
Abstract
A diversity of ion channels contributes to the active properties of neuronal dendrites. From the apical dendrites of hippocampal CA1 pyramidal neurons, we recorded inwardly rectifying K+ channels with a single-channel conductance of 33 pS. The inwardly rectifying K+ channels were constitutively active at the resting membrane potential. The amount of constitutive channel activity was significantly larger in the apical dendrites than in the soma. Activities of these inwardly rectifying K+ channels were inhibited by Ba2+ (200 μm) and tertiapin (10 nm), both of which are believed to block G-protein-coupled inwardly rectifying K+ (GIRK) channels. Intracellularly applied GTPγS (20 μm) during dual dendritic recordings significantly increased constitutive channel activity. Baclofen (20 μm), an agonist for the G-protein-coupled GABAB receptor, also significantly increased the level of channel activity. Therefore, these channels are GIRK channels, which are constitutively active at rest in the apical dendrites of CA1 pyramidal neurons and can be further activated via G-protein-coupled neurotransmitter receptors.
- K+ channels
- inward rectifier
- hippocampus
- dendrites
- G-protein-coupled receptors
- single-channel analysis
Introduction
Ion channels dynamically regulate neuronal excitability. Among all of the ion channel species, K+ channels comprise the most diverse group (Coetzee et al., 1999). The CA1 pyramidal neurons, for example, possess a variety of voltage- and/or Ca2+-dependent K+ channels, each contributing to the firing properties of the cell in a unique way (Storm, 1990). Many of the voltage-dependent K+ channels in CA1 pyramidal neurons are expressed in their dendrites and play important roles in regulating excitability and synaptic plasticity (Hoffman et al., 1997; Bernard et al., 2004; Chen and Johnston, 2004; Frick et al., 2004).
Whereas the voltage- and Ca2+-dependent K+ channels regulate the firing behavior of the cell, G-protein-coupled inwardly rectifying K+ (GIRK) channels provide a mechanism for slow inhibitory modulation of the overall excitability of the cell (Andrade et al., 1986; Luscher et al., 1997). Many inhibitory neurotransmitters and neuromodulators hyperpolarize the CA1 pyramidal neurons by activating GIRK channels, including GABA (via activation of GABAB receptors), serotonin (via activation of 5-HT1A receptors), and adenosine (via activation of A1 receptors) (Luscher et al., 1997).
Among the four subtypes of GIRK channel genes, GIRK1, GIRK2, and GIRK3 are expressed at high levels in CA1 neurons (Karschin et al., 1996). The GIRK2 subunit, in particular, is an essential part of GIRK channels in CA1 neurons. The absence of this subunit leads to concurrent downregulation of GIRK1 proteins in CA1 (Signorini et al., 1997) and to a total loss of the postsynaptic response to slow inhibitory neurotransmission (Luscher et al., 1997). Mice without the GIRK2 subunit also have a high propensity for epilepsy (Signorini et al., 1997). The GIRK channels are therefore critical regulators of neuronal excitability.
It is worth noting that the majority of excitatory and inhibitory inputs to the CA1 pyramidal neurons make synaptic contact onto their dendrites (Megias et al., 2001). This suggests that GIRK channels may localize in the dendrites to be close to the G-protein-coupled receptors. Furthermore, dendritic localization of GIRK channels could facilitate their participation in regulating synaptic integration of excitatory inputs (Takigawa and Alzheimer, 2002). The dendritic localization of GIRK channels has been shown in neocortical neurons with whole-cell recordings from isolated dendrites (Takigawa and Alzheimer, 1999). Less is known about GIRK channels in the dendritic membrane of CA1 pyramidal neurons, although immunoelectron microscopy studies have located GIRK1 proteins in the dendritic shaft and spines of these cells (Drake et al., 1997). In particular, no single-channel data are available for GIRK channels in intact CA1 neurons, nor is the spatial localization of these channels directly measured with dendritic recordings.
In this report, we characterize the biophysical properties of single GIRK channels and determine their spatial localization, pharmacology, and receptor activation with recordings from the dendrites of CA1 neurons in adult hippocampus.
Materials and Methods
Slice preparation. Acute hippocampal slices (400 μm thick) were prepared from 6- to 8-week-old Sprague Dawley rats and stored in a submerged chamber containing oxygenated external solution (described below). Pyramidal neurons of the hippocampal CA1 region were visualized with an Olympus (Tokyo, Japan) 60× water-immersion objective and a Zeiss (Oberkochen, Germany) Axioskop with infrared differential interference contrast. All of the channel recordings were done at room temperature (∼22-24°C) for better resolution of single-channel events and better stability of the patch membrane.
Solutions and drugs. During experiments, the slices were continuously superfused with oxygenated external solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 25 dextrose. For cell-attached recordings, the pipette solution contained the following (in mm): 140 KCl, 2 MgCl2, and 10 HEPES, pH 7.4. For whole-cell recordings, the pipette solution contained the following (in mm): 120 KMeSO4, 20 KCl, 10 HEPES, and 4 NaCl, pH 7.4. rTertiapin-Q (Alomone Labs, Jerusalem, Israel) was dissolved in a 0.1% BSA-supplemented pipette solution to a concentration of 2 μm; GTPγS was dissolved in water to make 20 mm aliquots; (R)-baclofen (Tocris Cookson, Ellisville, MO) was dissolved in water to make 5 mm aliquots and diluted to desired concentrations for experiments. Reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Data acquisition and analysis. Electrodes were pulled from thick-wall borosilicate glass pipettes and fire polished. The tips of the electrodes had similar diameters of ∼1 μm, with resistances of 5-7 MΩ when filled with pipette solution. Positive pressure was applied to the patch electrode while approaching dendrites in the slice. Tight seals of >10 GΩ were formed on release of the pressure. The patch membrane was held at various command potentials (Vcmd = approximately -120 to +40 mV) to record channel activities. Most of the records were 15 s in length, although 60 s records were used to construct the open probability (NPo)-Vm relationship in Figure 1C. After cell-attached recordings, the membrane patch was ruptured, and the resting membrane potential of the cell was recorded. This potential (Vrest = -66 ± 0.4 mV; n = 87) was then used to calculate the absolute voltage of the commands (Vm = Vrest - Vcmd). Liquid junction potential (-2 mV) was not corrected (Chen and Johnston, 2004).

Single-channel analysis of inwardly rectifying K+ channel(s) and channel distribution along the dendrites. A, Single inwardly rectifying K+ channel(s) recorded at 200 μm from the soma. Inset, An expanded stretch of record showing an idealized trace (smooth line; generated with HMMs in TAC software) overlaying with the original trace. Calibration: 1 pA, 20 ms. B, Current-voltage plot under control (Ctrl) condition (γ = 33 pS) in the presence of GTPγS (γ = 33 pS) or the presence of baclofen (γ = 31 pS). Some error bars are smaller than the symbols. Single-channel conductance was determined as the slope of the best-fitted line in the linear I-V range. C, Open probability plotted against membrane potential. D, The level of constitutive channel activity (measured at resting potential) was plotted against distance from the soma. The amount of channel activity was significantly larger in the dendrites compared with the soma. Recordings were binned for every 50 μm. Soma, NPo = 0.0038 ± 0.0012 (n = 5); 50 μm, NPo = 0.0085 ± 0.0051 (n = 6); 100 μm, NPo = 0.027 ± 0.0066 (n = 4); 150 μm, NPo = 0.031 ± 0.012 (n = 5); 200 μm, NPo = 0.018 ± 0.0053 (n = 4); 250 μm, NPo = 0.031 ± 0.010 (n = 5); 300 μm, NPo = 0.024 ± 0.0074 (n = 8). *p < 0.05 and **p < 0.01 compared with the soma. s.o., Stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum; s.l-m, stratum lacunosum-moleculare. Error bars represent SEM.
Recordings were performed using an Axopatch-1D voltage-clamp amplifier (Axon Instruments, Union City, CA) and an ITC-18 computer interface (InstruTech, Port Washington, NY). Data acquisition was controlled by custom software written in IGOR (WaveMetrics, Lake Oswego, OR). Channel records were analog filtered at 2 kHz, digitized at 10 kHz, and digital filtered at 1 kHz. For single-channel analysis, event detection was performed with custom-built hidden Markov models (HMMs) incorporated in TAC software (Bruxton, Seattle, WA). Subsequent analyses of channel conductance and open probabilities were performed using custom software written in IGOR. Single-channel conductance (γ) was calculated as the slope of the best line fit to the I-V plot in the linear range: I = g × (V - Vreversal).
Open probability of the channel(s) was calculated as follows: NPo = topen /ttotal.
Where appropriate, data are expressed as mean ± SEM. Significance of variation was tested with one-way ANOVA, which was followed by a t test between treatment groups.
Results
The inwardly rectifying K+ channels, including GIRK channels, owe their name to the fact that they pass inward current more efficiently than outward current (Nichols and Lopatin, 1997). To record activities of inwardly rectifying K+ channels in the cell-attached configuration, we used a KCl-based solution in the recording electrode (see Materials and Methods), creating a near-symmetric K+ concentration between the inside and the outside of the cell membrane (Fig. 1A). Under such conditions, the reversal potential for K+ is ∼0 mV, so that when the channels activate at resting and more hyperpolarized potentials, the driving force for K+ should be large enough to give rise to measurable inward current.
Single-channel events of inwardly rectifying K+ channels
At resting membrane potential (Vcmd = 0 mV; Vm = Vrest), cell-attached recordings revealed constitutive channel activities (Vm = -70 mV) (Fig. 1A). As the membrane was hyperpolarized, the channel current became larger in amplitude, in accordance with the increased driving force for K+. These K+ channels were inwardly rectifying, because they pass very little outward current at depolarized potentials (Fig. 1A,B).
Square-shaped channel-opening events were sometimes observed; however, most channel openings would be better described as “flickering.” To analyze these channels with brief openings, HMMs were built to perform event detection in the TAC software. The algorithm had a built-in inverse filter and was designed to minimize measurement errors introduced by analog filtering (Venkataramanan and Sigworth, 2002).
Single-channel conductance
Current amplitude during channel opening was variable, which likely resulted from the briefness of the opening and the randomness of sampling relative to transitions between open and closed states (Venkataramanan and Sigworth, 2002). HMMs with three to five states were implemented to fit our channel records (Fig. 1A, inset). Each of these states had a different current level, and the state with the largest amplitude should be the closest estimate of the real amplitude. We therefore only used those events to calculate single-channel conductance. This strategy for calculating single-channel conductance also assumes that the larger-amplitude events do not represent the simultaneous opening of multiple small-amplitude events. This is a reasonable assumption in our experiments, because the overall open probability was too low (see below) to observe simultaneous openings. The channels had a single-channel conductance of γ = 33 ± 1 pS (n = 11), which was determined in the linear range of the I-V relationship (Fig. 1B).
Open probability
From the same HMM fitting, we also calculated the NPo of the channel(s) at voltages between -110 and 0 mV, in which the channels conducted inward current (Vm = -106 mV, NPo = 0.018 ± 0.002; Vm = -86 mV, NPo = 0.018 ± 0.001; Vm = -66 mV, NPo = 0.019 ± 0.002; Vm = -46 mV, NPo = 0.015 ± 0.003; Vm = -26 mV, NPo = 0.013 ± 0.001; n = 5) (Fig. 1C). The low open probabilities within this voltage range indicate that these channels were not IRK channels (Liu et al., 2001) and that their gating was dependent on other factors in addition to voltage.
Distribution of channel activity along the apical dendrite
Because the open probability of the channel(s) was very low, we could not accurately determine the number of channels in the patches. Nevertheless, we looked at whether the level of channel activities changed with dendritic location. Constitutive activities of the inwardly rectifying K+ channels were recorded in every patch at resting membrane potentials. The level of such activities correlated with the distance from the soma (r = 0.38; p < 0.05). At dendritic locations of ≥100 μm, the level of constitutive channel activity was significantly higher than at the soma (p < 0.05; one-way ANOVA followed by a t test between the soma and each dendritic location) (Fig. 1D).
The constitutively active, inwardly rectifying K+ channels were inhibited by GIRK channel blockers
The minimal voltage dependence of activation, the flickering of the channels, and the low open probability led us to suspect that these inwardly rectifying K+ channels may be G-protein-activated inward rectifiers (GIRK channels) that were constitutively active in the absence of exogenous agonists (Soejima and Noma, 1984).
To test this hypothesis, we applied two pharmacological agents often used to directly block the GIRK channels: Ba2+ and tertiapin (Takigawa and Alzheimer, 2002). Ba2+ at a concentration of 200 μm blocked ∼70% of the channel activity (as measured by NPo at resting membrane potentials). At this concentration, Ba2+ is relatively specific for blocking inwardly rectifying K+ channels, including IRK and GIRK channels. But the channels we recorded were not purely voltage dependent (see above), suggesting that they are likely to be GIRK channels. The peptide toxin tertiapin also blocked the inwardly rectifying K+ channels in the dendrites (Fig. 2). In neurons, this toxin should specifically block GIRK channels, with little effect on IRK channels at concentrations up to 2 μm (Jin and Lu, 1998).

Pharmacology of the inwardly rectifying K+ channels. A, Trace examples with control solutions, Ba2+, or tertiapin in the recording pipette. The membrane was held at resting potentials. B, Both Ba2+ and tertiapin inhibited constitutive activities of the inwardly rectifying K+ channels. Control (Ctrl), NPo = 0.031 ± 0.007, 218 ± 9 μm from the soma (n = 11); 200 μm Ba2+, NPo = 0.011 ± 0.003, 228 ± 7 μm from the soma (n = 9); 2 mm Ba2+, NPo = 0.003 ± 0.001, 258 ± 20 μm from the soma (n = 4); 10 nm tertiapin, NPo = 0.015 ± 0.004, 215 ± 5 μm from the soma (n = 10); 50 nm tertiapin, NPo = 0.005 ± 0.001, 224 ± 8 μm from the soma (n = 8); 2 μm tertiapin, NPo = 0.001 ± 0.001, 223 ± 8 μm from the soma (n = 3). *p < 0.05 and **p < 0.01 compared with control. Error bars represent SEM.
GTPγS-stimulated activity of inwardly rectifying K+ channels in the dendrites
To test further whether the constitutively active, inwardly rectifying K+ channels were GIRK channels, we used intracellular application of GTPγS (Wickman et al., 1999; Han et al., 2003). In this set of experiments, we performed dual dendritic recordings and included GTPγS (0, 20, or 200 μm) in the whole-cell electrode. After establishment of the whole-cell configuration, we patched the same dendrite in the cell-attached configuration and recorded activities of inwardly rectifying K+ channels at the resting potential. The fluorescent Ca2+ indicator bis-fura-2 (100 μm) was also included in the whole-cell electrode to help visualize the dendrites and validate the establishment of the whole-cell configuration (Fig. 3A).

Activation of inwardly rectifying K+ channels by intracellularly applied GTPγS. A, Position of a whole-cell and a cell-attached electrode. The whole-cell electrodes contained 0, 20, or 200 μm GTPγS and bis-fura-2. The picture was taken after the experiment, showing the dendrite filled with bis-fura-2. B, Trace examples with no GTP or GTPγS in the whole-cell electrode, with or without Ba2+ in the cell-attached electrode. The membrane was held at resting potentials. C, Applying 20 μm GTPγS into the cell significantly increased the level of channel activity. GTPγS (200 μm) did not further increase channel activity. Ba2+ (200 μm) reduced channel activation with GTPγS by ∼67%. No GTP, NPo = 0.013 ± 0.006, 206 ± 8 μm from the soma (n = 5); 20 μm GTPγS, NPo = 0.127 ± 0.033, 206 ± 7 μm from the soma (n = 6); 200 μm GTPγS, NPo = 0.118 ± 0.026, 213 ± 8 μm from the soma (n = 4); 20 μm GTPγS with 200 μm Ba2+, NPo = 0.042 ± 0.015, 220 ± 16 μm from the soma (n = 4). *p < 0.05 compared with control; #p < 0.05 compared with 20 μm GTPγS. Error bars represent SEM.
Compared with control conditions in which no GTP was supplied via the whole-cell electrode, 20 or 200 μm GTPγS significantly increased activities of the inwardly rectifying K+ channels (Fig. 3B,C). The GTPγS-activated channels had the same single-channel conductance (γ = 33 ± 2 pS; n = 5) (Fig. 1B) as the constitutively active channels, indicating activation of the same channel type (amplitude histograms are presented in supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In addition, when 200 μm Ba2+ was included in the cell-attached electrode, the GTPγS-activated channel activities were significantly reduced (Fig. 3C) by an amount similar to the control condition (Fig. 2B). These results further support the hypothesis that these channels are GIRK channels.
Stimulation of GABAB receptors activated GIRK channels in distal dendrites of CA1 pyramidal neurons
GIRK channels are the final effectors of a diverse group of inhibitory neurotransmitters and neuromodulators, including GABA. In CA1 pyramidal neurons, focal application of the GABAB agonist baclofen to the apical dendrites elicits a larger GIRK channel-mediated hyperpolarization than when the agonist is applied close to the soma (Newberry and Nicoll, 1985). Immunohistochemistry studies have revealed localization of GABAB receptors in dendritic shafts of CA1 pyramidal neurons, whereas the distal dendrites in stratum lacunosum-moleculare (Fig. 1D) showed the strongest staining (Kulik et al., 2003).
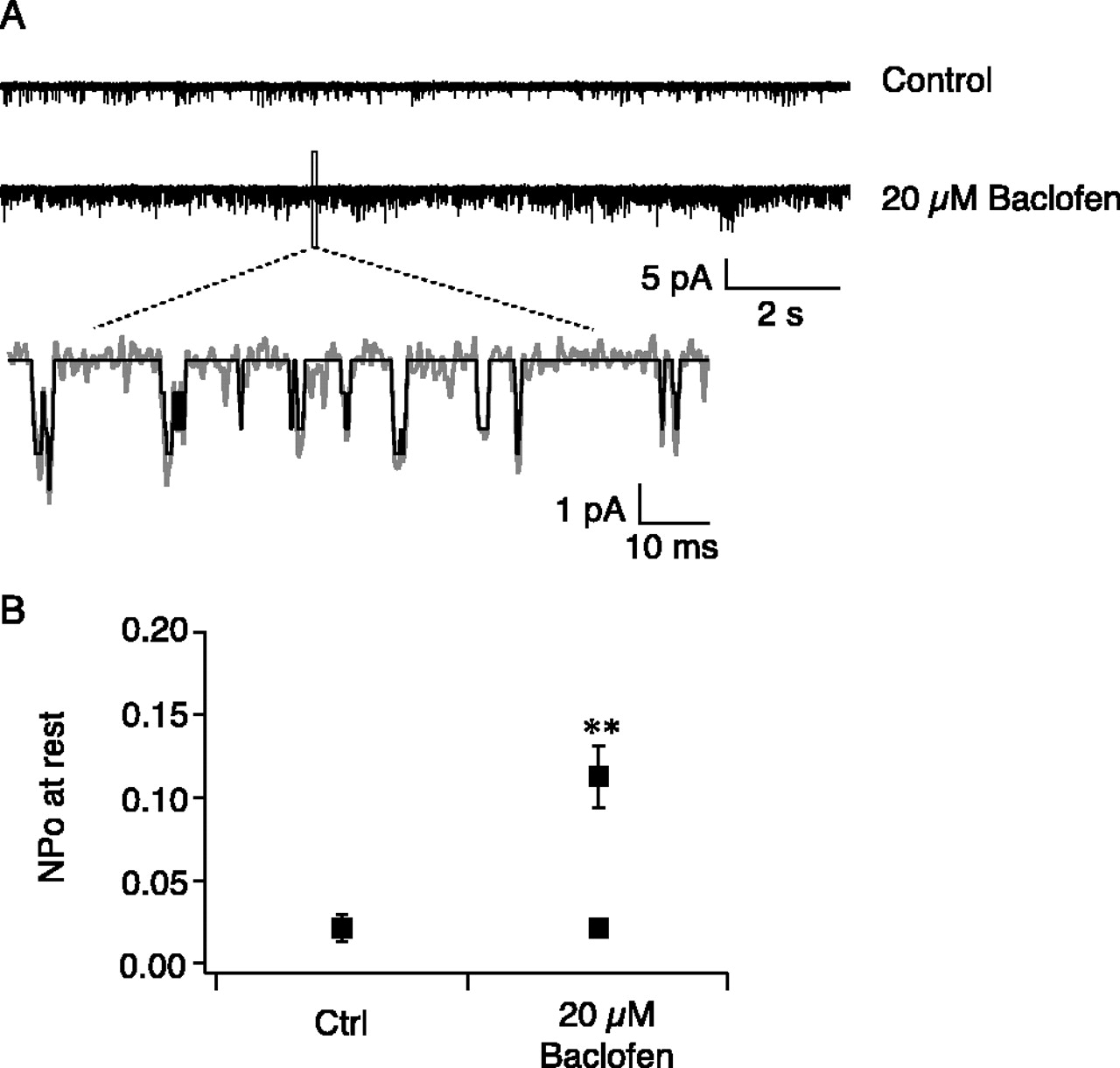
In the set of experiments illustrated in Figure 4, we tested whether the putative GIRK channels could be activated by baclofen. We included baclofen (20 μm) in the recording electrode and compared the level of channel activities at resting membrane potential with a separate group of control recordings. Because of the membrane-delimited nature of receptor-channel coupling (Soejima and Noma, 1984), the GABAB receptors and the GIRK channels must be in the same patch to exert an effect. The low probability of encountering GABAB receptors in the soma or proximal dendrites presumably led to the failure to uncover baclofen-stimulated GIRK activities at these locations (soma, NPo = 0.003 ± 0.001, n = 7; proximal dendrite 160 μm from the soma, NPo = 0.016 ± 0.002, n = 5). To increase the probability of getting both the GABAB receptors and the GIRK channels in the same patch, we performed these experiments in distal dendrites (280-330 μm from the soma). In 9 of 19 recordings with 20 μm baclofen in the electrode, the range of GIRK activity (NPo = ∼0.008-0.049) was within the range under control conditions (NPo = ∼0.008-0.061), and the average level of GIRK channel activity was not significantly different from control, suggesting that no GABAB receptors were present in these patches. However, in the other 10 recordings (of 19), each recording had a higher GIRK activity level than the largest under control conditions, whereas the average level of GIRK activities was significantly higher than control (Fig. 4). Single-channel conductance of baclofen-activated channels was very close to that of control (γ = 31 ± 1 pS; n = 5) (Fig. 1B). This indicates that, in these recordings, both GABAB receptors and GIRK channels were present in the same patch and that stimulation of GABAB receptors by baclofen led to opening of GIRK channels. In another eight recordings, 200 μm Ba2+ was included in the recording electrode in addition to 20 μm baclofen. None of these recordings had increased GIRK activity (NPo = 0.004 ± 0.0003; n = 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Stimulating GABAB receptors activated GIRK channels in distal dendrites. A, Trace examples with control solution or 20 μm baclofen in the recording pipette. The membrane was held at resting potentials. B, In 10 of 19 recordings, stimulation of GABAB receptors by baclofen led to increased GIRK channel activity. Control (Ctrl), NPo = 0.024 ± 0.007, 303 ± 4 μm from the soma (n = 8); 20 μm baclofen, NPo = 0.119 ± 0.017, 304 ± 9 μm from the soma (n = 10); NPo = 0.022 ± 0.004, 304 ± 8 μm from the soma (n = 9). **p < 0.01 compared with control. Error bars represent SEM.
Discussion
Constitutive activity of GIRK channels in the dendrites of CA1 pyramidal neurons
Using near-symmetric K+ in cell-attached recordings, we recorded inwardly rectifying K+ channels in the soma and apical dendrites of CA1 pyramidal neurons. These channels were constitutively active at resting membrane potentials with low open probability. The inwardly rectifying K+ channels in our recordings bore remarkable resemblance to the acetylcholine-activated inwardly rectifying K+ channels formerly recorded in isolated pacemaker cells of rabbit heart and later thought to be encoded by members of the GIRK family K+ channel genes (Sakmann et al., 1983; Soejima and Noma, 1984; Wickman et al., 1999). Those channels were among the first to be studied at the single-channel level and were found to be spontaneously active, with very low open probability in the absence of exogenous agonists. Later studies of the cloned and endogenous neuronal types of GIRK channels yielded similar results (Kofuji et al., 1995; Han et al., 2003). The inwardly rectifying K+ channels in our recordings had a single-channel conductance (33 pS) that fell into the conductance range of GIRK channels (Sakmann et al., 1983; Han et al., 2003) and were blocked by the inward rectifier blocker Ba2+ and the more-specific GIRK channel blocker tertiapin. They were also activated by intracellularly applied GTPγS and extracelluarly applied baclofen.
The voltage protocols in this study were designed to minimally activate voltage-dependent K+ channels in CA1 dendrites, because those channels have a more depolarized voltage activation range. Most of those channels inactivate during prolonged membrane depolarization (Chen and Johnston, 2004). CA1 neurons also express low levels of IRK genes (Karschin et al., 1996). However, our experimental condition did not reveal a significant contribution of IRK channels, because 2 μm tertiapin blocked ∼97% of total channel activity. Changes in ion composition of solutions and/or voltage protocols may be needed to better record IRK channels.
Distribution of GIRK channels along the apical dendrite of CA1 pyramidal neurons
GIRK channels were consistently recorded in every patch. Because of the very low open probability, actual numbers of channels were not determined. However, when the level of spontaneous channel activities at resting membrane potential was compared among different dendritic locations, there was a significant increase in such activities in the apical dendrites compared with the soma. This increase may come from increased channel numbers or increased open probability of the channels. Previous studies suggest that ambient adenosine may play a role in keeping these channels spontaneously active (Takigawa and Alzheimer, 2002). This seems unlikely to explain our results with cell-attached recordings in which the channels and receptors in the patch are only exposed to our pipette solutions. The level of channel activities remained stable for up to 10 min after seal formation (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), which argues against a role of prebound receptors, because the agonists are expected to unbind with an estimated off-rate of >0.9 min-1 (Newberry and Nicoll, 1985). We therefore favor the conclusion that the constitutive channel activity was agonist independent and may rely on resting GTP levels and spontaneous Gi-GTP turnover (Breitwieser and Szabo, 1988).
Functional implications
The best-known function of GIRK channels is to provide a common postsynaptic mechanism for slow inhibitory neurotransmission. We demonstrated the presence of GIRK channels in the dendrites of CA1 pyramidal neurons, which would make them physically near the G-protein-coupled receptors in the dendrites (Kulik et al., 2003) and readily accessible for receptor-mediated activation.
In addition to agonist-induced activation, the GIRK channels were also constitutively active independent of agonists. They might therefore contribute to the resting membrane conductance and play a role in synaptic integration and dendritic signaling of electrical events (Takigawa and Alzheimer, 2002). This functional role of GIRK channels is certainly facilitated by the presence of GIRK channels in the dendrites, in which most excitatory synaptic inputs are located.
Being part of the resting conductance in the dendrites also implies a role for GIRK channels in regulating the intrinsic excitability of neuronal dendrites, which strongly correlates with synaptic plasticity and temporal lobe epilepsy (Bernard et al., 2004; Frick et al., 2004). The low open probability of GIRK channels may prevent them from playing a role in the resting membrane potential and membrane conductance in the soma, in which the constitutive GIRK activities are minimal and the surface-to-volume ratio is low. However, in thinner structures such as distal and oblique dendrites, we speculate that this constitutive activity, even with low open probability, will contribute to the resting properties of dendrites.
Together, these findings suggest that dendritic GIRK channels could be important for regulating excitability and synaptic plasticity, as well as for gating behavioral states.
Footnotes
- Received October 20, 2004.
- Revision received March 1, 2005.
- Accepted March 2, 2005.
This work was supported by National Institutes of Health Grants NS37444, MH48432, and MH44754 and a fellowship from the Dart Foundation for summer research at the Marine Biology Laboratory. We thank Dr. Rick Gray for expert advice, Dr. Amiel Rosenkranz for suggestions on statistics, and Drs. Paul Pfaffinger and Darrin Brager for suggestions on this manuscript.
Correspondence should be addressed to Xixi Chen, Center for Learning and Memory, Institute for Neuroscience, University of Texas, Austin, TX 78712. E-mail: xixichen{at}mail.utexas.edu.
D. Johnston's present address: Center for Learning and Memory, Institute for Neuroscience, University of Texas, Austin, TX 78712.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253787-06$15.00/0