the activity for growth cone collapse by the ectodomain of Sema6A is dimerization dependent (Xu et al., 2000), we produced Fc-dimered AP-tagged recombinant proteins for the extracellular domain of Sema6A (AP-Sema6Aect-Fc) and Sema6B (AP-Sema6Bect-Fc) and examined their binding to L-cells that stably express the full-length plexin-A4 proteins (Fig. 6D). Both recombinant proteins bound to plexin-A4-expressing L-cells but not parental L-cells (Figs. 6D, 5B). The dissociation constant (KD) value for the interaction of AP-Sema6Aect-Fc with plexin-A4 estimated by Scatchard analysis was 3.56 nm (converted as a monomer) (Fig. 5C, D). The KD value for the interaction of Sema6A with plexin-A4 is in the same range as that for the interaction of other transmembrane semaphorins with plexins (Winberg et al., 1998; Tamagnone et al., 1999; Fujii et al., 2002). The Sema6A recombinant proteins bound to cultured SG neurons of wild-type but not PlexA4-/- embryos (Fig. 5E), suggesting that Sema6A binds to the native plexin-A4 proteins. We could not determine the KD value for the interaction of Sema6B with plexin-A4 because of the low affinity of AP-Sema6Bect-Fc recombinant proteins for plexin-A4.

Requirement of plexin-A4 for propagation of axon-repulsive activities of Sema6A. A, B, Growth cone collapse induced by Sema6A in SG axons of E13.5 plexin-A4 (A) and neuropilin-1 (B) mutant embryos. Percentages of collapsed growth cones were plotted against different concentrations of the Sema6A recombinant proteins (AP-Sema6Aect-Fc). The average percentage of growth cone collapse at each point was calculated for 15-20 SG explants (from 3 plexin-A4 littermates) and 5-10 SG explants (from 3 neuropilin-1 littermates). Vertical bars indicate SEM. *p < 0.001 (Student's t test). C-E, Effect of Sema6A on morphology of plexin-A4-expressing L-cells. C, Immunoblotting with anti-myc antibody of L-cell lines that stably express the myc-tagged full-length plexin-A4 proteins (Plexin-A4) or the cytoplasmic segment-deleted plexin-A4 proteins (Plexin-A4ΔC). D, Morphology of plexin-A4-expressing and plexin-A4ΔC-expressing cells without Sema6A (detected by immunohistochemistry with anti-myc antibody) and with the AP-Sema6Aect-Fc recombinant proteins (detected by AP histochemistry). Note that the addition of the Sema6A recombinant proteins induces contraction of plexin-A4-expressing but not plexin-A4ΔC-expressing cells. E, Quantitative analysis of cell size. Percentages of AP-Sema6Aect-Fc-positive cells smaller than 25 × 25 μm2 (625 μm2) were calculated. Vertical bars indicate SEM. *p < 0.001 (Student's t test). Scale bars: (in D) 100 μm.
Plexin-A4 propagates axon-repulsive activities of Sema6A and Sema6B
We next examined whether Sema6A and Sema6B function as repellents for SG axons, and also whether plexin-A4 mediates repulsive activities of these semaphorins.
The AP-Sema6Aect-Fc recombinant proteins induced growth cone collapse in SG axons of wild-type embryos in a dose-dependent manner (Fig. 6A): 30 nm of the proteins induced the complete collapse of growth cones. The EC50 value for Sema6A-dependent growth cone collapse was ∼5 nm, which closely coincided with the KD value of the interaction of Sema6A with plexin-A4 (Fig. 5D). In contrast, Sema6A-dependent growth cone collapse was completely abolished in SG axons of PlexA4-/- embryos. These results suggest that propagation of the activity of Sema6A requires plexin-A4. The Sema6A-dependent growth cone collapse was partially reduced in SG axons of PlexA4+/- embryos (Fig. 6A), suggesting that the propagation of Sema6A activities in SG axons depends on the dosage of the plexin-A4 gene. The Sema6A recombinant proteins induced growth cone collapse to the same extent in SG axons of wild-type, Nrp1-/-, and Nrp1+/- embryos (Fig. 6B), indicating that Sema6A can induce growth cone collapse of SG axons independently of neuropilin-1.
To address further whether plexin-A4 serves as a receptor for Sema6A, we performed a cell-contraction assay (Takahashi et al., 1999; Suto et al., 2003). We transfected L-cells with the full-length plexin-A4 cDNA or the cytoplasmic segment-deleted plexin-A4 (plexin-A4ΔC) cDNA and isolated cell lines that stably expressed the proteins (Fig. 6C). Addition of the AP-Sema6Aect-Fc recombinant proteins (15 nm) to the cultures induced contraction of the plexin-A4-expressing but not plexin-A4ΔC-expressing cells (Fig. 6D, E), although the recombinant proteins bound to both cells (Fig. 6D). The Sema6A recombinant proteins did not induce any morphological change in parental L-cells (data not shown). Moreover, addition of the Fc-dimered AP proteins (AP-Fc; 30 nm) did not induce contraction of parental L-cells, plexin-A4-expressing cells or plexin-A4ΔC-expressing cells (data not shown). These results indicate that the cytoplasmic segment of plexin-A4 is required for the propagation of Sema6A-induced cell-contraction activities into cells and suggest that plexin-A4 serves as a receptor for Sema6A.
The AP-Sema6Bect-Fc recombinant proteins did not induce any morphological changes in growth cones of SG axons. The result, as well as the low affinity of the recombinant protein for plexin-A4, leads us to speculate that a conformational change occurs in the proteins. Therefore, we adopted another assay system (the neurite outgrowth assay) to examine functions of Sema6B.We generated L-cell lines that stably express the full-length Sema6B proteins (L6B cells) by transfection of the Sema6B cDNA and placed small explants of SG of E13.5 wild-type or plexin-A4 mutant embryos on confluent monolayer sheets of L6B cells or parental L-cells. Neurite outgrowth from wild-type SG explants occurred on L-cell sheets but was suppressed on L6B cell sheets (Fig. 7A, B). In contrast, neurite outgrowth from plexin-A4-deficient SGs was not suppressed on L6B cell sheets (Fig. 7A, B). Moreover, the suppression of Sema6B-dependent neurite outgrowth was moderate for SGs of PlexA4+/- embryos (Fig. 7B). These results suggest the involvement of plexin-A4 in the propagation of axon-repulsive activity of Sema6B. When wild-type SG explants were placed on sparse cultures of L6B cells, neurites sprouted preferentially into cell-free spaces (Fig. 7C), suggesting that Sema6B suppresses neurite outgrowth in a contact-mediated manner.
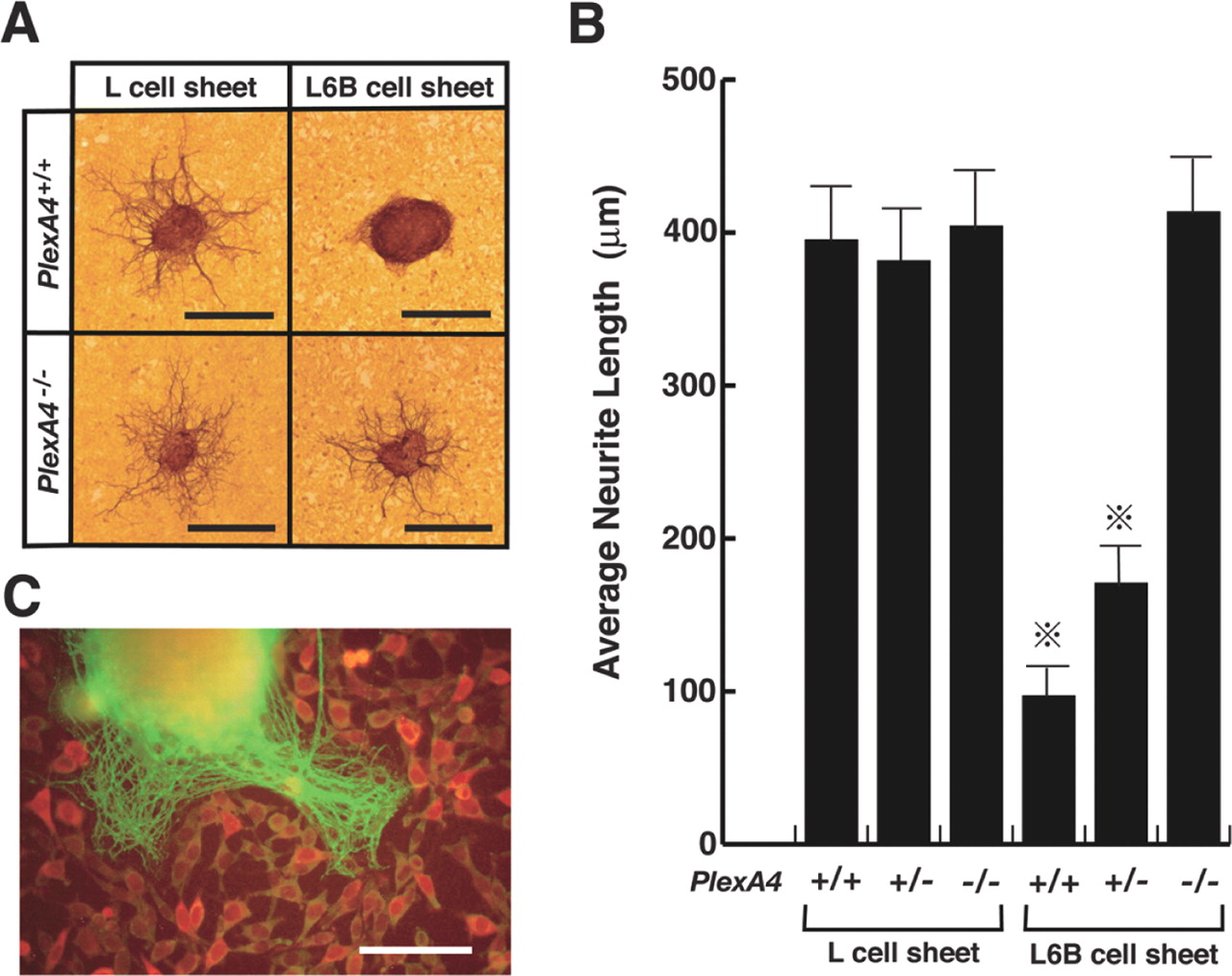
Requirement of plexin-A4 for propagation of neurite outgrowth-suppressive activity of Sema6B. A, Neurite outgrowth from SG explants of E13.5 wild-type (PlexA4+/+) and plexin-A4 homozygous (PlexA4-/-) embryos on monolayer sheets of L-cells or Sema6B-expressing cells (L6B cells; see Results), detected by immunostaining with an anti-β-tubulin antibody. B, Quantitative analysis on the neurite outgrowth of SG explants shown in A. Average length for the longest three β-tubulin-positive fibers for each SG explant was designated as the neurite length of a given explant. Neurite length was determined in 5-10 SG explants for each embryo. Averages of the neurite length for embryos (5 wild-type, 7 plexin-A4+/-, and 6 plexin-A4-/- embryos from 3 independent experiments) are given on the vertical axis. Vertical bars indicate SEM. *p < 0.001 (Student's t test). C, An SG explant cultured with L6B cells of low density is shown. Neurites sprouted from the explant, and Sema6B-expressing cells were detected by immunostaining with the β-tubulin antibody (green) and anti-myc antibody (red), respectively. Scale bars: A, 500 μm; C, 250 μm.
Discussion
Plexin-A4 propagates axon-repulsive activities of Sema3A but not Sema3F
It has been proposed that members of the plexin-A subfamily form receptor complexes with neuropilin-1 or neuropilin-2 to propagate axon-repulsive activities of secreted class 3 semaphorins (Takahashi et al., 1999; Tamagnone, 1999). In this study, we produced plexin-A4 null mutant mice and showed that DRG and SG axons taken from the mutant embryos partially lost their responsiveness to Sema3A. Sema3A binds to neuropilin-1 but not plexin-A4. Neuropilin-1-deficient DRG or SG axons completely lose their responsiveness to Sema3A (Kitsukawa et al., 1997) (Fig. 4B, D). In addition, we previously showed that the cytoplasmic segment of plexin-A4 protein was required to propagate Sema3A activities into cells (Suto et al., 2003). Therefore, we conclude that plexin-A4 forms a receptor complex with neuropilin-1 to mediate axon-repulsive activities of Sema3A, at least in DRG and SG axons.
The partial reduction in the responsiveness to Sema3A in plexin-A4-deficient axons indicates the requirement of other plexin-As for the propagation of Sema3A activities. DRG and SG neurons express plexin-A3, as well as plexin-A4 (Murakami et al., 2001). DRG and SG axons of plexin-A3 knock-out mice lose but partially their responsiveness to Sema3A (Cheng et al., 2001). These results suggest that, in DRG and SG axons, both plexin-A3 and plexin-A4 form receptor complexes with neuropilin-1 and function redundantly in the propagation of Sema3A activities.
One interesting result of this study is that SG axons of plexin-A4 knock-out mice did not lose their responsiveness to Sema3F, but rather became sensitive to the semaphorin. The result is in contrast to the previous finding that plexin-A3-deficient SG axons completely lose their responsiveness to Sema3F (Cheng et al., 2001). Because Sema3F activities are mediated by neuropilin-2/plexin-A receptor complexes, one explanation for the results is that, in SG axons, plexin-A3 but not plexin-A4 forms a receptor complex with neuropilin-2 to propagate Sema3F activities. The increase in the responsiveness to Sema3F in plexin-A4-deficient SG axons, however, suggests another possibility that plexin-A4 forms a receptor complex with neuropilin-2 for Sema3F but of low efficiency in Sema3F signaling. The removal of plexin-A4 from SG axons might lead more neuropilin-2 proteins to form a more efficient Sema3F receptor complex, neuropilin-2/plexin-A3, resulting in the increase in the response to Sema3F of the axons.
Plexin-A4 is a receptor for class 6 transmembrane semaphorins, Sema6A and Sema6B
Several studies have shown that plexins serve as direct receptors for semaphorins in not only invertebrates (Winberg et al., 1998; Fujii et al., 2002) but also vertebrates (Tamagnone et al., 1999; Artigiani et al., 2004; Gu et al., 2005). The present study provided evidence that plexin-A4 propagates activities of mammalian class 6 transmembrane semaphorins, Sema6A and Sema6B.
Sema6A bound to plexin-A4 with high affinity. Sema6A induced morphological change in cells that express the full-length plexin-A4 proteins but not the cytoplasmic segment-deleted plexin-A4 proteins. Plexin-A4-deficient SG axons completely lost their responsiveness to Sema6A. Moreover, SG axons of neuropilin-1 mutant embryos responded normally to Sema6A. These results suggest that plexin-A4 serves as a direct receptor for Sema6A and propagates the axon-repulsive activities of the semaphorin, independently of neuropilin-1. Sema6B also bound to plexin-A4 and suppressed growth of SG axons of the wild-type but not plexin-A4 mutant embryos, suggesting that plexin-A4 functions as a receptor for Sema6B, as well as Sema6A. The present results in combination with our previous result that plexin-A1 mediates activities of Sema6D (Toyofuku et al., 2004) suggest that plexin-As are dual semaphorin receptors that propagate activities of both class 3 and class 6 semaphorins.
Deficiency of plexin-A4 induces a wide variety of axon guidance defects in vivo
The present study showed that plexin-A4 deficiency induced various axon guidance defects in the PNS and CNS.
The cranial and spinal sensory nerves were defasciculated and overshot in plexin-A4 mutant embryos. The defects are fundamentally related to those of neuropilin-1 mutants (Kitsukawa et al., 1997) or Sema3A mutants (Taniguchi et al., 1997) but are more moderate than the phenotype in these mutants. Pathways and projections of peripheral sensory nerves were mostly normal in plexin-A3 mutant embryos (Cheng et al., 2001). The moderate phenotype of the PNS fiber projection in plexin-A4 and plexin-A3 mutants coincides well with the partial loss of the responsiveness to Sema3A of plexin-A4-deficient and plexin-A3-deficient DRG axons and further suggests redundant functions of these plexins in the signaling of Sema3A activities and axon guidance in vivo.
The projection of the oculomotor and trochlear nerves is defective in Sema3F mutants and neuropilin-2 mutants (Chen et al., 2000; Giger et al., 2000), suggesting the involvement of neuropilin-2-mediated Sema3F activities in the directional guidance of the nerves. In contrast, the present plexin-A4 mutants, plexin-A3 mutants (Cheng et al., 2001), Sema3A mutants (Taniguchi et al., 1997), and neuropilin-1 mutants (Kitsukawa et al., 1997) do not show any axon guidance defects in the oculomotor or trochlear nerves, although plexin-A3, plexin-A4, and neuropilin-1 are expressed in the nerves (Suto et al., 2003) (our unpublished observations). In these nerves, plexin-A1 and/or plexin-A2 may form receptor complexes with neuropilin-2 to propagate Sema3F activities. As discussed above, plexin-A3 is preferentially used to propagate Sema3F activities in SG neurons, although plexin-A4 is expressed in the neurons. These results suggest that members of the plexin-A subfamily form receptor complexes with neuropilin-1 or neuropilin-2 for class 3 semaphorins in a cell type-specific manner.
In plexin-A4 mutant embryos, several SG axons aberrantly grew into the prevertebrate muscles and connective tissues that express Sema6A and Sema6B. The results indicate the possibility that Sema6A and Sema6B function as short-distant repellents to suppress the invasion of SG axons into the tissues. However, because the projection of SG axons is also defective in neuropilin-1 and Sema3A mutant embryos (Kawasaki et al., 2002), more analyses are required to determine the functions of Sema6A and Sema6B in SG axon guidance.
Deficiency of plexin-A4 induced a pathfinding error for the anterior commissure. The anterior commissure is also defective in mice lacking neuropilin-2 (Chen et al., 2000; Giger et al., 2000) and Sema3F (Sahay et al., 2003) but develops normally in Sema3A-deficient mice (our unpublished observations). Plexin-A4 and neuropilin-2, but not neuropilin-1, are expressed in the anterior olfactory nucleus and the lateral telencephalon that send axons of the anterior commissure (Chen et al., 1997; Suto et al., 2003) (Fig. 3I). These results may mean that, in anterior commissure axons, plexin-A4 forms complexes with neuropilin-2 to serve as a Sema3F receptor and plays roles in the channeling of the axons. However, the phenotype of the anterior commissure in plexin-A4 mutant is different in several ways to that in neuropilin-2 or Sema3F mutant mice. In neuropilin-2 and Sema3F mutant mice, axons of the anterior commissure are defasciculated, but some of them decussate at a normal position within the forebrain (Giger et al., 2000; Sahay et al., 2003). In contrast, in plexin-A4 mutant mice, axons of the anterior commissure never decussated, and the acA tract of the anterior commissure shifted ventrally. Because Sema6A is expressed in the developing forebrain (our unpublished observations), plexin-A4-mediated Sema6A activities might play roles in the guidance of anterior commissure axons.
The barrel formation was abnormal in plexin-A4 mutant mice: the boundary of the barrels was indistinct. Plexin-A4 is expressed in the VPM of the dorsal thalamus (Suto et al., 2003), fibers from which terminate in the center of the barrels. Therefore, it is likely that plexin-A4-deficient VPM axons terminate diffusely in the somatosensory cortex, resulting in the indistinct boundary of the barrels. Because the barrels develop normally in Sema3A mutant mice (Ulupinar et al., 1999), a lack of plexin-A4-mediated Sema3A signals is not the cause of the abnormal barrel formation in plexin-A4 mutant mice. The thalamocortical projection is abnormal in Sema6A mutant mice (Leighton et al., 2001), suggesting that repulsive activities of Sema6A may define terminal arbors of VPM axons to form discrete barrels.
In zebrafish embryos, loss of function of plexin-A4 reduced the number of branching of the primary sensory axons of Rohon-Beard neurons and the central axons of the trigeminal ganglion neurons, suggesting that plexin-A4 promotes axon branching (Miyashita et al., 2004). In contrast, in the plexin-A4-deficient mice, the PNS sensory efferent sprouted more, and the terminals of the VPM axons of the trigeminal somatosensory pathway seemed to be sprouted. Moreover, no obvious defect was observed in the central axons of PNS sensory neurons in plexin-A4 mutant mice (our unpublished observations). Downstream signal pathways of plexin-A4 might be different between zebrafish and mice.
The present study showed that the responsiveness of axons to semaphorins in vitro and nerve fiber guidance in vivo depended on the dosage of the plexin-A4 gene. DRG and SG axons of plexin-A4 heterozygous embryos exhibited a slightly reduced response to Sema3A, Sema6A, and Sema6B. Embryos lacking a single plexin-A4 allele showed a moderate defect in the PNS fiber projection and the formation of the anterior commissure. The haploid-insufficient phenotypes suggest that axons contain plexin-A4 proteins at the minimum level required to propagate the activities of the semaphorins and that the responsiveness of given axons to semaphorins primarily depends on the expression level of the plexin.
Conclusion
The results obtained in the present study indicate that plexin-A4 is a multiple semaphorin receptor that mediates repulsive activities of secreted class 3 semaphorins by forming receptor complexes with neuropilins and transmembrane semaphorins of class 6 by direct interaction with the semaphorins and controls a variety of axon guidance events in a highly regulated manner.
Footnotes
This work was supported by grants from the Core Research for Evolutional Science and Technology of Japan Science and Technology Agency (H.F. and T.Y.), grants-in-aid from the Ministry of Education, Science, Sports, and Culture of Japan Ministry of Education (H.F. and T.Y.), and by The 21st Century COE Program (H.F.). We thank Dr. M. Tessier-Lavigne (Stanford University, Stanford, CA) for human Sema3F expression vector, Dr. J. Flanagan (Harvard Medical School, Boston, MA) for Aptag-4 expression vector, Dr. J. Miyazaki (Osaka University, Suita, Japan) for pCAGGS expression vector, Dr. S. Nagata (Osaka University) for pEF-Fc expression vector, Dr. T. Nagatsu (Fujita Health University, Toyoake, Japan) for anti-TH antibody, Dr. Y. Arimatsu (Mitsubishi Kagaku Institute of Life Sciences, Machida, Japan) for anti-β-tubulin antibody, and Dr. T. Kimura (Sumitomo Pharmaceutical Research Center, Osaka, Japan) for technical advice in the cloning of Sema6B cDNAs.
Correspondence should be addressed to Dr. Hajime Fujisawa, The 21st Century COE Program, Division of Biological Science, Nagoya University Graduate School of Science, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: fujisawa{at}bio.nagoya-u.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253628-10$15.00/0
↵* F.S. and K.I. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}