
Abstract
To investigate the function of laminin in peripheral nerve development, we specifically disrupted the laminin γ1 gene in Schwann cells. Disruption of laminin γ1 gene expression resulted in depletion of all other laminin chains known to be expressed in Schwann cells. Schwann cells lacking laminin do not extend processes required for initiating axonal sorting and mediating axon-Schwann cell interaction. They fail to downregulate Oct-6 and arrest at the premyelinating stage. The impaired axon-Schwann cell interaction prevents phosphorylation of β-neuregulin-1 receptors and results in decreased cell proliferation. Postnatally, laminin-null Schwann cells exhibit reduced phosphatidylinositol 3 (PI3)-kinase activity and activation of caspase cascades, leading to apoptosis. Injection of a laminin peptide into mutant sciatic nerves partially restores PI3-kinase activity and reduces apoptotic signals. These results demonstrate the following: (1) that laminin initiates axonal sorting and mediates axon-Schwann cell interactions required for Schwann cell proliferation and differentiation, and (2) that laminin provides a PI3-kinase/Akt-mediated Schwann cell survival signal.
- Schwann cell
- laminin
- proliferation
- apoptosis
- phosphatidylinositol 3-kinase
- myelin
Introduction
Myelination in the peripheral nervous system (PNS) is accomplished by Schwann cells. Schwann cell precursors are derived from neural crest cells and differentiate to immature Schwann cells. Before birth, Schwann cells destined to myelinate differentiate to promyelinating Schwann cells and extend cytoplasmic processes into axonal bundles, progressively separating these bundles to form a 1:1 ratio with individual axons (radial sorting) and enwrapping them with a myelin sheath (Webster, 1993; Mirsky and Jessen, 1999).
There is substantial evidence that Schwann cells require the formation of a basal lamina to properly ensheath and myelinate axons (Bunge, 1993). Laminins are major components of the basal lamina that appear to be especially important. In vitro studies using Schwann cell/neuronal coculture have shown that laminin deposition is required for myelination (Fernandez-Valle et al., 1993; Fernandez-Valle et al., 1994; Podratz et al., 2001). Both dystrophic mice (Xu et al., 1994; Sunada et al., 1995) and human congenital muscular dystrophy patients (Helbling-Leclerc et al., 1995) have mutations in their laminin α2 chain genes, resulting in muscular dystrophy and a dysmyelinating peripheral neuropathy. Schwann cell-specific disruption of β1 integrin, a component of many laminin receptors, also causes a dysmyelinating peripheral neuropathy with impaired radial sorting of axons (Feltri et al., 2002). Recently, we deleted laminin γ1 in the nervous system using the calcium/calmodulin-dependent protein kinase II α promoter to drive Cre expression (CaMKII/Cre:fLAMγ1 mice) (Chen and Strickland, 2003). These mice have laminin γ1 gene disruption in a subpopulation of Schwann cells and exhibit dysmyelinating phenotype. All of these results suggest that laminin is critical for PNS development, but the mechanistic details are unknown.
Laminins are heterotrimeric glycoproteins composed of an α-, β-, and γ-chain. Five α-chains, four β-chains, and three γ-chains have been identified, and 15 isoforms have been observed (Yin et al., 2003). Laminin γ1 is one of the most abundant chains and is present in 11 isoforms, including all known laminin isoforms expressed in the PNS (Patton et al., 1997; Previtali et al., 2003). To study the mechanism of laminin function in PNS development, we specifically disrupted the laminin γ1 gene in Schwann cells at their early development stages using the Mpz (myelin protein zero) promoter to drive Cre expression. Because laminin γ1 chain is a common component in all laminin isoforms expressed in Schwann cells, knocking out laminin γ1 depleted most if not all of the laminin isoforms in these cells. By doing so, this study revealed novel functions of laminin in the PNS and also uncovered the mechanism by which laminin regulates PNS development. Schwann cells that lack laminin have impaired axon interaction, which leads to impaired proliferation and differentiation. These cells also have reduced phosphatidylinositol 3 (PI3)-kinase signaling needed to maintain viability and therefore increased apoptosis. Our results, coupled with other studies (Feltri et al., 2002; Chen and Strickland, 2003), indicate a critical role for laminin in establishing a stable axon interaction that provides proliferation and differentiation signals and in maintaining Schwann cell viability.
Materials and Methods
Mice, genotyping, and analysis of Cre-mediated laminin γ1 gene recombination. Mice in which the exon 2 of the laminin γ1 gene was flanked by two loxP sites (fLAMγ1) and CaMKII/Cre:fLAMγ1 mice were generated as described previously (Chen and Strickland, 2003). mP0TOT(Cre) (P0Cre) transgenic mice have been described previously (Feltri et al., 1999). To obtain P0/Cre:fLAMγ1 mice, mice homozygous for the fLAMγ1 allele were crossed with mice heterozygous for the fLAMγ1 allele and hemizygous for the P0Cre transgene. fLAMγ1, P0Cre, Z/EG mice were generated by crossing fLAMγ1/+, P0Cre mice with a double reporter mouse line (Z/EG) from The Jackson Laboratory (Bar Harbor, ME) (Novak et al., 2000). This reporter mouse line expresses lacZ throughout embryonic development and adult stages. During Cre expression, the lacZ gene is removed, which then activates expression of the second reporter gene, enhanced green florescent protein (EGFP). Genotypes of the resulting offspring were identified by PCR analyses of tail genomic DNA as described previously (Chen and Strickland, 2003). For detection of Cre-mediated laminin γ1 gene recombination, genomic DNA were prepared from various tissues of fLAMγ1/fLAMγ1 (control) mice and P0/Cre:fLAMγ1 (mutant) mice and analyzed by PCR as described previously (Chen and Strickland, 2003).
Immunohistochemistry. Immunostaining were performed as described previously (Chen and Strickland, 2003). Primary antibodies were as follows: rat anti-laminin γ1 (1:500; Chemicon, Temecula, CA), rat anti-laminin α1 (1:500; Chemicon), rat anti-laminin β1 (1:500; Chemicon), chicken anti-neurofilament H (1:2000; Chemicon), rat anti-laminin α2 (1:500; Sigma, St. Louis, MO), rabbit anti-laminin α4 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-human myelin basic protein (MBP) (1:2000; Dako, Carpinteria, CA), rabbit anti-Oct-6 (1:200; a gift from Dr. Meijer, Erasmus University, Rotterdam, The Netherlands), rabbit anti-Krox-20 (1:50; Covance Research Products, Berkeley, CA), rat anti-bromodeoxyuridine (BrdU) (1:400; Abcam, Cambridge, UK), rabbit anti-S100 (1:1000; Sigma), and rabbit anti-activated caspase-3 and caspase-7 (1:100; Cell Signaling Technology, Beverly, MA). Fluorescein-conjugated, Rhodamine Red-X-conjugated, or Coumarin AMCA-conjugated secondary antibodies were used (1:1000; Jackson ImmunoResearch, West Grove, PA).
Semithin and electron microscopic analyses. Semithin sections and electron microscopic analyses of sciatic nerves were performed as described previously (Chen and Strickland, 2003). Nerves were fixed in 2.5% paraformaldehyde (PFA)/glutaraldehyde, and postfixed in 2% osmium tetroxide solution. The nerves were then embedded in resin, and ultra-thin cross sections were prepared and examined as described previously (Chen and Strickland, 2003).
BrdU incorporation assay. Female mice heterozygous for the fLAMγ1 allele and hemizygous for the mP0TOT(Cre) transgene were mated with male mice homozygous for the fLAMγ1 allele. BrdU incorporation assay of CaMKII/Cre:fLAMγ1 mice was as described previously (Chen and Strickland, 2003). The time of detection of a vaginal plug was counted as 0.5 d postcoitum (dpc). Pregnant mice at 13, 15.5, 17.5, and 19.5 dpc and postnatal day 5 (P5) mice were injected intraperitoneally with 100 μg of BrdU per gram of body weight. One hour later, the embryos or the sciatic nerves of P5 mice were dissected and frozen in dry ice, and the upper bodies or tails were used for genotyping. Whole embryos [embryonic day 13 (E13), E15.5, or E17.5] or sciatic nerve tangential cryosections (E17.5, E19.5, or P5) were prepared, fixed in cold methanol, denatured with 2N HCl for 20 min at 37°C, and neutralized in 0.1 m sodium borate, pH 8.5, for 10 min. Sections were coincubated with rat anti-BrdU (Abcam) and rabbit anti-neurofilament H (Chemicon). After staining with the appropriate secondary antibodies, the nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). Only cigar-shape nuclei inside the nerve tissues were counted, and double-labeled nuclei (both BrdU and DAPI) were determined. At each time point, six control and six mutant mice were analyzed. The differences in percentage of BrdU-incorporated nuclei between control and mutant nerves were analyzed by two-tailed Student's t test.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed using the In Situ Cell Death Detection kit (Roche Applied Science, Indianapolis, IN) according to the instructions of the manufacturer. Whole embryo (E15.5) or sciatic nerve tangential cryosections (P0 to adult) were prepared, fixed in 4% PFA in PBS, pH 7.4, for 20 min, and permeabilized in 0.1% Triton X-100/0.1% sodium citrate on ice for 2 min. E15.5 embryo sections were stained with anti-neurofilament antibodies to identify nerves. P15 sciatic nerve sections were stained with anti-S100, anti-laminin γ1, or anti-phosphoAkt antibodies, and nuclei were counterstained with DAPI. Double-labeled nuclei were determined. At each time point, six control and six mutant animals were analyzed. The differences in percentage of TUNEL-positive nuclei in control and mutant sciatic nerves were analyzed by two-tailed Student's t test.
Western blot analysis. Control and mutant mice at different ages were anesthetized. The sciatic nerves were exposed, immersed in PBS containing the phosphatase inhibitor Cocktail I and II (Sigma), and dissected. The nerves were homogenized in 50 mm Tris, pH 7.4, containing 150 mm NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxycholate, 1 mm EDTA, 1 mm EGTA, 10% glycerol, phosphatase inhibitor Cocktail I and II, and protease inhibitor cocktail (Sigma). Protein concentrations were determined by the method of Lowry (Bio-Rad, Hercules, CA). For immunoblotting of caspase-9, total protein of sciatic nerves were extracted in 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate cell extract buffer (Cell Signaling Technology) containing protease inhibitors. Proteins (15-20 μg) were fractionated on 4-15% SDS-PAGE, blotted onto polyvinylidene difluoride membrane (Millipore, Billerica, MA), and probed with primary antibodies. Antibodies against phosphorylated (p)-ErbB2, ErbB2, p-ErbB3, p-Akt, Akt, p-glycogen synthase kinase 3β (GSK-3β), GSK, and mouse caspase-9 were purchased from Cell Signaling Technology and used at 1:1000 dilution. Other antibodies used were anti-ErbB3 (1:200; Abcam), anti-β-actin (1:8000; Sigma), and rabbit anti-Oct-6 (1:1000; a gift from Dr. Meijer). After incubation with appropriate secondary antibodies (Amersham Biosciences, Piscataway, NJ), proteins were visualized by chemiluminescence according to the instructions of the manufacturer (Pierce, Rockford, IL).
Injections of laminin peptides. Injections were performed as described previously (Cosgaya et al., 2002). The laminin peptide EIKLLIS derived from laminin α1 chain (conserved in laminin α2 chain) and a control peptide with a scrambled amino acid sequence ILEKSLI (Tashiro et al., 1999) were purchased from American Peptide (Sunnyvale, CA). Laminin peptides (5 μg) were injected subcutaneously, starting from the caudal portion of the greater trochanter region and running parallel along the mutant sciatic nerves. The contralateral sciatic nerve served as control and was injected with control peptides. Injections were performed on postnatal day 1 and reinjected 2 d later for control and mutant mouse pups (20 of each genotype, three independent experiments). Nerves were extracted at postnatal day 5 (4 d of total treatment), and 20 μg of proteins was used for Western blot analysis. The differences of signal intensity of p-Akt and cleaved caspase-9 in Western blots were quantified and analyzed by two-tailed Student's t test.
Imaging analysis. Images of immunostained sections were acquired by using an AxioVision System (Zeiss, Oberkochen, Germany) and processed with Adobe Photoshop (Adobe Systems, San Jose, CA). The electron micrograph and Western blot film were digitized using a scanner (Microtek, Carson, CA). The signal intensity of the Western blot film was quantified by NIH Image.
Results
Generation of mice lacking laminin γ1 in Schwann cells
To specifically disrupt the laminin γ1 gene in Schwann cells, we created mice homozygous for a floxed laminin γ1 allele (fLAMγ1) (Chen and Strickland, 2003) and carrying a P0Cre transgene, mP0TOT(Cre), which activates Cre-mediated recombination specifically in Schwann cells between E13.5 and E14.5 (Feltri et al., 1999, 2002). P0/Cre:fLAMγ1 mice (referred to mutant mice hereafter) were born normally in accordance with the predicted Mendelian ratio. However, they showed tremor and progressive hindlimb paralysis during early postnatal stages. By the end of the fourth week, they had complete hindlimb paralysis, and most did not live past 2 months as a result of emaciation.
To determine the specificity of Cre-mediated laminin γ1 gene recombination, genomic DNA from various tissues was analyzed by PCR (Fig. 1A). Recombination only occurred in the mutant peripheral nerves (e.g., the sciatic nerve) but not in other tissues of mutant mice or in mice homozygous for a fLAMγ1 allele but without the Cre transgene (f/f, referred to control mice hereafter). To confirm the elimination of laminin γ1 expression in mutant peripheral nerves, transverse sciatic nerve sections at P0 were stained for laminin γ1. Laminin γ1 was present in control nerves but was absent in mutant nerves in the endoneurium outside Schwann cells; in mutant nerves, laminin γ1 was still present in blood vessels and the perineurium (outer region), in which it is presumably produced by fibroblasts (Fig. 1B). Additional analyses showed that laminin γ1 gene was disrupted at approximately E13.5-E14.5, and the protein expression of most laminin chains known to be expressed in Schwann cells were not detectable (α1, α2, α4, β1, and γ1) (see Fig. 2). These results demonstrate that the laminin γ1 gene was specifically disrupted in Schwann cells at approximately E13.5-E14.5, resulting in the depletion of all laminins.

Schwann cells lacking laminin γ1 expression are severe hypomyelination and fail to extend processes to initiate axonal sorting. A, PCR analysis of genomic DNA from various tissues of wild-type, homozygous fLAM γ1 mice (f/f; control) and P0/Cre:fLAM γ1 mice (f/f, P0-Cre; mutant). The primers used amplified the wild-type (1.3 kb), unrecombined (3.2 kb), and recombined (2.3 kb) fLAM γ1 alleles. B, Transverse sections of control and mutant sciatic nerves at P0 were double stained for laminin γ1 (red) and MBP (green). In the mutant nerve, laminin γ1 expression was absent in the endoneurium (asterisks) and only remained in the perineurium (arrowheads), and MBP expression was not detected. C, Transverse semithin sections from P28 sciatic nerves show that mutant nerves have large unsorted axonal bundles and few Schwann cells with myelin sheaths near the perineurium (arrows). D, Electron micrographs of P1 sciatic nerves show that mutant Schwann cells (arrows) do not extend cytoplasmic processes and leave axons unsorted. E, Electron micrograph of P28 sciatic nerves show that mutant nerves have large bundles of unsorted axons with some Schwann cells located outside (SC). F, Higher magnification of the boxed region in E shows that the mutant Schwann cell closely associated with unsorted axonal bundles lacks a continuous basal lamina (compare fuzzy materials indicated by arrows and denuded areas indicated by arrowheads) and does not extend processes between axons. Scale bar: B, 76 μm; C, 18.5 μm; D, 5.6 μm; E, 1.7 μm; F, 0.3 μm. wt, Wild type; ct, control; mt, mutant; Lnγ1, laminin γ1.

Disruption of laminin γ1 gene expression resulted in depletion of all other laminin chains known to be expressed in Schwann cells. Adjacent transverse sections of control and mutant sciatic nerves at P0 were stained for different laminin subunits, including α1, α2, α4, β1, and γ1. In the mutant nerves, disruption of laminin γ1 gene expression resulted in concurrent depletion of other laminin chains. Note that the laminin α1 chain, which is expressed in the perineurium in mature nerves, and the laminin α4 chain, which is expressed at low level in adult nerves, are both nearly undetectable at the P0 stage. Scale bar, 25 μm.
Mutant Schwann cells are severely hypomyelinated and fail to extend processes to initiate radial sorting of axons and mediate axon-Schwann cell interaction
We compared the onset of myelination in mutant and control sciatic nerves at P0 by staining transverse sections for MBP, a myelin marker. In control nerves, MBP was expressed normally, whereas at this time, no MBP expression could be detected in mutant nerves (Fig. 1B). At late postnatal stages (P28), myelination was complete in control nerves. In contrast, mutant sciatic nerves showed large unsorted axonal bundles and a few Schwann cells with normal myelin sheaths (Fig. 1C, arrows). In mice lacking β1 integrin in Schwann cells, perineurial cells are abnormally located along microfasciculations in the center of the nerve (Feltri et al., 2002). In agreement with this observation, laminin γ1 expression was not detectable in the endoneurium outside mutant Schwann cells before P5 (Figs. 1B, 2) but appeared at later stages in the endoneurium of some centrally located Schwann cells in mutant sciatic nerves (P28) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Therefore, these centrally located Schwann cells with normal myelin sheaths in mutant nerves might obtain laminin from nearby perineurial cells or escape recombination of the laminin γ1 gene and thus undergo normal differentiation. To address this question, we crossed the mutant mice with an EGFP reporter mouse line Z/EG (lacZ/EGFP) (Novak et al., 2000) and obtained mice that are homozygous for the floxed laminin γ1 allele and are also hemizygous for both P0-Cre transgene and the Z/EG reporter transgene. In mice carrying the reporter gene, during Cre-mediated recombination, EGFP will be expressed. We analyzed the sciatic nerves from these mice at P28 and found that the few Schwann cells that have laminin γ1 surrounding them expressed the reporter gene EGFP. This result indicated that these Schwann cells have undergone Cre-mediated laminin γ1 gene recombination since the reporter gene was expressed, but they have obtained laminin γ1 from other cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, it is also possible that some Schwann cells might escape Cre-mediated laminin γ1 gene recombination or undergo incomplete (monoallelic) recombination.
We next compared the morphology of control and mutant Schwann cells at P1 and P28 by electron microscopy. In control sciatic nerves at P1, most Schwann cells had extended processes to segregate axons, and some axons had formed a 1:1 relationship with individual Schwann cells (Fig. 1D). In contrast, mutant Schwann cells at P1 did not extend processes (Fig. 1D, arrows) and left axons unsorted. Ultrastructural analysis of mutant sciatic nerves at late postnatal stages showed that Schwann cells closely associated with unsorted axonal bundles (Fig. 1E, SC) lacked a continuous basal lamina (Fig. 1F, compare fuzzy materials indicated by arrows and denuded areas indicated by arrowheads) and did not extend their cytoplasmic processes between axons, thus failing to segregate, interact, and myelinate axons.
Mutant Schwann cells fail to downregulate Oct-6 and arrest in premyelinating stages
During the deposition of the basal lamina, the essential step for Schwann cell differentiation is the upregulation of the transcription factor Oct-6 (Bermingham et al., 1996; Jaegle et al., 1996). Oct-6 expression in Schwann cells is transient and peaks in the promyelinating stage and progressively downregulated during postnatal development. Oct-6 function is required in promyelinating Schwann cells for timely differentiation into myelinating Schwann cells (Jaegle et al., 1996). We examined Oct-6 at different developmental stages to detect whether their expression is altered by laminin deficiency. In both control and mutant peripheral nerves, Oct-6 was observed at a low level at approximately E15 (data not shown), became readily detectable at E17.5, and peaked at approximately P3, even in the absence of laminin γ1 (Fig. 3A). During postnatal development, most control Schwann cells gradually downregulated Oct-6 (Fig. 3B, control panel, D); however, mutant Schwann cells were unable to downregulate Oct-6, indicating that these Schwann cells were arrested at the premyelinating stage (Fig. 3B, D).

Mutant Schwann cells fail to upregulate Krox-20 and downregulate Oct-6. A, Whole embryo sections at E17.5 and longitudinal sciatic nerve sections at P3 were stained for Oct-6 (green) and laminin γ1 (Lnγ1; red), and images of Oct-6/laminin γ1 at P3 were merged. E17.5 embryo sections were stained for neurofilament (NF; blue) to identify nerves. These images indicate that initiation of Oct-6 does not require laminin γ1. B, Transverse control and mutant sciatic nerve sections at P15 and P28 were stained for Oct-6 (green) and neurofilament (red), and the images from mutant nerves were merged. During postnatal development, Oct-6 fails to be downregulated in mutant Schwann cells. C, Whole embryo sections at E19.5 and transverse sciatic nerve sections at P5 were stained for Krox-20 (green) and laminin γ1 (red), and the images of Krox-20/laminin γ1 were merged. E19.5 embryo sections were stained for neurofilament (blue) to identify nerves. Although initiation of Krox-20 does not require laminin γ1, high level expression of Krox-20 in Schwann cells is impaired during the postnatal development. Scale bar, 50 μm. D, Oct-6 expression in control and mutant sciatic nerves at P3, P15, and P28 (n = 15 per genotype per age) was assessed by Western blots with antibodies against Oct-6. β-Actin was the loading control. Mutant Schwann cells were unable to downregulate Oct-6 and showed aberrant consistent expression of Oct-6. c, Control; m, mutant.
Krox-20 is another transcription factor essential for Schwann cell myelination, which is expressed continuously and specifically in myelin-producing Schwann cells (Topilko et al., 1994). Several studies suggest that Oct-6 and Krox-20 cross-regulate each other (Zorick et al., 1999; Nagarajan et al., 2001). Oct-6 is required for timely expression of Krox-20. High level expression of Krox-20, in turn, is required for downregulation of Oct-6 after the peak of myelination and for the activation of the major myelin genes, including P0 and MBP. Therefore, we examined Krox-20 expression in mutant peripheral nerves. At approximately E15, Krox-20 was detected at very low levels in both control and mutant nerves (data not shown). At E19.5, normal Krox-20 expression was observed in the absence of laminin γ1. In contrast, during postnatal development, when control Schwann cells progressively upregulated Krox-20, only a few Schwann cells in mutant nerves expressed high levels of Krox-20 (Fig. 3C), indicating that upregulation of Krox-20 is impeded in mutant Schwann cells.
Together, these results suggest that laminins are not required for the induction of Oct-6 and Krox-20 but are critical for the maintenance of Krox-20 expression and downregulation of Oct-6 and are therefore necessary for differentiation of myelinating Schwann cells.
Laminin mediated axon-Schwann cell interactions are critical for Schwann cell proliferation
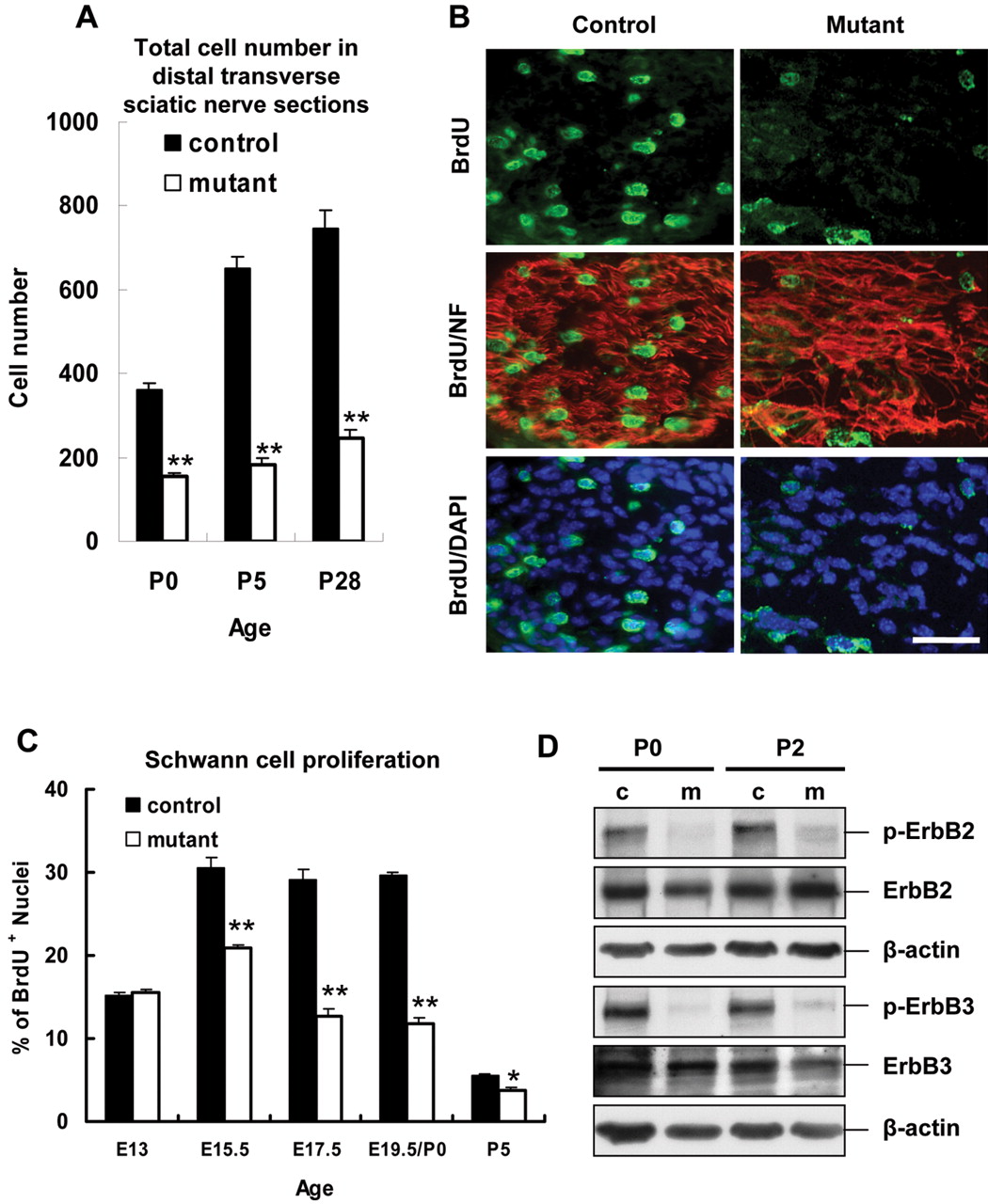
By gross observation, mutant sciatic nerves were thinner than controls (Fig. 1C), and this could be attributable to reduced total cell number inside mutant nerves. We stained transverse sections from similar distal parts of mutant and control nerves at various ages with DAPI to measure the total number of nuclei. The cell number in mutant sciatic nerves compared with controls was diminished to 43% at P0, 28% at P5, and 33% at P28 (Fig. 4A).

Impaired interactions of laminin γ1-null Schwann cells and axons result in severe reduction of Schwann cell proliferation. A, Plot of the total cell number in distal transverse sections (mean ± SEM) at various ages. Total cell number in distal transverse sections is significantly reduced in mutant sciatic nerves (open bars) at all ages compared with control nerves (filled bars) (n = 6 per genotype per age; **p < 0.001). B, Longitudinal sections of control and mutant sciatic nerves at E19. 5 were triple stained for BrdU (green), neurofilament (red), and DAPI (blue) after a 1 h pulse of BrdU, and the images of BrdU/neurofilament and BrdU/DAPI were merged. Mutant Schwann cells show reduced nuclei BrdU incorporation compared with control Schwann cells. Scale bar, 60 μm. C, Plot of the percentage (mean ± SEM) of BrdU-positive nuclei at various ages. The percentages of BrdU-incorporated nuclei are significantly reduced in mutant nerves (open bars) at E15.5, E17.5, E19.5/P0, and P5 compared with control nerves (filled bars) (n = 6 per genotype per age; *p < 0.01; **p < 0.001). D, The response of Schwann cells to NRG-1 at P0 and P2 was assessed on immunoblots with antibodies recognizing p-ErbB2 and p-ErbB3. β-Actin is the loading control. Mutant Schwann cells show severe reduction in response to axonal mitogens. c, Control; m, mutant.
During late embryonic and perinatal stages, Schwann cells proliferate vigorously to rearrange, sort, and ensheath axons (Stewart et al., 1993). Reduced Schwann cell proliferation could cause the decreased total cell number. To determine whether Schwann cells proliferate normally, we compared BrdU-incorporating nuclei in control and mutant nerves at various ages. During the peak of Schwann cell proliferation (E17.5-E19.5) (Stewart et al., 1993), control nerves showed extensive cell proliferation, whereas mutant nerves had few proliferating cells (Fig. 4B). Statistical analyses showed that the percentage of BrdU-incorporating nuclei was similar before laminin γ1 gene disruption (E13) but significantly decreased in mutant nerves compared with controls at E15.5 (20.8 vs 30.6%), E17.5 (12.7 vs 29.2%), E19.5/P0 (11.8 vs 29.6%), and P5 (3.8 vs 5.6%) (Fig. 4C), indicating that mutant Schwann cell proliferation is reduced.
Because axons are a major source of Schwann cell mitogens (Wood and Bunge, 1975; Morrissey et al., 1995), impaired axon-Schwann cell interaction could result in reduced Schwann cell proliferation. β-Neuregulin-1 (NRG-1) is a major axon-derived Schwann cell mitogen and can interact with and stimulate the phosphorylation of receptor tyrosine kinases ErbB2 and ErbB3 on Schwann cells (Morrissey et al., 1995). We measured the phosphorylation level of ErbB2 and ErbB3 in sciatic nerve extracts during early postnatal stages (Fig. 4D). We found that, although ErbB2 and ErbB3 receptor levels were not significantly changed, the phosphorylation of both proteins was greatly reduced, suggesting that the inability of mutant Schwann cells to be exposed to axonal mitogens is a major cause of decreased proliferation. This result indicates that laminin-mediated axon-Schwann cell interactions are essential for proliferation.
A previous study using a CaMKII-Cre transgene to direct conditional laminin γ1 gene knock-out (Chen and Strickland, 2003) showed a similar but much less severe peripheral nerve phenotype compared with this mouse line (P0/Cre:fLAMγ1 mice). In the CaMKII/Cre:fLAMγ1 mice (mice that are homozygous for a floxed laminin γ1 gene and also hemizygous for CaMKII-Cre transgene), Schwann cell proliferation was not affected during embryonic stages, in contrast to the P0/Cre:fLAMγ1 mice. We compared the patterns of laminin γ1 disruption in these two mouse lines during embryonic stages. We found that, in the endoneurium of P0/Cre:fLAMγ1 mice, laminin γ1 immunoreactivity was completely absent from E15.5 to E19.5/P0, and cell proliferation was dramatically reduced [Fig. 5 (quantitation in Fig. 4C)]. However, in the CaMKII/Cre:fLAMγ1 mice, laminin γ1 immunoreactivity was absent in some small patches in the endoneurium and Schwann cell proliferation was not significantly reduced [Fig. 5 (quantitative result by Chen and Strickland, 2003)]. This dramatic difference in the patterns of laminin γ1 gene disruption in these two mouse lines may explain the difference in Schwann cell proliferation.
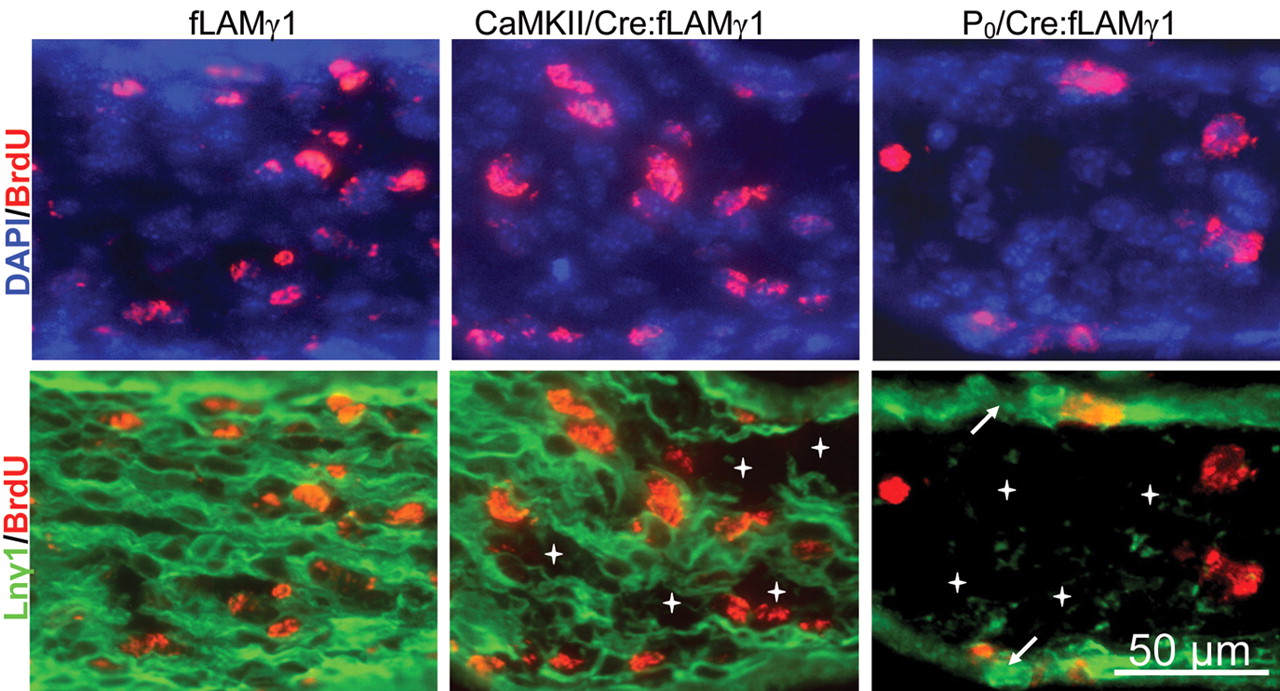
Schwann cell proliferation is not impaired in CaMKII/Cre:fLAMγ1 mutant mice. Longitudinal sections of control (fLAMγ1), CaMKII/Cre:fLAMγ1, and P0/Cre:fLAMγ1 mutant sciatic nerves at E19.5 were triple stained for BrdU (red), laminin γ1 (green), and DAPI (blue) after a 1 h pulse of BrdU, and the images of BrdU/laminin γ1 and BrdU/DAPI were merged. Laminin γ1 expression was disrupted in some small patches in the sciatic nerves of CaMKII/Cre:fLAMγ1 mutant mice (stars), and Schwann cell proliferation was not impaired [quantitative analysis was shown previously (Chen and Strickland, 2003)]. However, laminin γ1 expression was almost completely disrupted in the Schwann cells in P0/Cre:fLAMγ1 mutant sciatic nerves (stars), and cell proliferation was dramatically reduced (quantitative analysis shown in Fig. 4). Laminin γ1 expression in the epineurium of P0/Cre: fLAMγ1 mutant sciatic nerves was normal (arrows).
Together, our results show that complete ablation of laminin expression during embryonic stages dramatically reduces Schwann cell proliferation.
Schwann cells lacking laminin γ1 expression undergo apoptosis at postnatal stages
Extracellular matrix molecules such as laminin are often essential for maintenance of cell viability (Meredith et al., 1993; Chen and Strickland, 1997). Disruption of laminin γ1 may affect Schwann cell survival. We performed TUNEL to determine the extent of Schwann cell apoptosis at various ages (Fig. 6A, B). There was little apoptosis at E15.5 and E17.5 (data not shown), and the percentages of apoptotic cells were similar in control and mutant nerves. However, in early postnatal stages, the percentage of dying cells in mutant nerves progressively increased, peaked at P15, and gradually declined as the nerves matured. In contrast, we seldom found apoptotic cells inside control nerves during these stages. TUNEL-positive cells in mutant nerves were significantly higher than in controls at P0 (1.8 vs 0.5%), P5 (3.3 vs 0.1%), P15 (7.1 vs 0.04%), and P28 (3.0 vs 0.2%) but not at E15.5 (1.1 vs 0.6%) and adult (0.5 vs 0.2%).

Schwann cells that lack laminin γ1 show an increased percentage of cell death. A, Representative transverse sciatic nerve sections of control and mutant mice at P15 were stained with TUNEL (red) and counterstained with DAPI (blue), and the images were merged. Apoptotic cells can be detected in mutant but not control nerves. B, Plot of the percentage of TUNEL-positive nuclei (mean ± SEM) at various ages. The percentages of TUNEL-positive nuclei are higher in mutant (open bars) than in control nerves (filled bars) at P0, P5, P15, and P28 (n = 6 per genotype per age; *p < 0.01; **p < 0.001). C, P15 mutant sciatic nerve sections were stained with DAPI (blue), S100 (red), and TUNEL (green), and images were merged. The staining indicates that the cell with TUNEL-positive nuclei is a Schwann cell. D, P15 mutant sciatic nerve sections were stained with DAPI (blue), laminin γ1 (Lnγ1; red), and TUNEL (green), and images were merged. The staining shows the TUNEL-positive cell does not express laminin γ1 (arrows), whereas the rest of TUNEL-negative cells still expressed laminin γ1 (arrowheads). Scale bars: A, 20 μm; C, D, 8 μm.
Mesenchymal cells are recruited to peripheral nerves to generate the perineurium at E15-E17 (Parmantier et al., 1999). To determine whether the apoptotic cells in the sciatic nerve were Schwann cells and lack laminin γ1 expression, we stained mutant nerve sections at P15 for laminin γ1, TUNEL, and a Schwann cell marker, S100. Cytoplasmic S100 staining showed that the apoptotic cells in mutant nerves were Schwann cells (Fig. 6C) that had lost laminin γ1 expression (Fig. 6D).
Because axonal survival signals are only important in early (before P6) but not for later postnatal developmental stages (Grinspan et al., 1996), it is unlikely that late postnatal apoptosis (P15-P28) (Fig. 6B) is attributable to a lack of proper Schwann cell/axon relationship. This delayed apoptosis may indicate that laminin is required for long-term survival of Schwann cells, which has been suggested previously (Meier et al., 1999). Consistent with this observation, the CaMKII/Cre:fLAMγ1 mice, in which laminin disruption in Schwann cells happens later and is incomplete (at approximately E17.5-E19.5; data not shown) (Fig. 5) compared with that in P0/Cre:fLAMγ1 mice (Cre expression at approximately E13.5-E14.5) (Feltri et al., 1999) showed a later onset of Schwann cell apoptosis (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Schwann cell apoptosis continued through adulthood in CaMKII/Cre:fLAMγ1 mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) but not in P0/Cre:fLAMγ1 mice (Fig. 6B), probably because most Schwann cells die before P0/Cre:fLAMγ1 mice reach adulthood, and fewer Schwann cells remain within the sciatic nerve. Therefore, significant increases in apoptosis cannot be detected.
Together, our results coupled with previous studies indicate that loss of laminin could be a direct cause of apoptosis. However, we cannot rule out the possibility that impaired ErbB signaling in early postnatal development (Fig. 4D) may contribute to part of increased apoptosis because the axon-derived neuregulin signal still regulates Schwann cell survival to some extent in early postnatal stages (Grinspan et al., 1996).
Laminin γ1-null Schwann cells show reduced PI3-kinase activities and elevated apoptosis/caspase signaling
The PI3-kinase/Akt pathway plays a critical role in controlling the balance between cell survival and apoptosis (Burgering and Coffer, 1995). Several in vitro studies have shown that this pathway is important for Schwann cell viability (Cheng et al., 2000; Maurel and Salzer, 2000). To investigate whether the laminin γ1-null Schwann cells have reduced PI3-kinase/Akt signaling, we assessed PI3-kinase activity by measuring the phosphorylation level of Akt/PKB protein in sciatic nerve extracts at various ages (Fig. 7A). Akt phosphorylation progressively increased in control nerves during postnatal development. However, in mutant sciatic nerves, Akt phosphorylation decreased from P0 to P28 compared with control, with a maximum decrease at approximately P15. We stained nerve sections for both S100 and p-Akt and confirmed that the majority of PI3-kinase activity was from Schwann cells (data not shown).

Laminin γ1-null Schwann cells show impaired PI3-kinase activity and elevated apoptosis/caspase signaling. A, PI3-kinase activity of control and mutant sciatic nerves at P0, P5, P15, P28, and adult (n = 15 per genotype per age) were assessed on immunoblots with antibodies against p-Akt, the downstream effector of PI3-kinase. Mutant sciatic nerves compared with control nerves show reduced PI3-kinase activity at P0, P5, P15, and P28. B, Akt kinase activity of control and mutant sciatic nerves at P5 and P15 were assessed on immunoblots with antibodies recognizing p-GSK-3β. Mutant Schwann cells have reduced Akt kinase activities at P5 and P15 as judged by decreased phosphorylation of GSK-3β. C, Endogenous level of activated (cleaved) caspase-9 in control and mutant sciatic nerves at P5 and P15 was assessed by immunoblots with antibodies recognizing both full length (49kDa) and the large fragment of mouse caspase-9 after cleavage at Asp353 (37 kDa) and Asp368 (39 kDa). The mutant sciatic nerves show activation of caspase-9 at both time points. D, Longitudinal sciatic nerve sections of control and mutant mice at P15 were stained with activated capase-3 and caspase-7 antibodies (red) and counterstained with DAPI (blue), and the images were merged. Mutant sciatic nerves show increased activated caspase-3 and caspase-7. c, Control; m, mutant. Scale bars, 20 μm.
During activation, Akt phosphorylates and inactivates several downstream targets, including GSK-3 and caspase-9, to execute its anti-apoptotic effect. GSK-3 promotes apoptosis, and its activity can be inhibited by Akt-mediated phosphorylation at Ser21 of GSK-3α and Ser9 of GSK-3β (Cross et al., 1995). To determine the Akt-mediated anti-apoptotic effect, we examined the endogenous level of p-GSK-3β in sciatic nerve extracts at P5 and P15, the peak of apoptosis in mutant nerves. In mutant sciatic nerves, GSK-3β protein was comparable with controls, but its phosphorylation level was reduced at both ages (Fig. 7B).
Caspase-9 is inhibited by Akt-mediated phosphorylation (Cardone et al., 1998) and is the key initiator of the intrinsic apoptotic pathway (Budihardjo et al., 1999). During apoptotic stimulation, procaspase-9 (49 kDa in mice) is processed into a large active subunit (37 or 39 kDa) by self-cleavage. Cleaved caspase-9 activates other effector caspases, including caspase-3 and caspase-7, and initiates a caspase cascade, leading to programmed cell death. We examined P5 and P15 sciatic nerves for the large active fragment of caspase-9 by Western blotting (Fig. 7C) and the activation of downstream effector caspase-3 and caspase-7 (Fig. 7D) by immunostaining. At these ages, activated caspase-9, caspase-3, and caspase-7 could be detected in mutant nerves but not in controls (Fig. 7C,D). These results further suggest that PI3-kinase/Akt-mediated anti-apoptotic effects are impaired in Schwann cells lacking laminin γ1, resulting in elevated GSK-3 activity and the initiation of a caspase cascade to promote apoptosis.
Laminin-induced PI3-kinase/Akt activation is required for Schwann cell survival
The increased apoptosis and decreased PI3-kinase activities in mutant sciatic nerves are temporally correlated (Fig. 8A). This suggests that the disruption of laminin γ1 in Schwann cells may cause impaired PI3-kinase/Akt signaling, thus leading to apoptosis. To test this possibility, we performed a rescue experiment by injecting a laminin peptide into mouse sciatic nerves. It has been shown that a short peptide derived from laminin containing the sequence EIKLLIS, which may represent the integrin-activating site of laminin, can activate PI3-kinase/Akt-mediated survival signaling in neuronal cell culture (Tashiro et al., 1999; Gary and Mattson, 2001).

Restoration of PI3-kinase activity in mutant sciatic nerves suppresses caspase-mediated death signaling. A, The levels of p-Akt in mutant sciatic nerves from Figure 7A were quantified (normalized with total Akt level) and compared with control nerves to obtain the percentage of decreased p-Akt level. The percentages of increased TUNEL-positive nuclei in mutant sciatic nerves were obtained by subtracting the average percentages of TUNEL-positive nuclei in control from mutant nerves in Figure 6 B. A plot of the percentages of increased TUNEL-positive nuclei (black bars) and decreased p-Akt level (black circles) in mutant sciatic nerve against the ages of mice shows that reduced PI3-kinase activities correlated temporally with increased Schwann cell death. B, Control sciatic nerve or laminin peptide (lp)- and control peptide (cp)-injected mutant sciatic nerve extracts were immunoblotted with antibodies recognizing Akt or phosphorylated Akt. Caspase signaling was evaluated on the same blot using antibodies against caspase-9. β-Actin served as a loading control. Compared with control nerves, the PI3-kinase activities were partially restored in mutant sciatic nerves injected with the laminin peptide, resulting in the reduction of activated caspase-9 level. C, Plot of the signal intensity (mean ± SEM) of p-Akt and cleaved caspase-9 from quantitative analysis of Western blots from B. The signal intensity of p-Akt and cleaved caspase-9 in laminin peptide-injected (Ln) mutant sciatic nerves increased 75% and decreased 50%, respectively, compared with contralateral control (con) nerves (n = 20 mice per genotype per lysate, 3 independent experiments; *p < 0.05; **p < 0.01).
Laminin peptide was injected into one sciatic nerve of mutant mice, whereas the contralateral nerve was injected with a scrambled control peptide ILEKSLI (Tashiro et al., 1999). Injection of laminin peptide but not the control peptide along the mutant sciatic nerves partially restored Akt phosphorylation (increase 75% compared with contralateral side; normalized with total Akt) and also partially suppressed the activation of caspase-9 (decrease 50% compared with contralateral side; normalized with β-actin) (Fig. 8B, C). The control peptide was not toxic because injection into control nerves did not induce apoptosis. Together, these data indicate that the disruption of laminin γ1 in Schwann cells, at least in part, accounts for the impaired PI3-kinase/Akt signaling and increased Schwann cell apoptosis.
Discussion
Using the Cre/LoxP system, we specifically disrupted the laminin γ1 gene in Schwann cells during their early developmental stages. Disruption of laminin γ1 gene resulted in complete depletion of all laminins in Schwann cells. These mice have revealed novel mechanistic insights into laminin function in Schwann cell development. First, laminin is essential for differentiation of Schwann cells; mutant Schwann cells fail to downregulate Oct-6 and are arrested in the premyelinating stage. Second, laminin guides Schwann cells to extend processes and enwrap axons, which is essential for axon-Schwann cell interaction, axonal sorting, and myelination. Impaired axon-Schwann cell interaction prevents exposure of the cells to axon-derived mitogens and causes severe reduction of proliferation. Third, laminin provides a PI3-kinase-mediated signal to maintain Schwann cell survival. Disruption of laminin results in reduced PI3-kinase activity, leading to apoptosis. Injection of a laminin peptide into mutant peripheral nerves partially restores this signaling pathway and suppresses caspase-mediated death signal.
Laminin γ1 and β1 integrin in Schwann cell development
Many phenotypes in mice lacking laminin γ1 in Schwann cells (Fig. 1C,E) are similar to those observed in mice lacking β1 integrin in Schwann cells (Feltri et al., 2002), indicating that β1 integrin plays a major role in mediating laminin signaling. However, proliferation is reduced in Schwann cells lacking laminin γ1 but is not significantly affected in β1 integrin-null Schwann cells. There are three likely possibilities for why laminin γ1 affects proliferation: (1) laminin might act as a direct Schwann cell mitogen because this effect has been observed in vitro (McGarvey et al., 1984; Baron-Van Evercooren et al., 1986); in this case, laminin γ1 would use receptors other than β1 integrin; (2) the laminin basal lamina could act as a scaffold to attract and bind growth factors and influence Schwann cell proliferation; and (3) the proliferation effect might be secondary to axonal sorting and provision of axon-derived growth factors (Fig. 4D). We think the third possibility is most likely based on our results (Fig. 4D) and the following observations. In early postnatal stages (P1), Schwann cells lacking β1 integrin send abnormally shaped, thick cytoplasmic processes to ensheath groups of axons. The formation of unsorted axonal bundles in later stages partly results from the retraction of processes (Feltri et al., 2002), at which time Schwann cells may have the opportunity to encounter to axon-derived mitogens. However, Schwann cells lacking laminin γ1 are unable to extend cytoplasmic processes (Fig. 1D, E) and fail to interact with axonal mitogens. Based on these observations and our analyses from P0/Cre:fLAMγ1 mice, we suggest that laminins have two major functions in PNS development (Fig. 9). First, immature Schwann cells synthesize and deposit high levels of laminins. Laminin deposition guides Schwann cells to extend processes to separate axonal bundles and enwrap individual axons, which initiates axon-Schwann cell interaction. During axonal sorting, immature and premyelinating Schwann cells are able to interact with and be exposed to axon-derived signals, which are required for their proliferation and differentiation. β1 integrin on Schwann cells interacts with laminins to stabilize Schwann cell processes and facilitate the completion of radial sorting and ensheathment of axons. Second, after Schwann cells differentiate, laminins in the Schwann cell basal lamina provide a PI3-kinase/Akt-mediated survival signal to maintain Schwann cell viability and axon-Schwann cell unit integrity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed mechanism for the function of laminins in PNS development. Laminin deposition guides Schwann cells to extend processes that initiate axonal sorting and mediate axon-Schwann cell interaction. Axon-Schwann cell interaction allows Schwann cells to be exposed to axon-derived signals for proliferation and additional differentiation. β1 integrins expressed on Schwann cells interact with laminins to stabilize the cytoplasmic processes to facilitate completion of the radial sorting of axons. After Schwann cell differentiation, laminins in Schwann cell basal lamina provide PI3-kinase/Akt-mediated survival signal to maintain Schwann cell viability.
Schwann cells express several potential laminin receptors, including α6β1 and α6β4 integrins and dystroglycan (Previtali et al., 2003), and α6β1 is thought to be the major laminin receptor in Schwann cells. The β1 integrin-null Schwann cells can ensheath axons prenatally and myelinate axons after birth with some delay (Feltri et al., 2002) in contrast to laminin γ1-null Schwann cells that do not exhibit these processes. The postnatal myelination difference could be attributable to compensation of β1 integrin by another laminin receptor, for example α6β4 or dystroglycan, both of which are expressed in postnatal Schwann cells (Previtali et al., 2003). The prenatal ensheathment difference is harder to explain, because only α6β1 integrin is observed before birth (Previtali et al., 2003). This suggests that an unidentified laminin receptor expressed in embryonic Schwann cells is involved during the ensheathment of axons.
The defects in the PNS observed in mice lacking laminin γ1 (this report) are more severe than those found in dystrophic mice (dy2J/dy2J or dy3K/dy3K), which have a mutation or complete deficiency in their laminin α2 gene, resulting in a lack of laminin-2 (α2 β1 γ1) (Xu et al., 1994; Nakagawa et al., 2001). However, in the laminin α2 mutant mice, laminin-1 and laminin-8 are upregulated, which can partially compensate for the loss of laminin-2 (Patton et al., 1997; Previtali et al., 2003; Yang et al., 2005). Because laminin-8, laminin-1, and laminin-2 all contain the γ1 chain, compensation in the laminin γ1-depleted mice is not possible and results in a more severe phenotype. Consistent with this observation, combined deficiency of laminin 2/8 (dy2J/α4null mice) caused more severe defects than those in dystrophic mice (Yang et al., 2005), and the severity is similar to our mutant mice (this report). Yang et al. also provided evidence that laminins are important for Schwann cell proliferation. As with dy2J/α4null and Lnα2/α4-DKO mice, our mutant mice also show severe defects in Schwann cell proliferation (Fig. 4C).
Neuregulin-ErbB and PI3-kinase pathway in Schwann cell survival
At E12-E13 in mice, the survival of Schwann cell precursors depends on axon-derived β-neuregulin 1/Erb B pathway (Dong et al., 1995; Riethmacher et al., 1997). After this stage, Schwann cells establish an autocrine loop but also require laminin for long-term survival (Meier et al., 1999). Because P0/Cre-mediated laminin γ1 disruption occurs between E13.5 and E14.5 and because there was no significant cell death at approximately this stage (Fig. 6B), it is unlikely that the neuregulin 1/Erb B signaling pathway is affected in mutant nerves.
The PI3-kinase activity in mutant Schwann cells was severely reduced (Fig. 7A). This reduced PI3-kinase activity may be a cause or a consequence of the impaired differentiation of mutant Schwann cells because the PI3-kinase pathway is important for Schwann cell differentiation and myelination (Maurel and Salzer, 2000; Ogata et al., 2004). However, disruption of laminin γ1 may also contribute to the reduction of this survival signaling pathway and result in apoptosis based on the following observations. At P0/P1, both control and mutant Schwann cells are at similar differentiating stages (premyelinating stage), but the mutant Schwann cells had reduced PI3-kinase activity and increased apoptosis. Additionally, mutant Schwann cells infused with laminin peptides showed partial restoration of PI3-kinase activity and reduced apoptosis.
Krox-20 and Oct-6 in laminin γ1-mediated effects
We observed reduced Krox-20 expression and increased Schwann cell death in mutant sciatic nerves (Figs. 3, 6). Because Krox-20 can suppress c-Jun-mediated TGFβ-induced Schwann cell apoptosis (Parkinson et al., 2004), increased Schwann cell apoptosis could result from the failure of Krox-20 to inhibit c-Jun activation. However, the phosphorylation of c-Jun at postnatal stages between control and mutant sciatic nerves was similar (data not shown), suggesting that the TGFβ pathway did not play a major role in the increased apoptosis of mutant Schwann cells.
The observation of laminin-independent initiation of Oct-6 and Krox-20 (Fig. 3) raises interesting questions: can laminin directly induce myelinating signals, or is the main function of laminin in early Schwann cell development to initiate radial sorting and ensheathment of axons? Radial sorting and ensheathment of axons are prerequisites for myelination. Whether laminin produces a signal to induce myelin gene expression and what this signal is are future questions of interest.
Footnotes
- Received December 10, 2004.
- Revision received March 14, 2005.
- Accepted March 18, 2005.
This work was supported by National Institutes of Health Grants NS-35704 and NS-38472 (S.S.) and NS-045630-01 (M.L.F.). We thank Y. F. Chien and Z. Lin for assistance in mouse genotyping, E. C. Sphicas in the Bio-Imaging Resource Center at The Rockefeller University for technical support in electron microscopy, and members of the Strickland laboratory for helpful suggestions.
Correspondence should be addressed to Dr. Zu-Lin Chen, Laboratory of Neurobiology and Genetics, The Rockefeller University, 1230 York Avenue, New York, NY 10021. E-mail: chenz{at}rockefeller.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254463-10$15.00/0