

Abstract
Alzheimer's disease (AD) is often accompanied by extrapyramidal signs attributed to nigrostriatal dysfunction. The association between amyloid deposition and nigrostriatal degeneration is essentially unknown. We showed previously that the striatum and the substantia nigra of transgenic mice harboring familial AD (FAD)-linked APPswe/PS1ΔE9 mutants exhibit morphological alterations accompanied by amyloid-β (Aβ) deposition (Perez et al., 2004). In the present study, we further investigated the interaction between Aβ deposition and dopaminergic nigrostriatal dysfunction, by correlating morphological and biochemical changes in the nigrostriatal pathway with amyloid deposition pathology in the brains of 3- to 17-month-old APPswe/PS1ΔE9 transgenic mice and age-matched wild-type controls. We show that Aβ deposition is pronounced in the striatum of APPswe/PS1ΔE9 mice at 6 months of age, and the extent of deposition increases in an age-dependent manner. Tyrosine hydroxylase (TH)-positive dystrophic neurites with rosette or grape-like cluster disposition are observed adjacent to Aβ plaques and display multilaminar, multivesicular, and dense-core bodies as well as mitochondria. In addition, an age-dependent increase of TH protein levels are shown in nigral cells in these mutant mice. Using HPLC analysis, we found a reduction in the dopamine metabolite DOPAC in the striatum of these mice. These findings show a close association between amyloid deposition and nigrostriatal pathology and suggest that altered FAD-linked amyloid metabolism impairs, at least in part, the function of dopaminergic neurons.
Introduction
Alzheimer's disease (AD), the most common type of dementia in the elderly, is characterized by a progressive memory loss and cognitive decline. The histopathological hallmarks of AD include neuronal loss accompanied by amyloid deposition and formation of neurofibrillary tangles in brain (Whitehouse et al., 1985; Arnold et al., 1991; Braak and Braak, 1991). Amyloid deposits are composed of ∼4 kDa Aβ peptides that are liberated from larger amyloid precursor proteins (APPs) through the concerted proteolysis of BACE1 (β-site APP cleaving enzyme 1) and the γ-secretase complex. Familial, early-onset forms of the disease [familial AD (FAD)] are caused by mutations in genes encoding APP, presenilin1 (PS1), and presenilin2 (PS2) (Selkoe, 2001). Presenilin plays a central role in γ-secretase-mediated cleavage of APP, and expression of FAD-linked PS1 variants elevates production of highly fibrillogenic and neurotoxic Aβ42 peptides, which are deposited in the AD brain (Price and Sisodia, 1998).
In addition to cognitive deterioration, 35-40% of AD patients exhibit extrapyramidal motor features or parkinsonism (Lopez et al., 1997; Scarmeas et al., 2004). Some of these motor impairments are attributed to biochemical and structural changes in the dopaminergic substantia nigra (SN) basal ganglia system (Yanker and Mesulam, 1991; Murray et al., 1995; Liu et al., 1997; Burns et al., 2005). The nigrostriatal pathway exhibits neurofibrillary tangles, Aβ plaques, neuropil threads, neuronal loss, and a decrease in dopamine (DA) in AD (Rudelli et al., 1984; Ditter and Mirra, 1987; Gibb et al., 1989; Oyanagi et al., 1989; Braak and Braak, 1990; Uchihara et al., 1992; Selden et al., 1994a,b; Storga et al., 1996; Brilliant et al., 1997; Schneider et al., 2002; Lyness et al., 2003; Klunk et al., 2004) as well as in patients with AD/parkinsonism (Liu et al., 1997; Burns et al., 2005), suggesting that Aβ plays a critical role in dopaminergic dysfunction in these disorders.
During the past decade, numerous lines of transgenic (tg) mice have been developed to mimic, at least in part, some of the pathological lesions in AD (Suh and Checler, 2002; Oddo et al., 2003; Götz et al., 2004). Of particular interest is the finding that transgenic mice that coexpress FAD-linked mutant human APP and human wild-type (wt) α-synuclein exhibit enhanced accumulation of α-synuclein, neuronal degeneration, and motor deficits reminiscent of Parkinson's disease (PD). These findings suggest that elevated levels of Aβ peptides generated from the mutant APP might promote the induction of PD-like pathologies (Masliah et al., 2001). In agreement with these observations, we reported preliminary evidence that APPswe/PS1ΔE9 transgenic mice exhibit Aβ deposition in the striatum, dopaminergic dystrophic striatal neurites, and degenerative-like changes in substantia nigra dopaminergic neurons (Perez et al., 2004).
In the present study, we further investigate the mechanism(s) that underlie Aβ-induced nigrostriatal dysfunction. To this end, we performed quantitative morphologic and biochemical analysis of the nigrostriatal dopamine projection pathway in 3- to 17-month-old APPswe/PS1ΔE9 transgenic mice. In addition, we analyzed striatal lesions using ultrastructural methods. Together, the present findings of amyloid-associated nigrostriatal degeneration suggest that striatal Aβ deposition plays a critical role in nigrostriatal neuropathology that may lead to extrapyramidal motor impairments.
Materials and Methods
Transgenic mice
FAD-linked APPswe/PS1ΔE9 transgenic mice that coexpress PS1ΔE9 and a chimerical mouse-human APP695 with mutations (K595N, M596L) linked to Swedish FAD pedigrees (APPswe) have been described previously (Borchelt et al., 1996a; Lee et al., 1997; Lazarov et al., 2002). Transgene expression is driven by the mouse prion protein promoter (Borchelt et al., 1996b). A total of 35 heterozygous APPswe/PS1ΔE9 tg and wt mice between 3 and 17 months of age were used. The background strains for APPswe line C3-3 are (C3H/HeJ × C57BL/6JF3) × C57BL/6J n1, and C3H/HeJ × C57BL/6J F3 for PS1ΔE9 line S9. All animals were housed in an air-conditioned room and maintained on a 12 h light/dark cycle. Food and water were available ad libitum. Animal care and procedures were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Tissue preparation
All mice were deeply anesthetized with an intraperitoneal injection of ketamine (285 mg/kg) and xylazine (9.5 mg/kg), perfused transcardially with ice-cold saline solution (0.9% NaCl), and brains were rapidly removed from the skull and sectioned longitudinally in a frozen stainless steel brain blocker. One hemisphere was dissected for neurochemistry, whereas the other was immersion fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m phosphate buffer for 3-4 h at 4°C for histology and quantitative analysis. After fixation, the brains were cryo-protected in 30% sucrose at 4°C until they sank and were then cut on a sliding microtome in 40-μm-thick coronal or horizontal sections and stored at -4°C in a cryoprotectant solution (30% glycerol, 30% ethylene glycol in 0.1 m PBS) before use. For neurochemical analysis, the other hemisphere was placed in a stainless steel blocker, and the striatum was rapidly sectioned into 1 mm coronal slabs on a glass plate placed on wet ice. Striatal tissue was frozen on dry ice and stored at -80°C until analysis. For electron microscopy, brains were transcardially fixed with the same solution used for immersion fixation and cut at 50 μm on a Vibratome 1500 (Vibratome, St. Louis, MO).
Immunohistochemistry and immunofluorescence. Fixed tissue was processed as free-floating sections and singly labeled with either a polyclonal antibodies raised against TH (1:1000 dilution; Chemicon, Temecula, CA) or a monoclonal antibody raised against amyloid-β (Aβ; 10D5; 1:10,000 dilution; a gift from Elan Pharmaceuticals, San Francisco, CA) or dual stained for TH and Aβ. Sections were washed several times in Tris-buffered saline (TBS) before incubation with 0.1 m sodium periodate (to inhibit endogenous peroxidase activity) in a TBS solution for 20 min. After several rinses in a solution containing 0.25% Triton X-100 in TBS, the tissue was placed in the blocking solution containing TBS with 0.25% Triton X-100 and 3% of either goat or horse serum, according to corresponding secondary antibodies for 1 h. Sections were subsequently incubated in rabbit anti-human TH or mouse anti-Aβ for 24 h in a medium containing TBS 0.25% Triton X-100 and 1% goat normal serum or horse normal serum. After washes with 1% goat or horse normal serum in TBS, sections were incubated with biotinylated goat anti-rabbit or horse anti-mouse secondary antibodies (Vector Laboratories, Burlingame, CA) for 1 h. After several washes in TBS, the tissue was incubated for 60 min with an avidin-biotin complex (1:500; Elite kit; Vector Laboratories). Tissue was rinsed in 0.2 m sodium acetate, 1.0 m imidazol buffer, pH 7.4, and developed in an acetate-imidazol buffer containing 2.5% nickel sulfate, 0.05% 3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma, St. Louis, MO), and 0.0015% H2O2. The reaction was terminated using an acetate-imidazol buffer solution. A second series of sections were dual labeled for TH using a nickel chromagen, followed by incubation with the monoclonal 10D5 monoclonal antibodies raised against Aβ and detected using a nova red substrate kit (Vector Laboratories). This dual staining results in an easily identifiable two-colored profile: dark-blue TH-positive profiles and red Aβ-containing plaques. For Aβ visualization and quantification, a third series of sections was processed for confocal imaging. For this purpose, sections were immunolabeled using 10D5 antiserum (1:1000) and visualized with cyanine 3-conjugated donkey anti-mouse antibodies (1:300; Jackson ImmunoResearch, West Grove, PA). Finally, sections were mounted on glass slides, dehydrated in graded alcohols, cleared in xylenes, and coverslipped. In addition, several striatal sections from 3- to 6-month-old and 10- to 12-month-old APPswe/PS1ΔE9 tg mice groups were double stained with 10D5 antibodies and thioflavine-S and coverslipped in Apathy's Mounting Medium (made in Gur Laboratory). All sections were processed at the same time using the same chemical reagents to avoid batch-to-batch variation during immunostaining.
Transmission electron microscopy. Vibratome sections from three APPswe/PS1ΔE9 tg mice (1.5, 7.5, and 9.5 months old) and three wt mice (7, 13, and 16 months old) were postfixed in 1% OsO4 for 45-60 min, dehydrated, and embedded in Epoxy resin. The ultrathin sections (50-70 nm) were obtained with a Reichert-Jung Ultracut E (Leica, Nussloch, Germany) and then stained with 2% uranyl acetate and lead citrate. Striatal sections were viewed and photographed with a JEOL (Peabody, MA) transmission electron microscope.
HPLC. The striatum of 3- to 6-month-old (n = 6) and 10- to 12-month-old (n = 4) APPswe/PS1ΔE9 tg mice and age-matched controls (n = 12) were processed for determination of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) using HPLC. Tissue was placed in an antioxidant solution (0.4N per-chlorate, 1.343 mm EDTA, and 0.526 mm sodium metabisulfite) and homogenized using an ultrasonic homogenizer (BioLogics, Gainesville, VA). A small amount of the homogenate was used for protein determination (BCA Protein Reagent Kit; Pierce, Rockford, IL). The remaining suspension was spun at 14,000 × g for 20 min in a refrigerated centrifuge. The samples were separated on a Microsob MV C-18 column (5 μm; 4.6 × 250 mm; Varian, Walnut Creek, CA) and simultaneously examined for DA, DOPAC, and HVA using a 12-channel coulometric array detector (CoulArray 5200; ESA, Chelmsford, MA) attached to a Waters (Milford, MA) 2695 Solvent Delivery System, under the following conditions: flow rate, 1 ml/min; detection potentials, 50, 175, 350, 400, and 525 mV; and scrubbing potential, 650 mV. The mobile phase consisted of a 10% methanol solution in distilled water containing 21 g/L (0.1 m) citric acid, 10.65 g/L (0.075 m) Na2HPO4, 176 mg/L (0.8 m) heptanesulfonic acid, and 36 mg/L (0.097 mm) EDTA, pH 4.1. Unknown samples were quantified against a six-point standard curve with a minimum r2 of 0.97. Quality control samples were interspersed with each run to ensure HPLC calibration.
Data analysis
Imaging and quantification of amyloid burden. For quantification of Aβ burden, immunofluorescence images of 3-, 6-, 10-, and 12-month-old (n = 14) APPswe/PS1ΔE9 tg mice were taken with an Olympus (Tokyo, Japan) Optical Fluoview confocal laser-scanning microscope and volume of Aβ-immunoreactive (IR) deposits were analyzed using Multiscan 500PS equipped with MetaMorph 4.1 (Universal Imaging, Downing-town, PA), as described previously (Lazarov et al., 2002). Z series of 10 μm depth were imaged from each section with 1 μm intervals between images. These images were converted in all planes into binary images after calibration of pixel size. Single components were considered noise and were removed uniformly from all planes. Images were converted to grayscale by threshold, and the burden volume was estimated.
Stereologic analysis. The optical dissector method was used to determine the number of TH-IR neurons in the SN pars compacta (SNpc) of 3- to 6-month-old (n = 5) and 10- to 13-month-old (n = 5) APPswe/PS1ΔE9 tg mice, as well as age-matched wt mice (n = 12), as described previously (Ma et al., 1999; Jaffar et al., 2001). The SNpc was manually outlined under low magnification (10×), and at least 15% of the outlined region was systematically analyzed using a random sampling design. The estimated number of TH-IR neurons in the SNpc was performed by an observer blinded to age and genotype using MicroBrightField (Williston, VT) stereological software, an Olympus BX-60 microscope coupled with LEP MAC5000 (BioVision Technologies; Exton, PA) computer-controlled x-y-z motorized stage under a 100× planar oil-immersion objective (1.4 numerical aperture). The thickness of each section was assessed empirically, and upper and lower “guard zones” for each section were established between 3 and 4 μm before measurements. The coefficients of error were calculated according to the procedure of Gundersen et al. (1988) as estimates of precision. Values of <0.10 were accepted (West, 1993).
Optical density measurements of TH immunoreactivity in the striatum and substantia nigra. Quantification of the relative optical density (OD) of striatal TH immunoreactivity was performed using a densitometry software program (Image 1.60; Scion, Frederick, MD), as described previously (Mufson et al., 1997; Ma et al., 1999; Jaffar et al., 2001). The striatum and all of the neurons in the SN were outlined manually, and OD and area measurements were automatically analyzed in gray-scale images, using a computer program. Previous studies have shown that OD measurements reflect changes in protein expression parallel to those obtained using a biochemical protein assay such as Western blot (Mufson et al., 1997).

Photomicrographs of the striatum displaying TH-IR Aβ-IR plaques and dystrophic neurites in APPswe/PS1ΔE9 tg mice and wt mice. A-C, Confocal images showing age-related progression of striatal Aβ-IR plaques in APPswe/PS1ΔE9 tg mice. A, Lack of Aβ immunoreactivity at 3 months of age compared with the increase of Aβ deposition between 6 (B) and 12 (C) months of age. D, Higher-power image showing detail of an Aβ-IR plaque from a 12-month-old APPswe/PS1ΔE9 tg mice. E-G, Coronal striatal images showing single and double staining for thioflavine-S (E) and Aβ (F) in a 12-month-old APPswe/PS1ΔE9 mice. G, Merged image showing the colocalization of both markers in the plaque core (yellow), whereas the periphery was only Aβ positive (red). H-I, Bright-field photos of horizontal sections of the striatum from a 13-month-old wt mouse (H) showing the absence compared with the presence of numerous TH-IR dystrophic neurites (arrows) in a 15-month-old APPswe/PS1ΔE9 tg mouse (I). J, Higher-power image of a cluster of TH-IR dystrophic neurites shown in I. K, Horizontal section of the striatum of a 15-month-old APPswe/PS1ΔE9 tg mouse illustrating Aβ-IR plaques (red) surrounded by TH-IR dystrophic neurites (blue). L, Higher-power image of an Aβ-IR plaque from K showing swollen TH-IR dystrophic neurites. M-R, Examples of TH-IR dystrophic neurites in the striatum of 4- (M), 7.5- (N), and 10- (O-Q) month-old APPswe/PS1ΔE9 tg mice showing rosette-like patterns (M-O) and other aberrant profiles (P, Q). R, S, Higher-power image of Aβ-IR plaques (red) surrounded by TH-IR dystrophic neurites (blue) in the striatum of a 4- and a 17-month-old mutant mouse, respectively. Scale bars: A-C, 60 μm; D, 20 μm; E-G, 30 μm; H, I, 40 μm; J, K, S, 10 μm; L-Q, 7 μm.
Statistical analysis. Because the intensity levels of Aβ load were consistently below detectable threshold in the wt group, these data were excluded from statistical analysis. In contrast, to compare differences in Aβ load in young and old APPswe/PS1ΔE9 mice, we performed a quadratic regression model with intensity of Aβ load (log transformed) as the dependent variable and age and its quadratic term (age2) as the independent variables. The best-fitted curve from the model (solid line) and the corresponding 95% predictive band (dotted lines) are presented in Figure 4 A. The predictive band covers the area within which 95% of the Aβ load levels of the APPswe/PS1ΔE9 group are expected to fall (Kleinbaum et al., 1988). The data obtained from HPLC, stereologic, and optical density analyses were compared between groups using a nonparametric test (SigmaStat 3.0; Aspire Software International, Leesburg, VA). The data were evaluated using the Mann-Whitney rank-sum test, a powerful nonparametric test, which is used as an alternative to the parametric t test (Siegel, 1956). This test minimizes the effect of outliers in small samples. Because the median is the value used in the nonparametric Mann-Whitney rank-sum test, the observations are graphically presented in median values. The median values are more robust and informative than the means from these groups. Descriptive statistics were also presented in Table 1. The level of statistical significance was set at 0.05 (two-sided).
Morphological and biochemical measurements in the nigrostriatal system of APPswe/PS1 Δ E9 tg and wt mice
Results
Age-dependent increase in Aβ deposition in the striatum of APPswe/PS1ΔE9 transgenic mice
To examine the distribution of amyloid deposits in the striatum, brain sections were immunolabeled with antibodies against Aβ (Fig. 1A-D). Amyloid deposits were readily detected in the striatum of transgenic mice harboring APPswe/PS1ΔE9 by the age of 6 months (Fig. 1B); however, an occasional Aβ-IR plaque was observed in the striatum of 4-month-old mice (Fig. 1R). The extent of Aβ deposits increased with age and was most pronounced at 12 months of age (Fig. 1C,D). The distribution of the Aβ-IR plaques in the striatum of APPswe/PS1ΔE9 tg mice did not display a regional topographic pattern. Double staining of amyloid deposits for Aβ and thioflavine-S showed overlap in the plaque core, whereas Aβ immunoreactivity was seen at the periphery of the plaque (Fig. 1E-G). Amyloid deposits were not detected in the striatum or in any other brain areas in nontransgenic mice at any age.

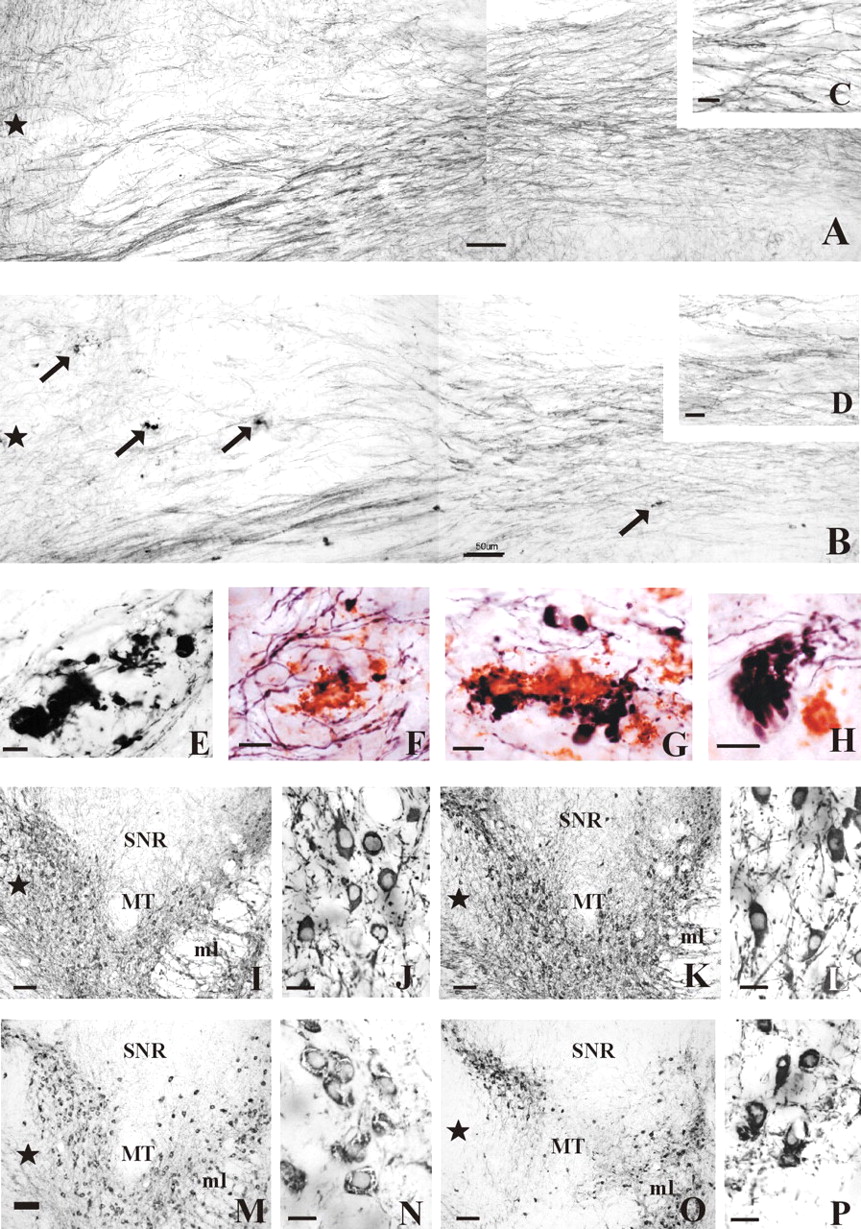
Photographs of the nigrostriatal bundle and the SN of APPswe/PS1ΔE9 tg and wt mice. A, B, Photomontages of horizontal sections showing TH-IR fibers within the nigrostriatal bundle of a 16-month-old wt (A) and 17-month-old APPswe/PS1ΔE9 tg (B) mouse. Note the presence of numerous TH-IR dystrophic neurites (arrows) and the relative decrease in the density of TH-IR fibers in bundle in the APPswe/PS1ΔE9 (B) compared with the wt (A) mouse. C, D, Insets showing detail of the TH-IR fibers of the nigrostriatal bundle from A and B, respectively. E, Example of swollen TH-IR dystrophic neurites observed in the nigrostriatal bundle in a 17-month-old mutant mice. F-H, Higher-power image of Aβ-IR plaques and/or TH-IR dystrophic neurites observed in the nigrostriatal bundle of a 17-month-old transgenic mice. Note the different shapes and sizes of the dystrophic neurites (blue) adjacent to the Aβ-IR profiles (red) (F, G), as well as an example of the grape-like neurite morphology (H). I, M, Photomicrographs of horizontal sections of nigral TH-IR neurons of a 7-month-old wt and a 7.5-month-old APPswe/PS1ΔE9 tg mouse, respectively. Note the strong TH neuronal immunoreactivity in the wt mice (I) compared with that seen in APPswe/PS1ΔE9 tg mice (M). J, N, Higher-power images of the TH-IR neurons shown in I and M, respectively. K, O, TH-IR neurons in the SN of a 16-month-old wt mouse and a 17-month-old APPswe/PS1ΔE9 tg mouse, respectively. L, P, Higher-power images of the SN neurons seen in K and O, respectively. Note the decrease of TH-IR processes in O as well as the shrunken appearance of TH-IR neurons with blunted dendritic processes in the SN (P). Stars indicate the rostral direction. SNR, Substantia nigra, pars reticulata; ml, medial lemniscus; MT, medial terminal nucleus accessory optic tract. Scale bars: A, B, I, K, M, O, 50 μm; C, D, E-H, 10 μm; J, L, N, P, 20 μm.
Aβ deposits in the striatum are coincident with TH-immunoreactive neurites
To evaluate the impact of Aβ deposition on the development of neuritic pathology in the striatum of transgenic mice, we dual stained brain sections with antibodies risen against Aβ and TH. Immunohistochemical analysis of striatal sections revealed that the majority of Aβ-IR deposits are surrounded by swollen, distended TH-IR dystrophic neurites (Fig. 1K,L,R,S) that displayed, in general, a rosette or grape-like cluster pattern (Fig. 1M-O). In the striatum of 3- to 6-month-old APPswe/PS1ΔE9 tg mice, TH-IR fibers appeared in the vicinity of the plaque but without morphological alterations. However, at the age of 15-17 months, we observed large and numerous dystrophic TH-IR neurites that surrounded the amyloid deposits (Fig. 1K,L,S). In addition, we observed a decrease in the density of TH-IR fibers innervating processes in the striatum at the age of 15-17 months (Fig. 1I). It should be noted that TH-IR dystrophic neurites in close proximity to amyloid deposits were also observed along the nigrostriatal bundle and within the neuropil of the SN of old APPswe/PSΔE9 tg mice (Fig. 2B,E-H). In addition, a striking reduction in the size of the nigrostriatal bundle and TH-IR neurons was observed at this age (Fig. 2A-D). The SNpc neurons appeared hollow and rounded with blunted dendritic processes (Fig. 2K,L,O,P).
Ultrastructural analysis of striatal profiles in APPswe/PS1ΔE9 transgenic mice
At the ultrastructural level, we observed variably sized striatal Aβ plaques, exhibiting fibrillar and/or amorphous scattered electron-dense material, as well as dystrophic neurites, by 7 months of age (Fig. 3B). Dystrophic neurites also appeared adjacent to Aβ plaques or in isolated groups removed from the plaque (Fig. 3B,C). Dystrophic neurites of diverse sizes showed an electron-dense or clear cytoplasm containing electron-dense multilaminar, multivesicular, and dense-core bodies, as well as mitochondria (Fig. 3C,D). In addition, myelinated axons presented signs of degeneration, including membrane whorls between myelin lamina, as well as an electron-dense cytoplasm (Fig. 3E,F). Although there was an occasional membrane whorl in the wt mice, the morphology and extent of these whorls were more conspicuous in APPswe/PS1ΔE9 transgenic mice (Fig. 3E,F).

Electron photomicrographs of the striatum from 7- and 7.5-month-old wt and APPswe/PS1ΔE9 tg mice, respectively. A, Striatal micrographs of a 7.5-month-old wt mouse showing normal ultrastructure of the cells and neuropil. B, Higher-magnification micrograph detailing a portion of a Aβ-plaque surrounded by dystrophic neurites (arrowheads) in the striatum of a 7.5-month-old APPswe/PS1ΔE9 mouse. Note the presence of Aβ branches within the striatal neuropil (black arrows). C, D, Examples of the dystrophic neurites in the striatum of a 7.5-month-old APPswe/PS1ΔE9 tg mouse showing multivesicular (white arrows), multilaminar (arrowheads), and dense core bodies (black arrows), as well as mitochondria. E, Electron micrograph from the striatum of a 7.5-month-old transgenic mouse showing a dystrophic neurite (white dashed circle) within an axon bundle. Note the presence of membrane whorls between the myelin lamina in some axons (arrows) as well as some with electron-dense cytoplasm (arrowheads), whereas in wt mice (F), these axonal changes were not observed. N, Nucleus; P, plaque. Scale bars: A, E, 4 μm; B, C, D, 2 μm; F, 1 μm.
Quantitative analysis of striatal plaque deposition in APPswe/PS1ΔE9 transgenic mice
To verify our observation of age-dependent amyloid deposition in the striatum of APPswe/PS1ΔE9 tg mice, the volume of amyloid immunoreactivity was quantified in immunostained brain sections of 3- to 12-month-old APPswe/PS1ΔE9 tg mice, as described previously (Lazarov et al., 2002). Striatal Aβ-IR deposition in APPswe/PS1ΔE9 tg mice increased exponentially, reaching a plateau at 10 months of age (Fig. 4A). No Aβ-IR plaques were detected in the striatum of 3-month-old mice (Figs. 1A, 4B). The estimated volume of Aβ-IR deposits in the striatum was sevenfold (285,242 mean) higher at 10 months than in 6-month-old APPswe/PS1ΔE9 tg mice (Fig. 4B).
TH-IR neuron numbers are unchanged in the substantia nigra of APPswe/PS1ΔE9 transgenic mice
To examine possible alterations in the numbers of TH-IR neurons in the SNpc of APPswe/PS1ΔE9 tg mice as a consequence of amyloid deposition in the striatum, the number of TH-IR neurons in cohorts of young and old mice was determined using unbiased stereology. Quantitative analysis revealed no difference in TH-IR neuronal density in the SNpc between the tg and wt mice, suggesting that amyloid deposition-associated neuropathology in the striatum does not lead to neuronal cell death in the SNpc at the time points examined (Fig. 5, Table 1).
TH-IR alterations in the substantia nigra of APPswe/PS1ΔE9 transgenic mice
TH immunoreactivity was examined in the striatum and the SNpc of transgenic mice at the age of 3-6 and 10-12 months using OD measurements with the aid of Scion image system. OD measurements revealed no difference in extent of TH immunoreactivity in the striatum of mutant and nontransgenic mice (Mann-Whitney rank-sum test; p > 0.05) (Fig. 6A, Table 1). However, a significant increase in extent of TH immunoreactivity could be detected in the SNpc of 10- to 12-month-old APPswe/PS1ΔE9 tg mice compared with 3- to 6-month-old mice (p = 0.016). A similar age-dependent increase in TH immunoreactivity did not occur in the SNpc of nontransgenic mice (Fig. 6B, Table 1). Notably, and for reasons that are presently not clear, the optical density of TH immunoreactivity was significantly lower in the SNpc of APPswe/PS1ΔE9 mice compared with nontransgenic mice at 3-6 months of age (p = 0.008) (Fig. 6B, Table 1). Furthermore, the average (184.350 μm2) cell size of nigral TH-IR neurons, as measured by the area of the somata (μm2), was reduced in APPswe/PS1ΔE9 tg mice at 3-6 months of age compared with wt mice (p = 0.048) (Fig. 6C, Table 1).
Altered dopamine metabolism in the striatum of APPswe/PS1ΔE9 transgenic mice
To evaluate possible alteration in the metabolism of dopamine in neurons as a consequence of striatal amyloid deposition, we determined the levels of DA, DOPAC, and HVA in the striatum using HPLC in 3- to 6-month-old and 10- to 12-month-old mutant and nontransgenic mice. These analyses revealed no age-dependent change in levels of DA and HVA in the striatum of APPswe/PS1ΔE9 tg mice (Fig. 7). However, we observed a significant reduction in the levels of DOPAC in the striatum of 10- to 12-month-old APPswe/PS1ΔE9 tg mice, compared with 3- to 6-month-old mice (Mann-Whitney rank-sum test; p < 0.05) (Fig. 7B, Table 1). Of interest is the observation that, at 3-6 months of age, striatal DOPAC levels were significantly higher in APPswe/PS1ΔE9 tg mice compared with nontransgenic mice (Fig. 7B, Table 1).
Discussion
The present study reports the novel observations of an age-related deposition of Aβ, dopaminergic dystrophic neurites, and defects in dopamine metabolism within the striatum in transgenic mice expressing FAD-linked APPswe and PS1ΔE9 (Borchelt et al., 1996a, 1997; Lee et al., 1997; Lazarov et al., 2002). In addition, dopaminergic neurons in the SN exhibit morphological phenotypic alterations indicative of neurodegeneration. The APPswe/PS1ΔE9 tg mouse is a well accepted model of the development of Aβ amyloidosis similar to that seen in AD and is characterized by age-associated acceleration of Aβ plaque deposition and neuritic pathology in the hippocampus and in the cerebral cortex (Lazarov et al., 2002). Our results show an early onset of Aβ deposition in the striatum of APPswe/PS1ΔE9 tg mice between the ages of 4 and 6 months and an exponential increase in amyloid burden until the age of 10-12 months. However, the degree of the Aβ deposition in the striatum was comparatively lower than that observed in the hippocampus of these mice (Lazarov et al., 2002). Nevertheless, coexpression of FAD-linked APP and PS1 transgenes driven by the mouse prion promoter not only produces an accelerated and early Aβ deposition in the cerebral cortex and the hippocampus (Lazarov et al., 2002) but also in the striatum (present study). The striatum is crucial for motor function control, and extracellular Aβ diffuse and compact plaques have been described in AD patients with or without extrapyramidal signs (Rudelli et al., 1984; Suenaga et al., 1990; Brilliant et al., 1997; Gearing et al., 1997).

Graphic presentation of the quantitative analyses of amyloid load of 3- to 12-month-old APPswe/PS1ΔE9 tg mice. A, Graph showing a quadratic regression representation of Aβ load (log-transformed) and its corresponding 95% predictive band (Pred. Band). Note that the increase in striatal Aβ load was exponential, reaching a plateau at 10 months (mos) of age. B, Histogram of the mean Aβ load in the striatum showing a higher level of Aβ deposition between 6 and 10 months, whereas at 3 months, no Aβ deposition was detected. Error bars represent SD.

Histogram showing the median estimated number of TH-IR neurons in an SNpc of one hemisphere of 3- to 6-month-old and 10- to 12-month-old APPswe/PS1ΔE9 tg and wt mice. Mann-Whitney rank-sum test did not reveal any significant differences in the number of nigral TH-IR neurons between the examined groups (p < 0.05). mos, Months.
In addition to an age-related increase in striatal Aβ plaques, we found a corresponding increase in the development of dopaminergic dystrophic neurites and dysfunction of dopamine metabolic activity in the striatum of APPswe/PS1ΔE9 tg mice. Numerous swollen TH-positive dystrophic neurites were observed surrounding Aβ-IR plaques in the striatum as well as within the nigrostriatal bundle. The number and the size of these dystrophic neurites increased with age in APPswe/PS1ΔE9 tg mice. Similar neuritic pathology has been reported in the hippocampus and cerebral cortex of different APP overexpressing tg mice (Wong et al., 1999; Diez et al., 2000; Jaffar et al., 2001; Boncristiano et al., 2002; German et al., 2003; Hu et al., 2003; Aucoin et al., 2005) but not in the striatum. In AD and in tg mice overexpressing APP (Brendza et al., 2003), dystrophic neurites were associated with thioflavine-S-positive plaques, supporting the suggestion that the toxic properties of Aβ are dictated by the protein conformation of these fibrils. In contrast, several studies have also suggested that Aβ promotes aberrant neuritic sprouting in vivo [AD (Masliah et al., 2003); APP/PS1 mice (Phinney et al., 1999; Hernandez et al., 2001)] and in vitro (Koo et al., 1993). Although the mechanism(s) underlying the interaction between Aβ and the formation of dystrophic neurites are unknown, their development has been related to deficits in axonal transport (Pigino et al., 2003; Stokin et al., 2005). In the striatum of AD patients, dystrophic neurites have also been described adjacent to compact Aβ deposits, suggesting that these deposits may induce the formation of these dystrophic neurites in AD (Suenaga et al., 1990). In contrast, it remains controversial whether dystrophic neurites are associated with neighboring diffuse striatal Aβ plaques in patients with AD/PD (Brilliant et al., 1997). Together, these observations suggest that amyloid conformation plays a critical role in neuritic dystrophy in the striatum, as reported in the cortex of tg mice overexpressing APP (Brendza et al., 2003). In this regard, Aβ antibody treatment reduces the size and number and straightens the geometry of dystrophic neurites in PDAPP tg mice, suggesting that neuritic damage is secondary to Aβ deposition and is partially reversible (Lombardo et al., 2003; Brendza et al., 2005). These data, together with the presence of TH-positive dystrophic neurites associated with compact Aβ plaques in the striatum and in the nigrostriatal bundle of APPswe/PS1ΔE9 tg mice, suggest that Aβ may play a critical role in promoting striatal dopaminergic dystrophic neurites, leading to dopaminergic dysfunction in the basal ganglia.
The electron microscopy analysis of the striatal neuropil in APPswe/PS1ΔE9 tg mice revealed the presence of subcellular degenerative structures, such as dystrophic neurites and Aβ plaques (present study). It is interesting to note that these structural alterations in the striatum were not observed in 1.5-month-old APPswe/PS1ΔE9 tg mice, coinciding with the lack of Aβ deposition in the striatum (present study). The fine structure of the dystrophic neurites in the striatum of APPswe/PS1ΔE9 tg mice was similar to those described in the cerebral cortex and hippocampus of APP-overexpressed mice (Masliah et al., 1996; Phinney et al., 1999; Luth et al., 2003) and AD (Masliah et al., 1996). Dystrophic neurites in the striatum, similar to that seen in the hippocampus and cortex of other APP-overexpressing tg mice are located mostly in close proximity to Aβ plaques and contain multilaminar, multivesicular, and dense-core bodies, as well as mitochondria (present study and Phinney et al., 1999; Masliah et al., 1996; Luth et al., 2003). Vesicular organelles have been ultrastructurally identified as part of a lysosomal system in APP-overexpressing tg mice and AD, which have been implicated in different endocytic pathways that are altered in this disease (Cataldo et al., 2000, 2004; Mathews et al., 2002). Moreover, Aβ-independent lysosomal pathology is found in single PS1 tg mice (Cataldo et al., 2004). The presence of multilaminar and multivesicular bodies as well as dense-core bodies in dystrophic neurites in our mice, similar to neuritic reactions and phagocytic pathways seen in AD, suggest that similar pathologic mechanisms underlie striatal fiber dystrophy seen in APPswe/PS1ΔE9 tg mice.
Despite the age-related increase in amyloid and TH-IR dystrophic neurites in the striatum, no significant changes in OD for TH immunoreactivity were observed in the striatum of 3- to 12-month-old APPswe/PS1ΔE9 mice. The factors underlying this finding remain unknown. It is possible that swollen TH-IR dystrophic neurites contain increased amounts of TH protein, masking the differences in OD measurements of TH immunoreactivity between APPswe/PS1ΔE9 tg and nontransgenic mice. Alternatively, the striatal levels of TH are not affected by the deposition of Aβ at these ages. Although the present investigation did not reveal alteration in striatal DA and HVA as reported in 12- to 18-month-old APPswe/PS1ΔE9 tg mice (Szapacs et al., 2004), there was a significant decrease in DOPAC in the striatum of these mice between 3 and 12 months of age (present study). Notably, Szapacs et al. (2004) reported no differences in striatal DOPAC levels in APPswe/PS1ΔE9 tg mice at 12 and 18 months of age. In addition, our findings, showing increased striatal DOPAC levels in APPswe/PS1ΔE9 tg mice at 3-6 months of age versus wt mice, suggest a developmental and/or compensatory upregulation of this DA metabolite. Future investigations are required to determine which of these mechanism(s) underlie the higher levels of striatal DOPAC seen in 3- to 6-month-old tg mice. In contrast, because DOPAC is the product of presynaptic oxidative deamination of DA by monoamine oxidase (MAO) (Greenawalt and Schnaitman, 1970) located in the outer mitochondrial membrane, it is possible that MAO or mitochondrial dysfunction are factors underlying striatal DOPAC changes in APPswe/PS1ΔE9 tg mice. Interestingly, mitochondrial dysfunction is implicated in the pathogenesis of AD (Blass, 2000; Castellani et al., 2002; Zhu et al., 2004; Takuma et al., 2005).
In the present study, we did not find reductions in the number of the TH-IR neurons in the SNpc of APPswe/PS1ΔE9 tg mice, a finding that further implies that, in transgenic mice harboring FAD-linked variants, axopathology rather than neuronal cell death is more pronounced in the nigrostriatal system after FAD-linked altered amyloid metabolism. Thus, the number of TH-positive perikarya in the SNpc was similar between 3- to 6-month-old and 10- to 12-month-old APPswe/PS1ΔE9 tg mice. In contrast, morphologic changes were observed in dopaminergic neurons of the SNpc in older mice between the ages of 15 and 17 months, in which the cell bodies appeared hollow, shrunken with blunted dendrites. In this regard, the SN has been described as being morphologically abnormal in AD patients (Ditter and Mirra, 1987; Gibb et al., 1989, Uchihara et al., 1992) as well as in patients with AD and parkinsonian signs (Murray et al., 1995; Liu et al., 1997; Burns et al., 2005). Interestingly, loss of DA SNpc neurons is not a necessary precondition in the pathology of AD with parkinsonism (Murray et al., 1995; Liu et al., 1997), suggesting that, symptomatically, our transgenic mice might represent a closer model of AD/parkinsonism than idiopathic PD. However, mice overexpressing APP and α-synuclein display Aβ plaques, aberrant intracellular synuclein accumulation, and striatal cell loss (Masliah et al., 2001), suggesting a role for synuclein in neuronal degeneration. It has been suggested that mice do not live long enough to show extensive neuronal loss or that protective factors, including transgene-derived soluble APP derivatives, could counter the neurotoxic effects of Aβ as implicated in APPswe tg mice (Stein and Johnson, 2002).
Although there were no significant differences in the number of dopaminergic neurons in the SN of APPswe/PS1ΔE9 mice, a statistical increase in the relative OD levels of TH-IR per neuron was detected compared with nontransgenic mice. Moreover, dopaminergic neuron size was smaller in APPswe/PS1ΔE9 tg mice compared with nontransgenic mice at 3-6 months of age. The increase of TH protein in the soma of nigral neurons may be indicative of changes in the steady-state levels of TH-mRNA or a result of a dysfunction in axonal transport. In this regard, studies have suggested that defects in axonal transport may be responsible for cellular neurodegeneration in AD and in transgenic animal models (Gajdusek, 1985; Morfini et al., 2002; Pigino et al., 2003; Stokin et al., 2005).

A, B, Linear analysis of median OD measurements of TH immunoreactivity within the striatum (A) and the SN (B), as well as the median area of TH-IR neurons within the SN (C) in 3- to 6-month-old and 10- to 12-month-old APPswe/PS1ΔE9 tg and wt mice. A, The median OD of striatal TH immunoreactivity did not reveal significant differences between the examined groups, whereas there was a significant increase in the median OD of TH-IR neurons of the SNpc between APPswe/PS1ΔE9 mice and wt mice. B, Note that, at 3-6 months of age, the median OD of TH immunoreactivity in the SN was significantly lower than in 10- to 12-month-old APPswe/PS1ΔE9 mice. C, Statistical analysis revealed significant differences in the median area of TH-IR neurons within the SNpc at 3-6 months of age between APPswe/PS1ΔE9 tg mice and wt mice. Mann-Whitney rank-sum test, p < 0.05. mos, Months.
Finally, APPswe/PS1ΔE9 tg mice display a motor deficit (Berg et al., 2005), suggesting that Aβ deposition and/or Aβ play a critical role in dopaminergic nigrostriatal dysfunction, which mimic some of the neuropathological events observed in AD and AD/parkinsonism. Therefore, the APPswe/PS1ΔE9 tg mouse provides a unique tool to investigate the relationship between Aβ and degenerative process of the dopaminergic system and their repercussions during motor function. However, it is important to understand that APPswe/PS1ΔE9 tg mouse and other models of amyloidosis do not replicate the full neuropathological signature of human AD or AD/PD. Despite these caveats, the APPswe/PS1ΔE9 tg mouse provides a valuable model to investigate the impact that compact Aβ burden has on the structure and function of the nigrostriatal system.

A-C, Linear analysis of median HPLC measurements of DA (A), DOPAC (B), and HVA (C) within the striatum of 3- to 6-month-old and 10- to 12-month-old APPswe/PS1ΔE9 tg mice and age-matched wt mice. Statistical evaluation revealed a significant age-related decrease in the median levels of striatal DOPAC (B) but not in DA (A) or HVA (C). At 3-6 months (mos) of age, the levels of DOPAC in APPswe/PS1ΔE9 tg mice were significantly higher than in the wt mice. Mann-Whitney rank-sum test, p < 0.05.
Footnotes
This work was supported in part by National Institute on Aging Grants AG021494 (S.S.S.) and AG10688 (E.J.M.), the Shapiro Foundation (E.J.M.), and a departmental grant from the Rush University Medical Center (S.E.P.). We thank Joanne Wuu for statistical advice and Nicholas G. Campbell for technical assistance with HPLC.
Correspondence should be addressed to Dr. Elliott J. Mufson, Professor of Neurological Sciences, Alla V. and Solomon Jesmer Chair in Aging, Rush University Medical Center, 1735 West Harrison Street, Suite 300, Chicago, IL 60612. E-mail: emufson{at}rush.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510220-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}