

Abstract
In the CNS, receptor recycling is critical for synaptic plasticity; however, the recycling of receptors has never been observed at peripheral synapses. Using a novel imaging technique, we show here that nicotinic acetylcholine receptors (AChRs) recycle into the postsynaptic membrane of the neuromuscular junction. By sequentially labeling AChRs with biotin-bungarotoxin and streptavidin-fluorophore conjugates, we were able to distinguish recycled, preexisting, and new receptor pools at synapses in living mice. Time-lapse imaging revealed that recycled AChRs were incorporated into the synapse within hours of initial labeling, and their numbers increased with time. At fully functional synapses, AChR recycling was robust and comparable in magnitude with the insertion of newly synthesized receptors, whereas chronic synaptic activity blockade nearly abolished receptor recycling. Finally, using the same sequential labeling method, we found that acetylcholinesterase, another synaptic component, does not recycle. These results identify an activity-dependent AChR-recycling mechanism that enables the regulation of receptor density, which could lead to rapid alterations in synaptic efficacy.
- toxin
- turnover
- synaptic transmission
- neuromuscular junction
- trafficking
- acetylcholine receptor (AChR)
Introduction
The density of neurotransmitter receptors is an important parameter in regulating the efficacy of synaptic transmission. In both the central (O'Brien et al., 1998; Carroll et al., 1999a) and peripheral (Fambrough and Hartzell, 1972; Akaaboune et al., 1999; Sanes and Lichtman, 2001) nervous systems, alterations in synaptic transmission cause changes in postsynaptic receptor density. Such changes are important in many forms of synaptic plasticity, which lead to the strengthening or weakening of synaptic connections (Carroll et al., 1999b; Luscher et al., 1999). Several mechanisms at CNS synapses, including receptor recycling, are known to be involved in regulating synaptic plasticity (Nishimune et al., 1998; Noel et al., 1999; Ehlers, 2000; Kim and Lisman, 2001; Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Park et al., 2004). In peripheral cholinergic neuromuscular junctions, however, it is not known whether acetylcholine receptors (AChRs) are stable until they are removed and degraded in a one-way trip to the lysosomes, or if they are in constant movement between internal compartments and the plasma membrane. To address this issue, we have developed a novel imaging technique that enables us to study new aspects of AChR dynamics at neuromuscular junctions in vivo. By selectively labeling different AChR pools (preexisting, recycled, and new) within a single synapse in the living mouse, we were able to provide an estimate of the contribution of each pool to the neuromuscular junction (NMJ) over time. We found that a significant number of these receptors undergo recycling back to the NMJ, and this recycling is dramatically affected by alterations in synaptic activity.
Materials and Methods
In vivo imaging. All animal usage followed methods approved by the University of Michigan Committee on the Use and Care of Animals. Adult female mice (20-27 g, non-Swiss albino; Harlan Sprague Dawley, Indianapolis, IN) were anesthetized with an intraperitoneal injection of ketamine and xylazine (17.38 mg/ml). Sternomastoid muscle exposure and neuromuscular junction imaging were done as previously described in detail (Lichtman et al., 1987; van Mier and Lichtman, 1994; Akaaboune et al., 1999). Briefly, the anesthetized mouse was placed on its back on the stage of a customized epifluorescence microscope, and neuromuscular junctions were viewed under a coverslip with a water immersion objective (20× UAPO, 0.7 numerical aperture; Olympus BW51; Optical Analysis Corporation) and a digital CCD camera (EXi; Retiga, Burnaby, British Columbia, Canada). Mice were intubated and ventilated for the duration of the imaging sessions. For imaging at multiple time points, the mouse was sutured after each session and allowed to fully recover before the next imaging session.
Labeling of distinct AChR pools. After exposure of the sternomastoid muscle, AChRs were saturated first with fully substituted BTX-biotin (5 μg/ml, 1.5 h; Invitrogen, Eugene, OR) and then with streptavidin conjugated to Alexa 660 (10 μg/ml, 2.5 h; Invitrogen) to saturate all biotin sites. To ensure that all receptors and all biotin sites were saturated, distinct colors of tetramethyl-rhodaminated BTX conjugate (TMR-BTX) and streptavidin-Alexa (streptavidin-Alexa 488) were added to the sternomastoid muscle. Superficial synapses were then immediately imaged under the control of IPLAB software (Scanalytics). Hours or days later, the mouse was reanesthetized, and the sternomastoid muscle was reexposed and bathed with streptavidin-Alexa 488 to label the recycled AChRs and TMR-BTX to label the new AChRs. The muscle was washed, and the same synapses were relocated and imaged. In this way, we were able to distinguish the preexisting, new, and recycled receptor pools. To rule out the possibility that streptavidin-Alexa binds directly to receptors, sternomastoid muscles were lightly labeled with TMR-BTX (5 μg/ml, 2 min application) to identify synapses. Streptavidin-Alexa 488 (green) (10 μg/ml) was then applied to the sternomastoid muscle for 2.5 h. In these junctions, no green labeling was observed, indicating that streptavidin does not bind directly to receptors.
Denervation. The sternomastoid muscle was denervated by excising a 5 mm piece of nerve to prevent its regeneration. Ten days later, the mouse was anesthetized and labeled as described above.
Muscle stimulation. Denervated muscle was stimulated by placing stimulating electrodes at either end of the muscle (3 ms bipolar pulses of 10-13 V at 10 Hz for 1 s duration every 2 s for the entire 8 h period).
Continuous blockade of activity. To study the effect of muscle postsynaptic activity blockade on the insertion of recycled and new receptors, curare (2.5 mg/ml), a poison that binds reversibly to AChRs, was applied to the sternomastoid muscle to block neuromuscular transmission. For the duration of the experiment, a coverslip was placed over the exposed muscle to prevent it from drying. Every 2 h, additional curare solution was applied to the muscle. Animals were intubated with a ventilator for the duration of the experiment to prevent asphyxiation.
Cell culture. C2C12 cells (obtained from American Type Culture Collection, Manassas, VA) were cultured on laminin-coated dishes in medium containing 20% FBS and then switched after 2 d to 5% horse serum to induce differentiation into myotubes. Three to 5 d after differentiation, cultures were labeled and imaged as with muscles in vivo. We used the appearance of the cells under phase contrast illumination to distinguish receptor clusters on living muscle cells from ghost receptor clusters.
Immunocytochemistry. Longitudinal sections (20 μm) of fixed [2% paraformaldehyde (PFA)] sternomastoid muscle that was labeled 4 d earlier with BTX-biotin (5 μg/ml, 1.5 h) followed by a saturating dose of fluorescent or unlabeled streptavidin (10 μg/ml, 2.5 h) were blocked with 10% BSA and goat serum, permeabilized with 1% Triton X-100, and incubated with primary antibodies for 1-2 h at room temperature in blocking solution. The primary antibodies used were as follows: antibody to biotin [1:200, monoclonal antibody (mAb) 2F5, anti-biotin-Alexa 488/594; Invitrogen]; rat anti-acetylcholine receptor (1:500, mAb35 or mAb210; Sigma, St. Louis, MO); and rabbit anti-early endosomal antigen (EEA1, 1:300; Abcam, Cambridge, MA). The sections were then washed extensively in PBS and incubated for 1 h with secondary goat anti-rat antibody conjugated to either Alexa 594 or Alexa 488 (Invitrogen) and/or with secondary sheep anti-rabbit antibody conjugated to Texas Red (Abcam).
Confocal microscopy. The analysis of intracellular and extracellular colocalization was performed using a 60× oil-immersion lens and laser illumination on a confocal Zeiss (Thornwood, NY) LSM 510 microscope. A series of optical planes were collected in the z dimension (z-stack) and collapsed into a single image. Images presented in Results were produced and adjusted for brightness and contrast using Adobe (San Jose, CA) Photoshop CS.
Quantitative fluorescence imaging. In experiments involving multiple color fluorescent ligands (such as the one illustrated in Fig. 1), the camera gain was adjusted to give a nicely exposed image on each color channel, but because of differences in the extent of labeling of the different ligands and the quantum efficiency of different fluorophores, it was not possible to compare the relative number of molecules labeled with each ligand. When such comparisons were essential (see Figs. 7, 8), the fluorescence intensity of labeled receptors at neuromuscular junctions was assayed using a quantitative fluorescence imaging technique, as described by Turney et al. (1996) with minor modifications. This technique incorporates compensation for image variation that may be caused by spatial and temporal changes in the light source and camera between imaging sessions by calibrating all images with a nonfading reference standard. A key feature of the quantitative imaging approach used in the current study is that it involves three sequential labeling steps with the same ligands (BTX-biotin and streptavidin-Alexa 594). Thus, as long as we verified that labeling had reached saturation and that the image pixel intensity was not saturated, it was simple to get an accurate quantitative measurement corresponding to the number of AChRs in each pool.
Results
Recycling of AChRs at the neuromuscular junctions of live animals
Experiments such as the one illustrated in Figure 1 suggested that AChRs are recycled at the postsynaptic membrane of the neuromuscular junction. The sternomastoid muscle of this mouse was labeled with a saturating dose of biotin-labeled α-bungarotoxin (BTX-biotin, 5 μg/ml, 1.5 h), which binds with extremely high affinity to AChRs (Green et al., 1975; Fambrough, 1979). BTX-biotin-bound receptors were then saturated with streptavidin conjugated to a fluorescent Alexa dye (10 μg/ml, 2.5 h), which in turn forms a noncovalent, very high-affinity bond with biotin (Weber et al., 1989; Green, 1990). To test whether all receptors were saturated with BTX-biotin and all biotins were saturated with streptavidin-Alexa, immediately after the unbound BTX-biotin and streptavidin-Alexa 660 were washed away (pseudocolor, blue), the muscle was exposed to alternately colored streptavidin-Alexa [streptavidin-Alexa 488 (green), 10 μg/ml] and fluorescent α-BTX [TMR-BTX (red), 5 μg/ml]. The superficial synapses were then imaged (Fig. 1A). The absence of red and green fluorescence indicates that saturation was achieved (Fig. 1B,C). When the same synapse was exposed 24 h later to streptavidin-Alexa 488 and TMR-BTX, much of the original fluorescence from the streptavidin-Alexa 660 remained (Fig. 1D), but there was also significant fluorescence on the green (streptavidin-Alexa 488) and red (α-TMR-BTX) channels (Fig. 1E,F). The blue label at 24 h indicates that most receptors were still bound to both of the original ligands. The red label at 24 h indicates those receptors that appeared on the surface after the initial labeling period (whether newly synthesized or from an internal storage pool) as well as any surface receptors that had lost their BTX-biotin label. For the rest of this paper, all receptors that stain with fluorescent α-BTX after an initial saturating dose of BTX will be referred to as part of the pool of “new” receptors. The green fluorescence present at 24 h indicates receptors that remained bound to BTX-biotin but that had lost their streptavidi-n-Alexa 660 label. Given the expected high affinity of the streptavidin-biotin bond, we were surprised by the extent of loss of streptavidin. This matter is further considered below and in Discussion.
Dissociation of streptavidin from the AChR-bungarotoxin-biotin complex does not occur on the muscle surface
We performed a series of experiments to test whether the dissociation of streptavidin from biotin-BTX-AChR could occur on the cell surface. First, to estimate the intrinsic rate of dissociation of streptavidin from the biotin-BTX-AChR complex at the neuromuscular junction, mice were perfused with 2-4% paraformaldehyde in PBS, and the sternomastoid muscle was removed and placed in a dish. We then labeled these muscles with saturating doses of BTX-biotin and then streptavidin-Alexa, as described above. Three to 5 d later, the muscle was bathed with a second color of fluorescent streptavidin. In contrast to the result obtained for receptors at synapses in living animals at this time (where a significant amount of the signal intensity was attributable to receptors that had lost their original streptavidin-Alexa tag and were then able to be relabeled with new streptavidin-Alexa at a later time point) (see Fig. 1), no fluorescence from the second streptavidin fluorophore was observed. Thus, under these conditions, the rate of biotin-streptavidin dissociation is negligible. Because both the BTX-biotin and the streptavidin-Alexa were applied after fixation and extensive washing, this protocol likely accurately reflects the extremely slow intrinsic rate of biotin-streptavidin dissociation in the absence of processes associated with living cells.

Multiple AChR pools are present at synapses on sternomastoid muscles of living mice. A, In a living adult mouse, a neuromuscular junction of the sternomastoid muscle was labeled with a saturating dose of BTX-biotin followed by a saturating dose of streptavidin (strept)-Alexa 660 (blue, pseudocolor). B, C, The junction was then immediately bathed with a saturating dose of streptavidin-Alexa 488 (green) and TMR-BTX (red). The absence of any labeling (even when camera exposure was increased significantly) indicates that all receptors and biotin sites were saturated by the initial applications. D-G, The same neuromuscular junction imaged 24 h later, after application of new streptavidin-Alexa 488 (green) and TMR-BTX (red) to the sternomastoid muscle. The remaining preexisting receptors were still labeled with BTX-biotin-streptavidin-Alexa 660 (blue, pseudocolor). The streptavidin-Alexa 488 labeling indicates the presence of receptors that retained BTX-biotin sites but lost their streptavidin-Alexa 660. These free sites are unlikely because of the loss of streptavidin on the surface (see Figs. 2, 3, 4, 5) and therefore referred to as recycled receptors. The TMR-BTX labeling indicates receptors that were not bound to either BTX-biotin or streptavidin and are referred to as new receptors. Scale bar, 20 μm.
Another possibility is that factors released from living muscle may increase the dissociation of streptavidin on the muscle cell surface. To test this possibility, we took advantage of “ghost” clusters observed in aneural myotube cultures grown on laminin. C2C12 myotubes grown on laminin display large, complex AChR clusters that often remain bound to the laminin, even after the muscle cell that used to hold them on its surface has died (Kummer et al., 2004). Sequentially saturating these ghost clusters with BTX-biotin and streptavidin-Alexa 594 therefore enabled the analysis of AChR-BTX-biotin-streptavidin complexes isolated from muscle membranes but still surrounded by living muscle cells in a normal extracellular milieu. When these ghost clusters were labeled with either streptavidin-Alexa 488 or anti-biotin 488 5 d later, no green fluorescence was observed (Fig. 2A-C). Thus, there was no detectable dissociation of streptavidin over a 5 d period when receptors are surrounded by medium but not on living cells. We also tested for the dissociation of streptavidin from BTX-biotin on the surface of living myotubes. When receptor clusters on the myotube surface were labeled with BTX-biotin and saturated with streptavidin-Alexa 660 and then relabeled 12 h later with streptavidin-Alexa 488 (green) and BTX-Alexa 594 (Fig. 2D-G), considerable red staining but no green staining was observed. This indicated that, despite a significant loss of fluorescence (attributable to the turnover rate of AChR) over this period and significant insertion of new receptors, biotin-streptavidin dissociation did not occur under these conditions. We also found that there is no significant difference in the rate of internalization of receptors on cultured C2C12 myotubes that were labeled with either fluorescent bungarotoxin or BTX-biotin-streptavidin-Alexa (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Together, these results indicate that biotin-streptavidin dissociation does not occur in the normal extracellular environment, that recycling does not occur in aneural myotubes, and that streptavidin binding does not significantly increase internalization.
Although the previous experiments show that the dissociation of streptavidin from biotin does not occur from the surface of fixed muscle or AChR clusters on living or dead cultured myotubes, it is still possible that biological processes could occur at the surface of the NMJ of an intact muscle in vivo that might cause streptavidin-biotin dissociation from the surface of synapses (such as the secretion of proteases by infiltrating immune cells). To examine this possibility, we transplanted a portion of a sternomastoid muscle that was first fixed with 2% PFA and then labeled with BTX-biotin-streptavidin-Alexa 594 into the neck of a host mouse directly next to the living sternomastoid muscle. In this way, the BTX-biotin-streptavidin bound to immobilized receptors after fixation was exposed to a fully active and living sternomastoid muscle environment. As in the previous in vivo labeling studies described above, the mouse was then allowed to recover. Seven days later, we quantified the amount of biotin-streptavidin dissociation at the NMJs of the fixed and transplanted muscle. We found that the original fluorescence of the transplanted muscles decreased by <3%, and we found no evidence of new streptavidin-Alexa binding, compared with normal levels of new streptavidin-Alexa binding observed at NMJs in the live host muscle. These experiments indicate that extracellular proteases do not cause biotin-streptavidin dissociation on the surface of the muscle.
To further rule out the possibility of extracellular dissociation of streptavidin from biotin, we used the same sequential labeling protocol on another synaptic component in vivo. Because of the availability of the snake toxin fasciculin 2, which selectively and specifically labels acetycholinestrase (AChE), we monitored the dissociation of streptavidin from fasciculin 2-biotin over 4 d. AChEs were labeled with biotinylated fasciculin 2 (7 μg/ml, 1 h) and then saturated with streptavidin-Alexa 594 (10 μg/ml, 2.5 h). The mouse was allowed to recover, and after 4 d, the sternomastoid muscle was exposed to a new saturating dose of streptavidin-Alexa 488 (green). In sharp contrast to AChR recycling (Fig. 3A,B), we found that AChEs originally labeled with fasciculin 2-biotin-streptavidin-Alexa 594 showed barely detectable green labeling and therefore negligible biotin-streptavidin dissociation after 4 d (Fig. 3C,D). These results show that the dissociation of streptavidin from biotin does not occur on the surface of the synapses and rules out the possibility that extracellular proteases or other unknown cellular processes at the cell surface are responsible for such dissociation, suggesting that this dissociation likely occurs in intracellular compartments after being internalized.

Streptavidin does not dissociate from AChR-BTX-biotin clusters on muscle cells in culture. A, In C2C12 myotube cultures, ghost clusters (as described by Kummer et al., 2004) that were no longer associated with myotubes were labeled with BTX-biotin followed by streptavidin-Alexa 594. B, Five days later, the same clusters were treated with streptavidin-Alexa 488 (to identify receptors that retained BTX-biotin and lost streptavidin). C, Overlay of images in A and B. The absence of green labeling in B indicates that there was no dissociation of streptavidin from receptor-bungarotoxin-biotin complexes on ghost clusters after 5 d. D, AChRs on a living C2C12 myotube that were labeled with a saturating dose of BTX-biotin followed immediately by a saturating dose of streptavidin-Alexa 660 (pseudocolor blue) and then imaged 12 h later. E, F, The same cells were then incubated with streptavidin-Alexa 488 (green) to label receptors that retain BTX-biotin and lose streptavidin and BTX-Alexa 594 (red) to label new AChRs. The absence of green labeling in E indicates that no detectable streptavidin-biotin dissociation occurred over this period, whereas the red labeling indicates the addition of new receptors. G, Overlay of D-F. Scale bars, 20 μm.
In denervated muscle, receptors are known to turn over at an accelerated rate, and muscle stimulation has been shown to prevent this increase in receptor loss (Salpeter and Loring, 1985; Andreose et al., 1993; Caroni et al., 1993; Akaaboune et al., 1999). If dissociation does not occur on the surface but only after the AChR-BTX-biotin-streptavidin complex is internalized, one would predict that decreasing the rate of internalization of receptors would also decrease the streptavidin-biotin dissociation. To test this possibility, we labeled synaptic AChRs on denervated muscle with BTX-biotin followed by streptavidin and then directly stimulated the sternomastoid muscle by placing stimulating electrodes at either end of the muscle for the entire 8 h experiment. As predicted from the previous results of others, 8 h after stimulation, we found no evidence of receptor loss (supplemental Fig. 2 A, available at www.jneurosci.org as supplemental material). Furthermore, when we added a second dose of streptavidin-Alexa and then quantified the recovery of fluorescence, we did not see any evidence for new streptavidin-Alexa binding. In contrast, a loss of preexisting fluorescence and the binding of new streptavidin-Alexa were easily measured in control, unstimulated muscle over the same time window (supplemental Fig. 2 B, available at www.jneurosci.org as supplemental material). This result argues strongly that the dissociation of streptavidin from AChR-BTX-biotin does not occur spontaneously on the muscle surface but, rather, only after internalization.
Evidence for the intracellular dissociation of streptavidin from the AChR-bungarotoxin-biotin complex
If streptavidin dissociates from the AChR-BTX-biotin-Streptavidin-Alexa complex at an intracellular site, it should be possible to observe Alexa fluorescence in intracellular vesicles. It should also be possible to observe intracellular vesicles with biotin freed from streptavidin. To test the first possibility, muscles were initially labeled with a saturating dose of BTX-biotin (5 μg/ml, 1.5 h) followed by a saturating dose of fluorescent streptavidin-Alexa 488. After an appropriate waiting time (4 d), the mouse was perfused with 2% paraformaldehyde. The sternomastoid muscle was then dissected and cut in longitudinal sections parallel to the axis of the muscle fibers to allow visualization of both the cell surface and the region below it, using confocal microscopy.
In control muscles that were labeled with BTX-biotin-streptavidin-Alexa 488 and then immediately fixed and sectioned (rather than waiting 4 d), the antibody-mediated AChR labeling was intense at the surface receptors of the synapse, plus a number of small spots (puncta), whereas the streptavidin-Alexa labeling was also intense on the cell surface but not visible in puncta. This is the expected result because the AChR immunocytochemistry detects all receptors, including those present internally, whereas the streptavidin-Alexa staining procedure initially detects only surface receptors, and some time must elapse before they become internalized. However, when 4 d were allowed between labeling and fixation, we found that most of the initial streptavidin label colocalized with receptors on the surface and in intracellular puncta (Fig. 4A-F); z-stack analysis of the images confirmed that these colocalized fluorescent spots were beneath the muscle surface, indicating that AChR-BTX-biotin-streptavidin was internalized as a complex. A significant number of the intracellular AChR-containing puncta were also labeled only by the anti-receptor antibody. These could be receptors that have lost their streptavidin label and are either in the process of recycling or degradation or unlabeled newly synthesized receptors yet to be inserted. A small number of green fluorescent spots were also observed, which did not colocalize with receptors, perhaps corresponding to accumulation of fluorescence in degradative vesicles.

Lack of streptavidin (Strept) dissociation from AChE-fasciculin 2-biotin in vivo. A, Example of a living adult mouse neuromuscular junction previously labeled with BTX-biotin (to label AChRs) followed by a saturating dose of streptavidin-Alexa 594 (red) at time 0 and imaged at 4 d. Red signal indicates the preexisting receptors still remaining on day 4. B, Same neuromuscular junction imaged after application of streptavidin-Alexa 488 (green). Green signal indicates the AChRs that were recycled over 4 d. C, Example of an NMJ labeled with fasciculin 2-biotin (to label AChEs) followed by a saturating dose of streptavidin-Alexa 594 (red) at time 0 and imaged at 4 d. Red signal indicates the preexisting acetycholinestrase remaining on day 4. D, The same neuromuscular junction imaged after application of new streptavidin-Alexa 488 (green) and viewed with much higher camera gain than used in B. The weak green signal indicates that there is very little streptavidin dissociation after 4 d. When the same gain was used as in B, the fluorescence was almost undetectable. Scale bars, 20 μm.
It seemed likely that many of the puncta were intracellular vesicles. To test this possibility, muscle fibers were fixed and then processed so that AChR and the early endosome marker EEA1 could be detected immunocytochemically (Fig. 4G-O). The EEA1 labeling was present in a profusion of small spots located below the cell surface, whereas the AChR labeling consisted of intense labeling of the surface receptors of the synapse plus a modest number of small spots. Although many EEA1 spots were negative for AChR, most AChR-positive spots were also positive for EEA1. Thus, puncta of AChR staining represent reliable markers of internalized AChR. Given the high correlation between AChR labeling of puncta and EEA1 labeling of puncta, the presence of puncta that were labeled both for AChR and for streptavidin in the experiments shown in Figure 4A-F demonstrates that fluorescent Alexa from surface AChRs labeled with BTX—biotin-streptavidin-Alexa is internalized. The puncta that stained only with the antireceptor antibody but were negative for Alexa most likely represent newly synthesized receptors yet to be inserted, whereas the puncta that stained for Alexa but not for AChR seem likely to correspond to degradative vesicles that have completely consumed their AChR.
Having shown that the Alexa from AChR-BTX-biotin-streptavidin-Alexa complexes can be internalized, we next tested whether biotin that had lost streptavidin could also be observed inside of muscle fibers. We first showed that the anti-biotin antibody we were using can effectively stain AChR-BTX-biotin complexes (Fig. 5A) but not AChR-BTX-biotin-streptavidin-Alexa 488 complexes (Fig. 5B-D). Thus, if muscles are pretreated with BTX-biotin and streptavidin-Alexa, this antibody will label only receptor complexes that have been stripped of streptavidin. Confocal images of muscle sections saturated with BTX-biotin-streptavidin and fixed and processed 4 d later showed that staining with both the anti-AChR antibody and the anti-biotin antibody labeled the entire synaptic region, and both fluorescent labels were also found colocalized in small spots in the vicinity of the junction (Fig. 5E-J). Approximately 40% of the intracellular spots were labeled by both antibodies, and the rest were labeled only by the antireceptor antibody. An alternate approach was also used to confirm the presence of free biotin within puncta. Longitudinal sections of mouse sternomastoid muscle saturated 4 d previously with BTX-biotin followed with streptavidin-Alexa 594 or unlabeled streptavidin were fixed and permeabilized as described above and labeled with the antireceptor antibody and streptavidin-Alexa 488. The number of green fluorescent puncta was similar to the number obtained when the anti-biotin antibody was used.
In summary, we see AChR-BTX-biotin complexes that have been stripped of their streptavidin labels both in intracellular vesicles and on the cell surface in vivo. Because we have seen no indication that dissociation occurs on the cell surface, the likely sequence is that the AChR-BTX-biotin-streptavidin complex is endocytosed into vesicles; the streptavidin is stripped off the complex and degraded; and then the AChR-BTX-biotin complex is returned to the surface. For these and other reasons considered in Discussion, we will refer to the pool of receptors that can be relabeled with a second color of streptavidin-Alexa as “recycled receptors.”
Time-lapse imaging of synaptic AChR recycling
Having found that receptors are able to recycle, we next wanted to determine how quickly recycled receptors were incorporated into the postsynaptic membrane. To do this, sternomastoid muscles were labeled with a low dose of BTX-biotin (5 μg/ml, 2 min, to ensure that synaptic activity remained fully functional) (Lingle and Steinbach, 1988) followed by a saturating dose of streptavidin-Alexa 488. A dose of streptavidin-Alexa 594 (30 min) was then added to ensure the saturation of all biotin sites (Fig. 6A,B). Every 2 h, a fresh dose of streptavidin-Alex 594 was added to the muscle to label recycled receptors. Using high detector gain with a CCD camera, we were able to see a clear red fluorescence signal at 2 h. In all 10 synapses imaged in three mice, the fluorescence signal increased in intensity after each new streptavidin-Alexa 594 application (Fig. 6C-F). This result suggests that recycled receptors are rapidly inserted into the membrane.
Effect of synaptic activity on the recycled and new AChR pools
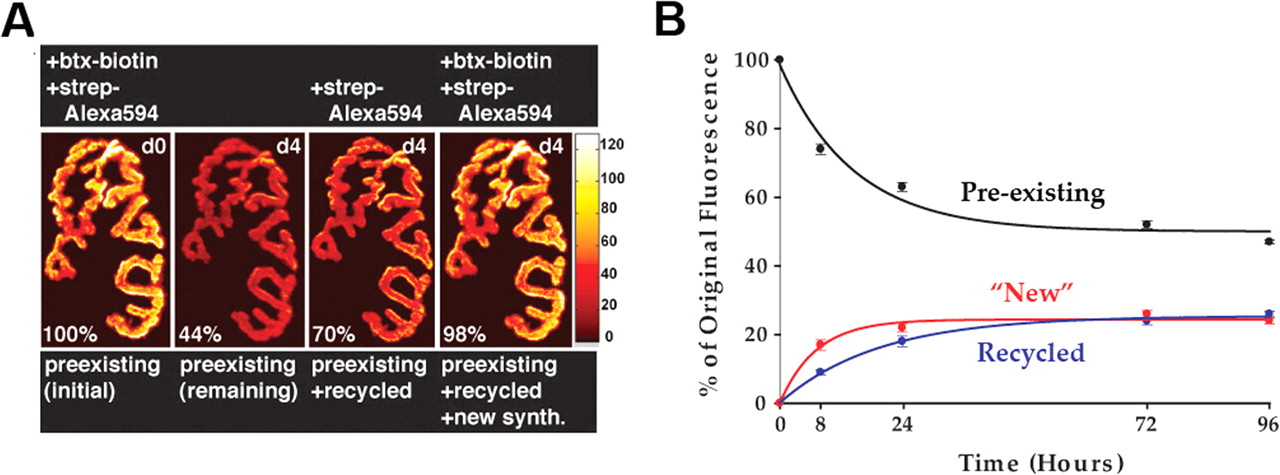
To quantitate the number of recycled and new receptors inserted into the same synapse over time, we used the same fluorophore in three distinct labeling steps, which allowed us to distinguish each receptor pool, and then we assayed fluorescence intensity using an established in vivo quantitative fluorescence method (Turney et al., 1996; Akaaboune et al., 1999). The sternomastoid muscles of six mice were saturated with an initial dose of BTX-biotin (5 μg/ml, 1.5 h) followed by streptavidin-Alexa 594 (red) (10 μg/ml, 2.5 h) to saturate biotin sites. The fluorescence intensity of superficial neuromuscular junctions was then assayed at time 0 and then at a second time point 8 h or 1, 3, or 4 d later (Fig. 7A,B). At 4 d, we found that junctions labeled at time 0 with a saturating dose of BTX-biotin-streptavidin-Alexa 594 had lost 53 ± 3% (SD; n = 30) of their original fluorescence. To quantitate the amount of receptors that were recycled over time, we applied a second saturating dose of streptavidin-Alexa 594 and measured the resulting increase in fluorescence intensity. We found that at 4 d, the fluorescence from the recycled receptors equaled 26 ± 5% (SD; n = 29) of the original fluorescence (Fig. 7A,B).

Confocal imaging of AChR-bungarotoxin-biotin-streptavidin (Strept) complexes in intracellular compartments. Sternomastoid muscles were labeled in living mice with BTX-biotin and saturated with streptavidin-Alexa 488. After 4 d, the muscles were fixed, sectioned longitudinally, and then processed for immunocytochemical detection of AChRs with an Alexa 594-conjugated secondary antibody. A, Example of a stack of 20 confocal images (0.5 μm intervals) of an NMJ that was initially labeled with BTX-biotin-streptavidin-Alexa 488 and collapsed onto a single plane. The small green fluorescence puncta could represent either the accumulation of fluorescent streptavidin still attached to AChR-BTX-biotin complexes or the accumulation of fluorescent streptavidin in intracellular compartments.B, Immunostaining with anti-AChR and secondary antibody conjugated to Alexa 594. These puncta could represent the presence of receptors that are newly synthesized, internalized and recycling, or internalized and in the process of degradation. C, Overlay of the Alexa 488 fluorescence in green and the anti-AChR fluorescence in red. D-F, Detail from the boxed regions in A-C. Arrows indicate colocalization of original streptavidin-Alexa 488 (green) and AChR (red) in intracellular puncta, showing that the AChR-BTX-biotin-streptavidin complexes are internalized. Red arrowheads indicate intracellular puncta that contained only receptors (likely unlabeled receptors that are in the process of insertion or BTX-biotin-labeled receptors in the process of recycling). Green arrowheads indicate intracellular puncta that contained only streptavidin-Alexa 488. G-O, Example of a neuromuscular junction on a muscle section (labeled as described above) immunostained for AChRs and EEA1. G-I, Eight confocal slices at 2 μm intervals were collapsed onto a single image plane. In the overlay image, green indicates anti-AChR immunostaining, and red indicates anti-EEA1 immunostaining. J-L, The boxed regions of G-I shown at higher power. M-O, z-stack of fluorescence intensity taken at various depths along the white lines in J-L. The top of the section is to the right, and the width of these panels corresponds to a total depth of 20 μm.
To quantitate the contribution of the new receptors added to these same synapses during the 4 d, the sternomastoid muscle was then labeled with a fresh saturating dose of BTX-biotin followed with a fresh saturating dose of streptavidin-Alexa 594. We found that an additional 24 ± 5% (SD; n = 29) of the original fluorescence was recovered (Fig. 7A,B). In summary, 4 d after a single transient application of BTX-biotin, the sum of remaining original (preexisting), new, and recycled receptors was very close to 100% of the original signal, and the contributions of newly inserted receptors and recycled receptors were nearly equal at ∼25% of original fluorescence each.

Confocal images of AChR-BTX-biotin complexes in intracellular compartments. A, Example of a neuromuscular junction labeled with BTX-biotin followed by anti-biotin-Alexa 488 or 594. This staining was a positive control to demonstrate that this antibody is able to effectively label biotin in complex with AChR. B, Example of a neuromuscular junction that was labeled with biotin-bungarotoxin followed by a saturating dose of streptavidin (strept)-Alexa 488 and then immediately fixed. C, When the same synapse was immunostained with anti-biotin-Alexa 594, no red fluorescence was observed. The absence of staining with anti-biotin indicates that this antibody can only interact with BTX-biotin when it has been stripped of streptavidin. D, Overlay of images in B and C. E, Example of a synapse on a muscle initially incubated with BTX-biotin-streptavidin and then fixed and sectioned 4 d later and immunostained with anti-AChR and an Alexa 594-conjugated secondary antibody. F, Same synapse as in E labeled with anti-biotin-Alexa 488. G, Overlay of images in E and F. H-J, Boxed areas from E-G at higher magnification and viewed at a camera gain sufficient to saturate the signal from synaptic AChRs to accentuate the signal coming from the internal spots. Arrowheads indicate spots of fluorescence that stained positive for AChR, but not for biotin, and arrows indicate spots where both labels were colocalized. Scale bar, 20 μm.
When the contributions of original, recycled, and new receptors were examined at earlier time points after the initial labeling (Fig. 7B), several interesting characteristics became apparent. At 1 d after a one-time bungarotoxin-AChR blockade, the contribution of recycled receptors was less [18 ± 5% (SD) of original fluorescence recovered; n = 25] than that of new receptors [22 ± 5% (SD) of original fluorescence recovered; n = 20; p < 0.005]. This effect was even more pronounced at 8 h with recycled receptors contributing 9 ± 3% (SD) of original fluorescence (n = 30) and new receptors accounting for 17 ± 7% (SD; n = 20; p < 0.0001) (Fig. 7B).
Receptors in the pools defined as preexisting and recycled both have BTX bound and so do not function, but the receptors in the new pool are unlabeled and therefore unblocked and functional. In the NMJ, it is known that a large safety factor exists that enables synapses to be fully functional with only 10-20% of receptors being unblocked (Lingle and Steinbach, 1988). Therefore, it would be expected that by 8 h, the 17% of the total receptors that are new and unblocked would be sufficient to restore superthreshold end plate potentials and muscle-spiking activity. It thus seemed likely that the changes in the rate of receptor recycling in transiently blocked NMJs (1.5 h one-time bungarotoxin treatment) are the consequence of the initial activity blockade by BTX and subsequent restoration of neuromuscular transmission by new unlabeled and unblocked AChRs.
To further examine the role of synaptic activity on the insertion of recycled and new receptors, we monitored the appearance of receptors in continuously blocked synapses, in which AChRs are reported to be lost at an accelerated rate (Salpeter and Loring, 1985; Martyn et al., 1992; Akaaboune et al., 1999). To test the effect of activity cessation on receptor trafficking, we chronically blocked synaptic transmission with curare (a poison that reversibly blocks AChRs) and so indirectly blocked muscle spiking. The longest this treatment could be performed without mortality was 8 h plus a 2 h treatment to label either recycled receptors or new receptors but not both. However, this was sufficiently long to resolve several interesting differences from the one-time labeling of AChRs with saturating bungarotoxin, in which blockade is transient and muscle activity recovers over time (Fig. 8). First, as expected from previous studies, the removal of original receptors was more pronounced (on average falling to 65% of the initial fluorescence at 8 h compared with 74% in animals receiving a one-time block with BTX). Second, the rate of appearance of new AChRs decreased to approximately one-third that observed when synaptic transmission was restored after a few hours but remained at observable levels [4.5 ± 2% (SD); n = 20]. Most dramatically, the contribution of the recycled receptors decreased to nearly undetectable levels [0.4 ± 1% (SD) of original fluorescence; n = 60] (Fig. 8A,B).

Time-lapse imaging of the appearance of recycled receptors. A, In a living adult mouse, a neuromuscular junction of the sternomastoid muscle was labeled with a low dose of BTX-biotin followed by a saturating dose of streptavidin (strept)-Alexa 488 (green). B, The junction was then immediately bathed with a saturating dose of streptavidin-Alexa 594 (red) and viewed at substantially higher gain. The absence of any red labeling indicates that all biotins were initially saturated with streptavidin-Alexa 488. C-F, The same neuromuscular junction imaged every 2 h for 8 h after application of new streptavidin-Alexa 594 (red) to the sternomastoid muscle before each time point. Scale bar, 20 μm. Similar results were obtained at 10 synapses from three mice.
Discussion
These results are based on the surprising finding that at sternomastoid muscle synapses in living mice, streptavidin dissociates unexpectedly rapidly from AChR-BTX-biotin complexes. The dissociation of purified streptavidin from biotin in test tube assays is extremely slow because of their extraordinarily high affinity (Chilkoti and Stayton, 1995). It therefore was a great surprise to us that at sternomastoid muscle synapses in living mice, streptavidin-binding sites rapidly appeared on the surface of muscle fibers after initial saturation with biotin-BTX followed by saturation with streptavidin-Alexa. One possible explanation was that biotin-BTX was able to label AChR inside the muscle fibers as well as on the muscle cell surface, but the streptavidin-Alexa could only bind to receptors on the cell surface of living muscle fibers. Were this the case, the appearance of new streptavidin-binding sites over time would represent the delivery of preexisting intracellular biotin-BTX-AChR complexes to the surface, rather than the creation of such complexes as a consequence of dissociation of the streptavidin from the biotin. Two lines of evidence rule out this possibility. First, according to this hypothesis, the rate of appearance of new streptavidin-binding sites after saturation with biotin-BTX and Alexa-streptavidin should have been much greater on C2C12 myotubes than on muscle fibers in living mice because cultured cells are known to have a much larger pool of intracellular AChRs (Devreotes and Fambrough, 1975), and these cultured myotubes rapidly internalize existing surface receptors and deliver new ones to the surface (Fig. 2, supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, there was no detectable appearance of new streptavidin-binding sites on the cell surface of C2C12 myotubes (Fig. 2). Second, the results of experiments presented in Figure 5 demonstrated that biotin-BTX applied to the surface of muscle fibers in living mice did not have access to intracellular AChR. In this series of experiments, free biotin was detected with an anti-biotin antibody in fixed, sectioned muscle fibers that were treated before fixation with saturating levels of biotin-BTX and then streptavidin-Alexa. The absence of any detectable free biotin in muscles fixed shortly after labeling (Fig. 5C) demonstrated that there was no intracellular AChR pool labeled only with biotin-BTX. We conclude from these results that streptavidin must be dissociating unexpectedly rapidly from AChR-BTX-biotin complexes.
Our observation that biotin-streptavidin dissociation occurs at an accelerated rate in vivo is not unprecedented: avidin-biotin complexes have been previously reported to dissociate after their intraperitoneal injection into live animals (Fraenkel-Conrat and Fraenkel-Conrat, 1952; Wei et al., 1971; Lee et al., 1972, 1973a,b). One potential molecular mechanism for enhanced dissociation comes from an older study that reported that, although the avidin-biotin complex is highly resistant to elevated temperature, acidic or basic pH, and proteases, the weak oxidizing environment provided by 0.3% H2O2 can readily cause dissociation (Gyorgy, 1943), which could allow the specific liberation of the streptavidin-Alexa tag from the AChR-BTX-biotin complex without affecting the AChR-BTX bond. A second potential mechanism for enhancing unbinding is suggested by recent studies conducted on purified proteins. When added to biotin-streptavidin complexes, two different classes of proteins have been demonstrated to have the ability to rapidly displace the streptavidin from the biotin (Subramanian et al., 1997; Morris and Raney, 1999). In both cases, the extra protein (a helicase in one study and a monoclonal antibody in the other) can be thought of as a chaperone, placing the streptavidin in a nonnative conformation that enhances unbinding. It seems plausible that the combination of low pH, an oxidizing environment, and perhaps some modification in the conformation of streptavidin by unknown factors within recycling vesicles could be responsible for the observed dissociation of streptavidin-Alexa from biotin. This mechanism would also be consistent with the absence of dissociation on the cell surface because this is a relatively reducing environment at nearly neutral pH.
The cellular location of dissociation of streptavidin from biotin
For several reasons, we believe that the dissociation of streptavidin from biotin takes place within intracellular vesicles rather than on the cell surface. First, we saw no evidence for streptavidin-biotin dissociation occurring on the cell surface of fixed muscle tissue (even when transplanted into an in vivo milieu), from AChR ghost clusters or from clusters of AChRs on living aneural muscle cells in culture. Second, we could readily observe AChR-BTX-biotin complexes that had been stripped of streptavidin in intracellular vesicles near synaptic sites in sternomastoid muscles. Third, when the rate of internalization of receptors was greatly decreased by electrical stimulation of the muscle, we could no longer detect dissociation of streptavidin from biotin. Finally, when the same labeling protocol was applied to AChE, a component of the synaptic cleft localized in the extracellular matrix rather in the plasma membrane, there was no evidence of streptavidin-biotin dissociation. This last observation implies that AChE is recycled very slowly or not at all. Insertion of AChE into the basal lamina requires collagen (ColQ) (Feng et al., 1999), so one possible explanation for the failure to recycle is that AChE-fasciculin 2-biotin-streptavidin complexes are internalized after detaching from ColQ and are therefore unable to insert back into the basal lamina. Whatever the reason for the lack of streptavidin unbinding from AChE complexes over time, these findings provide compelling evidence that local surface events are not responsible for the biotin-streptavidin dissociation observed with AChRs at the living neuromuscular synapse.

Quantification of original, recycled, and new receptors at the neuromuscular junction after transient activity blockade. These pseudocolor images provide a linear representation of the density of AChRs (white-yellow, high density; red-black, low density). A, Example of a mouse neuromuscular junction labeled with a saturating dose of BTX (btx)-biotin followed immediately by a saturating dose of streptavidin (strep)-Alexa 594, imaged, and then reimaged 4 d later. The total fluorescence intensity (a measure of the total number of AChRs) was expressed as 100% at the initial labeling (left panel, day 0) and normalized to this on each successive view. Second panel from left, Preexisting receptors remaining after 4 d when >50% of the initial fluorescence is lost. To quantitate the recycled receptors, streptavidin-Alexa 594 (red) fluorophore was then added to the muscle. Third panel from left, Sum of preexisting and recycled receptors after 4 d. New receptors were identified by next adding a saturating concentration of BTX-biotin followed by a saturating dose of streptavidin-Alexa 594 (red), as shown in the fourth panel. synth., Synthesized receptors. The fluorescence from these new receptors is added to the preexisting and recycled receptors to determine the total receptor density after 4 d. B, Summary of the amount of original receptors remaining after various times and the proportion of new and recycled receptors that were inserted into the synapse, obtained from many junctions with the approach shown in A. Each data point represents the mean percentage of original fluorescence intensity ± SEM (error bars).
A final reason for favoring vesicles as the site of the biotin-streptavidin dissociation is that a vesicular intermediate provides a ready explanation for the observation that the ability to recycle receptors to the cell surface can be dissociated from the ability to endocytose. Under two situations in which there was no synaptic activity (aneural cultures of C2C12 myotubes and in vivo synapses chronically blocked with curare), internalization of receptors was readily detected, as expected from previous results (St. John and Gordon, 2001), yet we saw no indication that there was any recycling (no vacant streptavidin binding sites on the cell surface). Regarding the failure to observe receptor recycling in cultured C2C12 myotubes, it has been shown previously that a large subsurface “hidden” pool of receptors exists in cultured chick and rat myotubes, but such a pool is absent in innervated muscle (Devreotes and Fambrough, 1975). This pool constitutes up to 30% of the total AChR complement of cultured myotubes and was found to derive partially from the surface of the muscle as opposed to newly synthesized receptors (Devreotes and Fambrough, 1975). As with AChRs at inactive synapses in living mice, these cultured muscle fibers are apparently lacking a signal that allows the internalized receptors to return to the cell surface.

Quantification of original, recycled, and new receptors at the neuromuscular junction after a transient and maintained activity blockade. A, Assessment of the extent of recycling 8 h after an initial transient blockade of activity with BTX-biotin. B, Assessment of the extent of recycling 8 h after a maintained blockade of activity with curare. C, Summary of results obtained from many synapses studied as in A and B. The data show the mean ± SEM (error bars). For all four conditions, the difference between the two experimental groups was significant (p < 0.001). These experiments were not performed beyond 8 h because of mouse mortality from the long-term curare treatment. It thus was not possible to measure recycling and new receptors at the same synapses. The insertion of new receptors was therefore measured in a parallel experiment using BTX-Alexa 594. strept, Streptavidin.
Both the in vitro and in vivo results could be readily explained if vesicles containing receptors available for recycling are delivered to the cell surface only when there is synaptic activity. An obvious candidate for mediating the activity-dependent signal is intracellular calcium because it is the most common signal that regulates exocytosis in both secretory and nonsecretory cells (Chavez et al., 1996). At CNS synapses, Ca2+ influx into the postsynaptic cell through NMDA receptors is involved in AMPA receptor recycling in cortical and hippocampal neurons (Carroll et al., 1999b; Luscher et al., 1999). Such alterations in AMPA and NMDA receptor numbers in the postsynaptic density are proposed to account for changes in synaptic strength (Malinow and Malenka, 2002; Malinow, 2003). Indeed, the activity-dependent cycling of AMPA receptors at the postsynaptic membrane underlies many aspects of synaptic plasticity (Malinow and Malenka, 2002; Malinow, 2003; Park et al., 2004). At the NMJ, AChRs allow the entry of calcium in addition to sodium and potassium, so it is conceivable that blocking the receptors could inhibit Ca2+ influx and thus block a signal required for receptor recycling.
What is the extent of AChR recycling at normally functioning synapses?
Our experiments have demonstrated that AChR recycling can occur at the neuromuscular junction of living mice. Although measurements of changes in fluorescence intensity over time were made with great precision, there are several important caveats to the interpretation of our results that cause us to be cautious about extrapolating these data to estimate the rates of internalization and recycling of native receptors when activity is unmodified.
First, only receptors that have been internalized have the possibility of recycling. If the internalization rate is slow, it is more difficult to detect recycling, even if the cellular mechanisms that cause recycling are fully functional.
Second, it is necessary to bind the bulky ligands α-BTX-biotin and streptavidin-Alexa to detect recycling. It is unclear to what extent the binding of streptavidin alters the life history of receptors. For instance, it is widely accepted that antibody binding to AChR can accelerate the rate of internalization and degradation (Fumagalli et al., 1982), perhaps as a consequence of cross-linking or because of other unknown factors. Streptavidin has four biotin-binding sites and so has the potential to cross-link biotins on multiple toxin molecules and thus indirectly cross-link receptors. However, the time course of the loss of fluorescence on cultured myotubes was not significantly different when receptors were directly labeled with α-BTX-Alexa or indirectly labeled with α-BTX-biotin and then with streptavidin-Alexa. This indicates that either steric factors prevented the streptavidin from cross-linking receptors labeled with biotin-BTX, or receptors cross-linked by streptavidin do not have an altered rate of internalization beyond any effect of α-BTX alone. It should be emphasized that recycling was readily detected at 8 h, but little or no detectable recycling occurred over the same period under three other experimental conditions (receptor clusters on cultured myotubes, synapses chronically inhibited with curare, and synapses on denervated muscle fibers that were chronically stimulated). Thus, binding of streptavidin to biotin-BTX is not sufficient to trigger recycling. Furthermore, at the stage that receptors recycle, they are no longer bound to streptavidin-Alexa, so the streptavidin is not necessary for the process to occur.
Finally, the amount of recycling we detect is a minimum estimate, and the actual amount could be greater. There are two possible ways that receptors could recycle without being detected by the methods we used. Any receptor that is endocytosed and returned to the cell surface without losing either its α-BTX-biotin or its streptavidin-Alexa will be counted as part of the preexisting pool, even though it has recycled. Similarly, any receptor that loses its α-BTX-biotin and then recycles will be counted as part of the new pool. At present, there are no available methods that would allow us to determine whether either of these two types of events occurs at a sufficient frequency to significantly increase the actual recycling rate above what we measured. Even without including any contribution from these potential sources of additional recycled receptors, we have demonstrated that, under one specific condition (recovery from a one-time blockade with BTX), at least 9% of the synaptic receptors on the cell surface were recycled over the first 8 h period, and ∼25% of the surface receptors were recycled over a 4 d period.
The existence of a recycled pool of AChRs requires that a number of earlier experiments on the life history of AChRs be reinterpreted. For instance, a substantial number of experiments using fluorescently labeled α-BTX have shown that the half-life of fluorescence loss at synapses in living mice is ∼7 d after a one-time blockade with bungarotoxin (Salpeter and Loring, 1985; Andreose et al., 1993; Caroni et al., 1993; Akaaboune et al., 1999, 2002). This had been interpreted as the half-life of receptors on the cell surface, but our results suggest that each receptor can recycle back to the cell surface (perhaps several times) before it is degraded, and junctional AChRs are therefore being internalized from the surface at a significantly faster rate than previously thought. The results reported here suggest that, despite the stability of the synaptic structure over the lifetime of the animal (Balice-Gordon and Lichtman, 1990), receptors are highly dynamic and are constantly exchanged between the plasmalemma and internal compartments, in addition to being frequently shuttled between junctional and extrajunctional regions (Akaaboune et al., 2002).
Together, our results suggest a model for the regulation of the postsynaptic receptor density at the neuromuscular junction in live animals involving two separate pathways: a receptor-recycling pathway and a new receptor pathway. When postsynaptic transmission is functional, both the recycled and the new receptor pathways contribute significantly to the postsynaptic density (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). In the absence of postsynaptic activity, however, internalized AChRs are shifted from a recycling pathway to a degradative pathway (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). The new receptor insertion pathway is also affected by the absence of postsynaptic activity but continues to deliver receptors at a reduced rate. We do not know, however, whether new and recycled receptors are added directly to the junction or are inserted into the perijunctional region and then migrate into the junction to be anchored. It would be of considerable interest to see whether different patterns of activity can alter the proportion of new and recycled receptors at synapses in live animals.
Footnotes
This work was supported by the University of Michigan, National Institute of Neurological Disorders and Stroke Research Grant NS047332 (M.A.), National Science Foundation Research Grant IBN-0077634 (R.I.H.), and National Institutes of Health Training Grant MH14279 (E.B.). We thank Daniel Goldman, Victoria Schlesinger, John Kuwada, John Oliva, Rafiqa Ameziane, and members of our department for helpful discussions about this work.
Correspondence should be addressed to Mohammed Akaaboune, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, 830 North University Avenue, Ann Arbor, MI 48109. E-mail: makaabou{at}umich.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259949-11$15.00/0
↵* E.B. and D.S. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}