

Abstract
One of the major modes of activity of high-density cultures of dissociated neurons is globally synchronized bursting. Unlike in vivo, neuronal ensembles in culture maintain activity patterns dominated by global bursts for the lifetime of the culture (up to 2 years). We hypothesize that persistence of bursting is caused by a lack of input from other brain areas. To study this hypothesis, we grew small but dense monolayer cultures of cortical neurons and glia from rat embryos on multi-electrode arrays and used electrical stimulation to substitute for afferents. We quantified the burstiness of the firing of the cultures in spontaneous activity and during several stimulation protocols. Although slow stimulation through individual electrodes increased burstiness as a result of burst entrainment, rapid stimulation reduced burstiness. Distributing stimuli across several electrodes, as well as continuously fine-tuning stimulus strength with closed-loop feedback, greatly enhanced burst control. We conclude that externally applied electrical stimulation can substitute for natural inputs to cortical neuronal ensembles in transforming burst-dominated activity to dispersed spiking, more reminiscent of the awake cortex in vivo. This nonpharmacological method of controlling bursts will be a critical tool for exploring the information processing capacities of neuronal ensembles in vitro and has potential applications for the treatment of epilepsy.
- bursting
- dissociated culture
- cortex
- epilepsy
- distributed stimulation
- multi-electrode array
- deep brain stimulation
Introduction
The mammalian cortex has been studied in vitro in the form of dissociated monolayer cultures for several decades. Such cultures retain many morphological, pharmacological, and electrical properties of cortical networks in vivo (Dichter, 1978) and allow much more detailed observation and manipulation than intact brains, at the molecular, cellular, and network levels (Droge et al., 1986; Emery et al., 1991; Curtis et al., 1992; Wilkinson, 1993; Bove et al., 1994, 1997; Rhoades et al., 1996; Canepari et al., 1997; Gross et al., 1997; Liu et al., 1997; Harsch et al., 1998; Honma et al., 1998; Jimbo et al., 1998, 1999; Turrigiano, 1999; Harsch and Robinson, 2000; Zhu et al., 2000; Keefer et al., 2001; Streit et al., 2001; Shahaf and Marom, 2001; Corner et al., 2002).
The most prominent feature of the electrical activity of highdensity dissociated cortical cultures is their propensity for synchronized bursting (Murphy et al., 1992; Gross et al., 1993; Wong et al., 1993; Kamioka et al., 1996; Canepari et al., 1997; Voigt et al., 1997; Gross and Kowalski, 1999). The cells in these cultures (see photographs in supplemental Fig. 1, available at www.jneurosci.org as supplemental material) begin firing after ∼4 d in vitro (DIV) and soon thereafter synchronize their activity globally across the culture. This synchronization takes the form of intense bursts of activity, 0.5-2 sec in duration, that recur several times per minute. During global bursts, a large fraction of cells in the culture rapidly increase their firing rates by a factor of ≥10. Bursting persists for the lifetime of the culture, although the fully synchronized bursts of young cultures are gradually replaced by more spatially localized bursts in maturity (Maeda et al., 1995; Corner et al., 2002). Globally synchronized bursting is an extremely robust phenomenon. Suppressing it using pharmacological agents like glutamate receptor blockers (Furshpan and Potter, 1989; Gross et al., 1993; Kamioka et al., 1996) also abolishes most or all other spontaneous electrical activity.
In vivo, bursting occurs during development and plays a role in establishing appropriate connections (Meister et al., 1991; Ben-Ari, 2001; Zhang and Poo, 2001; Leinekugel et al., 2002). However, this phase lasts only for days or, at most, weeks. The persistence into maturity of bursting in culture may then be interpreted as a sign that cultures are arrested in their development (Corner et al., 2002). Bursting in cultures has also been likened to spindles observed in the EEG of sleeping brains (Krahe and Gabbiani, 2004) as well as to epileptic activity (Furshpan and Potter, 1989; Litt and Echauz, 2002). Techniques that reduce bursting in culture are therefore of potential importance for the treatment of epileptic patients.
We hypothesize that the persistence of global bursts in dissociated cortical cultures is a result of deafferentation. Deafferentation has two effects. First, the lack of (thalamocortical) input might lead to increased strength of connections within the network. Indeed, Turrigiano (1999) showed that blocking the inputs to cortical neurons using TTX during development significantly increased the strength of excitatory connections. Second, the lack of structured input and presence of strong excitatory connections put the network in a highly unstable state in which positive feedback between excitatory cells can easily lead to synchronized bursts of activity (Corner and Ramakers, 1992). Latham et al. (2000) found that bursting results when too few cells in the network are tonically active. This autoregulation may be caused by slow afterhyperpolarization or regulation of intracellular Ca2+ (Darbon et al., 2002). We propose that substituting multielectrode stimulation for sensory input (Heck, 1995) has the same effect as an elevated tonic firing rate and therefore should reduce the predominance of global bursts, favoring more locally differentiated neuronal activity.
Materials and Methods
Cell culture. Neocortical cells were dissociated from the brains of embryonic day 18 rats and plated on multi-electrode arrays (MEAs). Timedpregnant Wistar rats were killed by CO2 inhalation according to protocols approved by the National Institutes of Health. Embryos were removed, chilled, and decapitated. The entire neocortex, excluding the hippocampus, was dissected under sterile conditions. Cortices were cut into 1 mm3 cubes in Segal's medium (Segal et al., 1998) containing (in mm): 5.8 MgCl2, 0.25 CaCl2, 1.6 HEPES, 90 Na2SO4, 30 K2SO4, 1 kynurenic acid, and 0.5 dl-2-amino-5-phosphonovaleric acid (APV), pH 7.3, using NaOH and 0.001% phenol red. After enzymatic digestion for 30 min by 2.5 U/ml papain (Roche 108014, Roche, Indianapolis, IN) in Segal's medium, cells were separated by six or nine trituration passes using a 1 ml pipette tip, in Neurobasal medium with B27 (Invitrogen, Carlsbad, CA) (Brewer et al., 1993), 0.5 mm Glutamax (Invitrogen), and 10% equine serum (Hyclone, Logan, UT). After every three passes, the cells already in suspension were transferred to a separate tube to reduce stress on them. Cells were centrifuged at 160 × g onto 5% bovine serum albumin in PBS, resuspended by very gentle trituration, and passed through a 40 μm cell strainer (Falcon, Bedford, MA) to remove large debris. Fifty thousand cells were plated in a 20 μl drop of Neurobasal on MEAs precoated with polyethyleneimine and laminin as described previously (Potter and DeMarse, 2001). This led to a plating density of 2500 cells per square millimeter in a monolayer. After 1 hr of incubation, 1 ml of Neurobasal was added to each culture dish. After 24 hr, the plating medium was replaced by a medium adapted from Jimbo et al. (1999): DMEM (Irvine Scientific, Santa Ana, CA) with 0.5 mm Glutamax and 10% equine serum, but no antibiotics or antimycotics. Cultures were maintained in an incubator with 5% CO2 and 9% O2 (Brewer and Cotman, 1989). We replaced one-half of the medium every 5-7 d. Glial growth was not suppressed, because glia are essential to long-term culture health. As a result, glia gradually formed a carpet over the neurons. Our use of Teflon-sealed dishes (Potter and DeMarse, 2001) allowed us to maintain the incubator at 65% relative humidity, making it an electronics-friendly environment. Thus we could perform all experiments inside the incubator, ensuring long-term stability of recording conditions. Experiments took place at 25-45 DIV. At this age, >90% of electrodes recorded spikes. Only cultures that fired at least three bursts in 10 min of pre-experimental screening were used.
To determine the fraction of inhibitory cells, we stained two cultures at 16 DIV for MAP2 and GABA, as described below. In the two cultures, 10 randomly selected images showed 29 of 499 neurons (5.8%) and 16 of 440 neurons (3.6%) with anti-GABA immunoreactivity (Fig. 1).

Two-photon images of immunocytochemically stained neurons. A, MAP2. B, GABA. The two images show the same field of view. Negative controls showed no visible signals. Images were taken with a Carl Zeiss LSM510 multiphoton microscope. Arrows indicate GABA-positive neurons. Scale bar, 20 μm.
Recording system. Electrical activity was recorded with a square array of 60 substrateembedded titanium nitride electrodes, 30 μm in diameter, with 200 μm spacing (Multi Channel Systems, Reutlingen, Germany). After 1200× amplification, signals were sampled at 25 kHz using a Multi Channel Systems data acquisition card, controlled through our Meabench software (available for free public download: http://www.its.caltech.edu/~wagenaar/meabench.html). Meabench's digital filtering system for reducing stimulus artifacts (Wagenaar and Potter, 2002) allowed us to detect action potentials as early as 2 msec after stimulation (except on the electrode used for stimulation, which remained saturated by stimulation artifacts for 50-150 msec). Spikes were detected on-line by thresholding at 5× rms noise and later validated based on the shapes of their waveforms (P. P. Mitra, personal communication).
Stimulation system. Stimuli were generated using our custom-made 60 channel stimulator (Wagenaar and Potter, 2004). We used biphasic rectangular voltage pulses, positive phase first, because these were found to be the most effective stimulus at any given voltage (Wagenaar et al., 2004). We used stimulus pulse widths of 400 μsec per phase and voltages between 100 and 900 mV. To prevent possible electrochemical damage to electrodes and nearby cells, higher voltages were not used. The stimulator was switched to high impedance output 100 μsec after each pulse using the built-in switches of our stimulator.
Experimental protocols. Before experimenting on any MEA, we probed each electrode in the array with voltage pulses between 100 and 900 mV, in random order. For each electrode, we determined the voltage V* at which the response was five times the spontaneous firing rate. Typically, 40-50 electrodes per dish were in sufficiently close contact with the culture to attain that level of response by voltages in the range tested. For each experimental series, we randomly selected either individual electrodes or groups of 2-25 electrodes from this pool.
We used the following three stimulation protocols. (1) “S” or singleelectrode stimulation: one electrode was stimulated repeatedly at its voltage V*. We used this protocol at 10 different frequencies between 0.05 and 50 stimuli per second (stim/sec). (2) “M” or multi-electrode stimulation: a group of 2-20 electrodes was stimulated cyclically at 2-20 stim/sec, such that each electrode received stimuli once per second, or 25 electrodes were stimulated cyclically at 50 stim/sec, each receiving 2 stim/sec. Each electrode was stimulated at its own previously determined V*. Five different group sizes with corresponding stimulation rates were tested with this protocol. (3) “FB” or closed-loop feedback stimulation: 10 electrodes were stimulated cyclically at 10 stim/sec (so that again each electrode was stimulated once per second), with voltages tuned continuously to maintain a constant tonic firing rate, as described below. With this protocol, we could stably maintain firing rates between spontaneous levels and 500 spikes per second array-wide (SPSA) using voltages not exceeding 900 mV (Fig. 2).

Performance of feedback protocol. A, Median firing rate (dish-wide) achieved versus target. Any firing rate (FR) between the spontaneous rate and 500 SPSA could be stably maintained. Higher rates were not achievable in this culture without exceeding the safe voltage limit of 900 mV. The dotted line marks equality of achieved and target rates. Data are from five series on different sets of electrodes; 45 DIV. B, Stimulus voltage used to control firing rate at different levels.
Experimental runs lasted 5 min each and were randomly interleaved with each other and with control runs in which we recorded spontaneous (unstimulated) activity. Protocols M and S were performed on n = 11 cultures from three platings; protocol FB was performed on n = 10 cultures from two platings. In all cases, each condition was tested 10 times on each culture, with a new random selection of electrodes each time.
Quantifying the level of bursting. Bursts come in different forms, so simply tallying up the number of bursts is not sufficient to describe the burstiness of a culture. It is essential to account for the size of bursts, measured in terms of number of participating neurons, aggregate number of spikes, or duration. Fortunately, we found that it is not necessary to identify individual bursts to quantify the level of burstiness of a recording. Instead, we used the following method: divide a 5 min recording into 300 1-sec-long time bins and count the number of spikes (total across all electrodes) in each bin. Compute the fraction of the total number of spikes accounted for by the 15% of bins with the largest counts. If the firing rate is tonic, this number, f15, will be close to 0.15. Conversely, if a recording is so bursty that most of the spikes are contained in bursts, f15 will be close to 1, because even at the highest burst rates observed during these experiments, bursts did not occupy >45 1-seclong bins (15%) in a 5 min recording. We then defined a burstiness index (BI), normalized between 0 (no bursts) and 1 (burst dominated) as BI = (f15 - 0.15)/0.85. (Statistical fluctuations make the BI deviate slightly from zero even in complete absence of bursts.)
Tuning the feedback. For the closed-loop stimulation protocol, we continuously monitored the actual firing rate of a culture and adjusted the stimulation voltages for each electrode to maintain the target firing rate, f0, as follows. Initially, we used a base voltage, V̄ = 200 mV, applied to all electrodes. We then measured the (culture-wide) firing rate, f̄, in 2 sec sliding windows and used this to update V̄ every 100 msec according to the following: 
where ϵ is a gain factor that determines how fast V̄ reacts to changes in f̄.Weset ϵ = 0.02, corresponding to a time constant of 5 sec. This ensured rapid feedback and prevented oscillations caused by overcompensation.
To account for variations in stimulation efficacy between electrodes, we measured individually the firing rates in the first 100 msec after each stimulus. For each electrode k, we used these measurements to maintain a running average, fk, of the firing rates after the most recent 20 stimuli to that electrode. Every 100 msec, we recalculated fine-tuning factors, αk, as follows: 
where  is a normalization factor to make 1 the average of all αk values. We then set the voltage for the next stimulus on electrode k to the following:
is a normalization factor to make 1 the average of all αk values. We then set the voltage for the next stimulus on electrode k to the following: 
Thanks to Meabench and our custom-made stimulator (Wagenaar and Potter, 2004), these adjustments could be made in real time without interrupting the stimulation process.
Because we wanted to control the tonic firing rate, updating of V̄ and αk was suspended during putative bursts. These were detected using a simple heuristic: any 100 msec windows that had a spike count higher than five times the target were considered potential bursts and thus excluded for the estimate of the tonic firing rate.
Immunostaining. The fraction of GABAergic neurons was determined as follows. Cultures were fixed with 4% paraformaldehyde at room temperature for 30 min. After treatment with 0.1% Triton X-100 in PBS for 20 min, they were incubated in 2% goat serum for 1.5 hr and then in the primary antibodies anti-MAP2 [mouse, 1:200; MAB378 (Chemicon, Temecula, CA)] and anti-GABA [rabbit, 1:100; AB131 (Chemicon)] overnight at 4°C. After washes, cells were incubated with secondary antibodies [Alexa Fluor 488 goat anti-mouse, 1:200; Alexa Fluor 594 goat anti-rabbit, 1:1000; and Hoescht, 1:1000 (all from Molecular Probes, Eugene, OR)] for 1 hr at room temperature. Fluorescence images were obtained from a Sony digital camera on a Nikon TE300 fluorescence microscope and a Carl Zeiss (Thornwood, NY) LSM510 multiphoton microscope.
Results
Spontaneous bursting
Before developing a method to control bursting, we needed to characterize the different kinds of bursting encountered in the spontaneous activity of cultured cortical networks. The frequencies of bursts as well as their sizes were highly variable between cultures from different platings and even between cultures within platings. Additionally, individual cultures showed large variations from day to day. Cultures spontaneously exhibited a wide range of bursting behaviors, from short single-cell bursts, to small local bursts involving two to five electrodes, to long global bursts (Fig. 3). Some cultures exhibited “superbursts”: stereotyped sequences of global bursts, separated by several minutes devoid of bursts (D. A. Wagenaar, Z. Nadasdy, and S. M. Potter, unpublished observations). Typically, global bursts were first observed at ∼7 DIV, after which burstiness increased steadily until they dominated the activity at ∼20-25 DIV. After that, burstiness fluctuated somewhat but remained high for as long as we observed (up to 45 DIV in these experiments).

Examples of different spontaneous bursting patterns, with array-wide firing rates (line graphs) as well as per-electrode firing rates (grayscale plots). A, Chaotic bursting. Insets below show spike raster plots for a large global burst, a single-channel burst, and a small local burst, at 20× magnification (recorded at 25 DIV). B, Spontaneously regular bursting (recorded at 39 DIV). C, Superbursts (recorded at 34 DIV). The inset shows spikes at 10× magnification.
Response to stimulation
The immediate response to stimulation at any electrode consisted of three phases (Wagenaar et al., 2004): (1) direct, nonsynaptically propagated responses, with very precise timing (typical jitter: 100 μsec), and latencies of 3-10 msec; (2) postsynaptic responses, mostly with latencies between 5 and 50 msec; (3) bursts, often evoked by strong or low-frequency stimuli. Such bursts were time-locked to the stimulus pulse with latencies characteristic of the local network around the electrode stimulated, usually in the range of 50-200 msec, but were otherwise similar to spontaneous bursts. Examples of early responses are shown in Figure 4.

Array-wide responses to stimulation. Each graph shows the responses on one electrode, represented according to the geometry of the array. The stimuli were delivered to the marked electrode. Vertical line indicates time of stimulation. Spikes were detected after artifact suppression (Wagenaar and Potter, 2002); TTX control confirmed the biological origin of all detected spikes.
During slow single-electrode stimulation (0.05 stim/sec), most or all stimuli entrained bursts as reported previously by Gross et al. (1993) and Maeda et al. (1995). At slightly higher frequencies (0.1-0.5 stim/sec), bursts were elicited less consistently, depending on stimulation electrode. At still higher frequencies (1-5 stim/sec), most stimuli did not elicit bursts, and in fact the burstiness began to drop below spontaneous levels. Increasing the stimulation rate (10-50 stim/sec) did not further reduce burstiness (Fig. 5A). The best burst control on average was achieved at 10 stim/sec: BI = 0.19 ± 0.02 (mean ± SEM; n = 105 runs using different electrodes in 11 cultures; range of means per culture, 0.04-0.55). This level of burstiness was significantly below the average spontaneous level BI = 0.48 ± 0.02 (n = 199 runs, same 11 cultures; range, 0.19-0.86).

A, Burstiness during single-electrode stimulation (protocolS) and spontaneous activity (no stimulation). Each row shows the array-wide firing rate (coded by the grayscale at right) as a function of time during one 5 min experimental run. In the 10 examples of spontaneous activity shown (bottom), bursts occurred irregularly approximately once per minute. In the 10 examples of stimulation at 0.05 stim/sec, bursts were perfectly aligned with stimuli, except in a few cases during which a spontaneous burst just preceded the stimulus. (The stimulating electrode was different in each of the 10 rows.) At 0.1-0.2 stim/sec, bursts underwent period doubling. Bursts during stimulation at 1-5 stim/sec were less frequent, but still mostly stimulus locked. In the 10-50 stim/sec runs, burst control was perfect for the first 45 sec, after which a spontaneous-like pattern returned. Data are from a culture at 39 DIV. Note that experimental runs were executed in random order. B, Bursting during multi-electrode stimulation (protocol M), same culture. Perfect and sustained burst control is attained at the higher stimulation frequencies. Note the increase in tonic firing rate (background shading) as the stimulation frequency is increased. C, Burstiness index as a function of stimulation frequency, for single-electrode stimulation (□) and multi-electrode stimulation (▪). Slow single-electrode stimulation elevates the burstiness over spontaneous (unstimulated) levels (○), whereas rapid stimulation reduces it. Values are mean ± SEM from n = 100 runs on 10 cultures. The most effective protocol tested, 50 stim/sec distributed across 25 electrodes, suppressed bursts completely (n = 60 runs, 6 cultures).
When stimuli were applied through a single electrode at high rates, the immediate response to stimulation (spikes recorded 2-20 msec after stimulus) decreased dramatically with increasing stimulation frequencies (Fig. 6). This was likely responsible for the lack of improvement of burst control at those high frequencies; however, the responses to infrequent stimulation through one electrode were not affected by rapid stimulation through another electrode, so this reduction of efficacy was caused by to a mechanism local to the stimulated electrode and not a networklevel fatiguing effect.

Stimuli presented to a single electrode (primary stimulation electrode; •) yielded much-reduced responses in the first 20 msec after stimulus when the stimulation rate was increased. (We focused on short-latency responses, because the majority of response spikes occurred at short latencies and because responses cannot be defined unambiguously beyond one interpulse interval, i.e., 25 msec for the highest stimulation frequency.) In fact, at a stimulation frequency of 40 stim/sec, the response was not much higher than the spontaneous firing rate (arrow at left). Each stimulation series lasted 5 min, and we discarded the responses recorded during the first 30 sec to measure the sustained response rate. Results are mean ± SEM from 53 electrode pairs in four cultures. During these experiments, we presented stimulus pulses to a second electrode every 5 sec. The responses in the first 20 msec after these latter stimuli (○) were not affected by the rate at which the first electrode was stimulated. Response strength in all cases was normalized to the results obtained from single-electrode stimulation at 0.2 stim/sec. The response strengths are plotted as a function of the frequency at which the primary electrode was stimulated. Inset and associated arrows indicate the stimulation protocol. Regardless of the frequency of the primary stimulation electrode, the secondary electrode was stimulated once every 5 sec.
Burst control by distributed stimulation
Based on the observation that rapidly stimulating single electrodes reduced the efficacy of those stimuli but not of stimuli to other electrodes, we proceeded to test whether better burst control could be achieved by distributing the stimulus load across several electrodes, using protocol M (see Materials and Methods). At intermediate frequencies (2-10 stim/sec), this protocol resulted in somewhat higher burstiness than single-electrode stimulation, but at frequencies above 10 stim/sec, multi-electrode stimulation resulted in greatly improved burst reduction (Fig. 5 B, C). At the highest stimulation rate tested, 50 stim/sec distributed across 25 electrodes, bursts were completely suppressed in all cultures tested. In all cases, a change of stimulation protocol rapidly affected burstiness, and bursting resumed as soon as stimulation was stopped (Fig. 7).

When switching between stimulation protocols, the activity pattern of a culture changed rapidly to match the new stimulation context. Here we show switches from rapid single-electrode stimulation to slow single-electrode stimulation, to rapid multi-electrode stimulation, and to no stimulation.
Burst control by closed-loop stimulation
Perfect burst control was achieved using protocol M, but only at very high stimulation rates and using a large number of electrodes. If good burst control could be attained using fewer electrodes or lower stimulation rates, this would have practical advantages. We noted that the bursts that occurred in protocol M at intermediate stimulation rates were mostly entrained by only one of the electrodes used in a given run, indicating that the calibration of stimulus efficacy performed before the experiment (see Materials and Methods) was not a very good predictor of efficacy in the context of much more intense multi-electrode stimulation (data not shown). Thus we hypothesized that the level of burst control attained by predefined voltage pulses could be improved further by tuning the stimulation voltages in real time to obtain a constant level of response. We used feedback control (protocol FB; see Materials and Methods) to regulate the median firing rate at nine fixed levels between 50 and 800 SPSA. Increasing the median firing rate over spontaneous levels reduced burstiness monotonically (Fig. 8). At the highest target rate of 800 SPSA, this protocol was significantly more effective than either single- or multi-electrode stimulation compared at the same stimulation rate (10 stim/sec). This held despite the fact that the spontaneous BI was 50% higher on average for those cultures on which we tested feedback stimulation compared with those tested with single- or multi-electrode stimulation. (This difference in spontaneous behavior was attributable to variability between cultures and not to our intervention.)

Burstiness during closed-loop control of tonic firing rate. A, After an initial period of ∼15 sec during which the feedback algorithm settles, burst control was perfect at the higher target firing rates (f.r.). A culture at 43 DIV was used. (This culture was not tested at 800 SPSA.) B, Burstiness index decreased monotonically with the target rate and was always below the spontaneous level (○). Values are mean ± SEM from n = 85 runs using different sets of electrodes on 10 cultures.
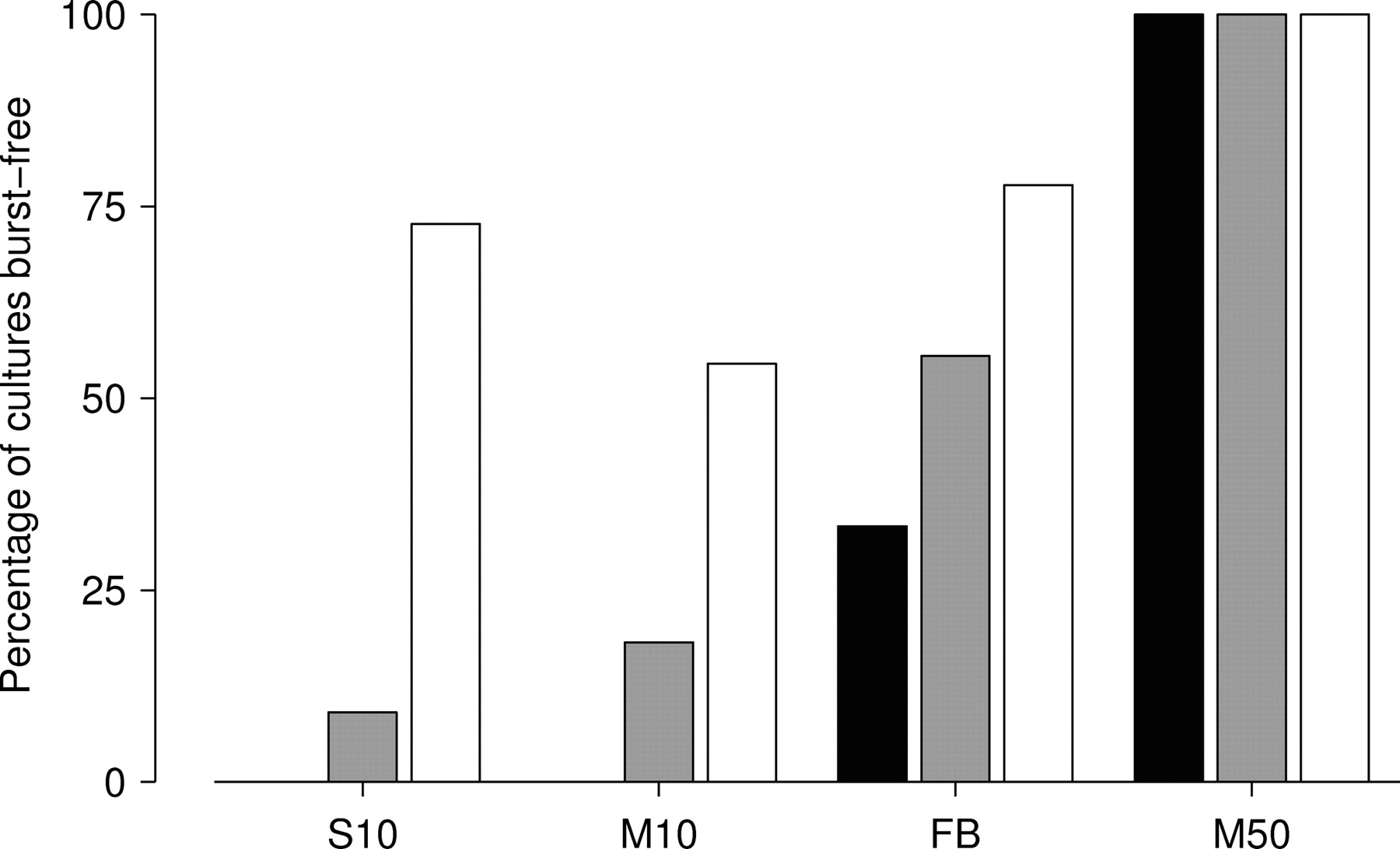
A final comparison of the various protocols tested was made by counting in what percentage of cultures each protocol suppressed bursts completely during a 5 min run (Fig. 9). Any run with BI < 0.05 was considered burst free for this assessment. The most intense protocol M stimulation (50 stim/sec distributed across 25 electrodes) suppressed bursts in all cultures, independent of the selection of stimulation electrodes. Although a set of electrodes could be found to suppress bursts at 10 stim/sec with fixed voltages in >50% of cultures (Fig. 9, white bars), a random selection of electrodes suppressed bursting in only one in five cultures (gray bars). Closed-loop feedback did much better: a random selection of electrodes suppressed bursting in >50% of cultures (Fig. 9, gray bar), and in 30% of cultures, all 10 random selections of electrodes tested suppressed bursts (black bar).

Assessment of the success rate of different burst-suppression protocols. Bars show the percentage of cultures in which each protocol successfully suppressed bursting, by random electrode selection (gray), by at least 1 of 10 selections tested (white), or by all of 10 selections tested (black). None of the cultures used in these experiments were burst-free in spontaneous activity. Protocols compared are as follows: protocol S at its optimal stimulation rate (10 stim/sec; S10); protocol M at the same rate (M10); protocol FB at its optimum (target 800 SPSA; FB); and protocol M at its optimum (50 stim/sec distributed across 25 electrodes; M50).
Discussion
Several years ago, Latham et al. (2000) showed that networks with a large fraction of intrinsically active neurons have a reduced tendency to burst. We extend this finding by demonstrating that increasing the tonic activity above spontaneous levels by highfrequency multisite electrical stimulation also reduces or suppresses bursting. Strikingly, complete suppression of bursts was achieved by a combination of stimuli that entrained bursts when applied singly. Rapid stimulation through single electrodes yielded fewer bursts than slow stimulation, not just per stimulus but per unit of time: stimulation at ≥5 stim/sec reduced burstiness to below spontaneous levels. Distributing the stimuli across ≥20 electrodes proved highly effective in reducing it even further, and with 50 stim/sec distributed across 25 electrodes, bursting was suppressed completely in all cultures tested, independent of the selection of electrodes. However, such a high stimulation rate may be undesirable in some applications, or that many electrodes may not be available. When the number of electrodes used for burst control was limited to 10, stimulation with closed-loop feedback was found to be the optimal solution. This protocol completely suppressed bursts in >50% of cultures using 10 stim/sec distributed across randomly selected groups of 10 electrodes. With careful selection of electrodes, fixed-voltage stimulation through single or multiple electrodes suppressed bursting in a similar fraction of cultures as feedback stimulation. Feedback stimulation, however, was far more robust: in 30% of cultures it worked regardless of electrode selection. Electrode independence was never seen for fixed-voltage stimulation at 10 stim/sec. To extend burst control beyond 5 min runs, such robustness is highly desirable.
Synchronized bursting is fundamentally a network phenomenon, emerging from the synaptic interactions between a large number of cells. Whether these cells would endogenously burst in the absence of synaptic input is probably not essential for this phenomenon. The cellular and network mechanisms of bursting and burst suppression are not yet understood in detail. There is some controversy about the origin of the refractory periods for spontaneous bursts. Opitz et al. (2002) reported synaptic depression immediately after population bursts, whereas Darbon et al. (2002) found no evidence of synaptic depression, no depletion of vesicles, and no desensitization of postsynaptic receptors.
It has been suggested that the persistence of global bursting in mature cultures is evidence that such cultures are in a state of arrested development as a result of lack of sensory input. Our experiments support this view, because we found that substituting for thalamic inputs with distributed electrical stimulation reduced bursting dramatically. Given that the developmental fine structuring of several primary cortical sensory areas in vivo is known to be determined by the pattern of inputs, it is tantalizing to ask whether persistently applied stimulation in vitro might similarly influence network topology.
In contrast to burst suppression by (partially) blocking excitatory synaptic transmission, e.g., using AP5, CNQX (Jimbo et al., 2000), magnesium, or kainic acid (Furshpan and Potter, 1989), distributed stimulation does not reduce the ability of the culture to respond to additional stimuli. Continuously applying distributed stimulation to suppress bursts is thus compatible with studies of use-dependent modification of activity in cultured networks. Additional stimuli can be superimposed on a background of burst-quieting stimuli, to tetanize particular pathways, or to probe network activity. Moreover, distributed stimulation mimics more natural modes of activation in which sensory signals are continuously coming in to the network. Bursts are known to have an effect on tetanus-induced synaptic plasticity (Maeda et al., 1998). Therefore, we suggest that burst suppression may lead to more stable connections and more predictable results of tetanization (Z. C. Chao, Wagenaar, and Potter, unpublished observations). We expect that burst control will make these networks more useful for the study of distributed information processing, robotic control, and network plasticity related to learning and memory (Potter, 2001; DeMarse et al., 2001; Potter et al., 2005).
If distributed stimulation so effectively reduces bursting in vitro, it might also work in vivo. Epileptic seizures in human cortex, although probably originating from very different causes, have a strikingly similar phenomenology: ensemble bursts extending over large areas of neural tissue (Lopes da Silva et al., 2003). Electrical stimulation has been used in several experimental therapies for epilepsy; stimulation of the vagus nerve is the most well known example (Penfield and Jasper, 1954; Hammond et al., 1995; Ben-Menachem et al., 1994; Fisher et al., 1997; Handforth et al., 1998; Koo, 2001). Alternatively, animal and modeling studies suggest that focal stimulation at the site of the seizure can terminate seizures after they have started (Lesser et al., 1999; Franaszczuk et al., 2003; Slutzky et al., 2003). In humans, focal stimulation in the cortex or hippocampus has indeed been found effective in a number of studies (Cooper et al., 1973, 1976, 1977; Lüders et al., 1988; Shulz et al., 1997; Velasco et al., 2000, 2001; Motamedi et al., 2002; Vonck et al., 2002). Stimulation through a single electrode offered protection against seizures, but only if the stimulus was strong enough that the entire seizure-prone area was reached (Motamedi et al., 2002; Kellinghaus et al., 2003), which was difficult in practice. Distributing stimulation across multiple electrodes might be attractive for several reasons. First, the amplitude of pulses delivered to each electrode could be much lower, reducing commensurately the risk of side effects (Wheless, 2001; Schachter, 2002), tissue damage (Shepherd et al., 1991; Tehovnik, 1996), or electrode damage. Second, the system would be more fault tolerant (Davis, 2000), because losing one or two electrodes from a large ensemble would hardly compromise efficacy. Third, unlucky placement of a single electrode can result in poor burst control, whereas with multi-electrode stimulation, the result is much less dependent on exact placement. Finally, if the electrodes were connected to a recording system equipped with seizure prediction software, stimulation parameters could be tailored to the predicted locus of impending seizures (Iasemidis, 2003). Our real-time controlled stimulator (Wagenaar and Potter, 2004) could be a starting point for developing such a system.
Footnotes
This work was partially supported by National Institutes of Health (NIH)-National Institute of Neurological Disorders and Stroke Grants NS38628 and NS44134, by NIH-National Institute of Biomedical Imaging and Bioengineering Grant EB00786, and by the Whitaker Foundation and the National Science Foundation Center for Behavioral Neuroscience. We thank Mark Booth for help with immunostaining and imaging, Sheri McKinney and Eno Ekong for technical assistance with cell culture, and anonymous reviewers for thoughtful comments.
Correspondence should be addressed to Steve Potter, Department of Biomedical Engineering, BME0535, Atlanta, GA 30332. E-mail: steve.potter{at}bme.gatech.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250680-09$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}