

Abstract
The development of abnormal primary sensory neuron excitability and neuropathic pain symptoms after peripheral nerve injury is associated with altered expression of voltage-gated sodium channels (VGSCs) and a modification of sodium currents. To investigate whether the β2 subunit of VGSCs participates in the generation of neuropathic pain, we used the spared nerve injury (SNI) model in rats to examine β2 subunit expression in selectively injured (tibial and common peroneal nerves) and uninjured (sural nerve) afferents. Three days after SNI, immunohistochemistry and Western blot analysis reveal an increase in the β2 subunit in both the cell body and peripheral axons of injured neurons. The increase persists for >4 weeks, although β2 subunit mRNA measured by real-time reverse transcription-PCR and in situ hybridization remains unchanged. Although injured neurons show the most marked upregulation,β2 subunit expression is also increased in neighboring non-injured neurons and a similar pattern of changes appears in the spinal nerve ligation model of neuropathic pain. That increased β2 subunit expression in sensory neurons after nerve injury is functionally significant, as demonstrated by our finding that the development of mechanical allodynia-like behavior in the SNI model is attenuated in β2 subunit null mutant mice. Through its role in regulating the density of mature VGSC complexes in the plasma membrane and modulating channel gating, the β2 subunit may play a key role in the development of ectopic activity in injured and non-injured sensory afferents and, thereby, neuropathic pain.
Introduction
Neuropathic pain occurs as a consequence of lesions or dysfunction of the nervous system and is characterized by spontaneous pain, allodynia, and hyperalgesia. After peripheral nerve injury, ectopic discharge in primary sensory neurons may directly initiate spontaneous pain (Wall and Devor, 1983; Price et al., 1989). Abnormal excitability in the injured primary sensory neurons occurs at the neuroma, along the axons, and in the dorsal root ganglia (DRGs), as well as in adjacent intact afferents (Wall and Devor, 1983; Wu et al., 2001; Ma et al., 2003; Amir et al., 2005). Spontaneous afferent inflow from both injured and non-injured A- and C-fibers may also generate activity-dependent changes in dorsal horn neurons (Amir and Devor, 2000; Gold, 2000; Wu et al., 2001).
Voltage-gated sodium channels (VGSCs) are responsible for the generation and propagation of action potentials in the membranes of most neurons and excitable cells. The VGSC is composed of a combination of pore-forming α subunits and auxiliary β subunits. Ten genes encoding the selective pore-forming α subunits have been identified, and both tetrodotoxin (TTX)-sensitive α subunits (Nav1.7, Nav1.1, Nav1.6, Nav1.2, and Nav1.3) and TTX-resistant α subunits (Nav1.8 and Nav1.9) have been identified in DRG neurons (Black et al., 1996; Amaya et al., 2000; Yu and Catterall, 2004). The mature channel in the plasma membrane is composed of an α subunit associated with a disulfide-linked β2 or β4 subunit and a noncovalently linked β1 or β3 subunit (Isom et al., 1992, 1995; Morgan et al., 2000; Yu et al., 2003). All known β subunits (β1-β4) are expressed in primary sensory neurons, and recent findings suggest that β3 and β4 may substitute for β1 and β2, respectively, in some sodium channels (Morgan et al., 2000; Yu et al., 2003).
Altered neuronal excitability after peripheral nerve injury may depend to a considerable extent on injury-induced modulation of VGSC expression and trafficking in primary sensory and dorsal horn neurons (Hains et al., 2004; Wood et al., 2004). Coexpression of β2 subunits with α subunit channels in cell expression systems and studies of null mutant mice have demonstrated the role of the β2 subunit in voltage dependence of channel activation and inactivation and augmentation of channel density at the plasma membrane (Isom et al., 1995; Chen et al., 2002). Based on these results, we investigated whether the β2 subunit may be involved in abnormal neuronal excitability in injured and non-injured neurons after peripheral nerve injury.
We have examined changes in β2 subunit expression in primary sensory neurons and their peripheral axons in two models of neuropathic pain: the spared nerve injury (SNI) model, in which the common peroneal and tibial nerves are severed leaving the sural nerve intact, and the L5 spinal nerve ligation (SNL) model (Kim and Chung, 1992; Decosterd and Woolf, 2000). Both models allow analysis of changes in injured and non-injured primary sensory neurons. To study whether altered β2 subunit expression has functional consequences, neuropathic pain-related behavior after SNI was investigated in β2 subunit null mutant mice (Chen et al., 2002).
Materials and Methods
Animals and surgery. Experiments were performed in 200-250 g adult male Sprague Dawley rats (Charles River France, L'Abresle, France). Adult wild-type C57BL/6 mice and β2 subunit null mutant mice (B6.129-Scn2btm1Lli, backcrossed over 12 generations with C57BL/6 mice) (Chen et al., 2002) were included in a subset of behavioral experiments. All procedures were approved by the Committee on Animal Experimentation of the canton of Vaud, Switzerland, in accordance with the Swiss Federal Law on Animal Welfare and the guidelines of the International Association for the Study of Pain (Zimmermann, 1983). Animals were housed under a 12 h light/dark cycle and had access to water and food ad libitum.
Surgical and injection procedures were performed under 1.5-2.5% isoflurane (Abott, Baar, Switzerland) general anesthesia as described previously (Decosterd et al., 2002). Briefly, after exposure of the sciatic nerve by an incision of the biceps femoris, the sciatic nerve was ligated by a 5.0 silk suture (Ethicon; Johnson & Johnson, Brussels, Belgium) and transected for the total axotomy model. For the SNI model, the common peroneal and tibial nerves were ligated with 5.0 silk suture and transected, anda3mm portion of the nerves was removed. To produce an SNL, L4 and L5 transverse processes were exposed and an L5 hemilaminectomy was performed to access the L4 and L5 spinal nerves. The L5 spinal nerve was then tightly ligated with 6.0 silk thread. Muscle and skin were closed in two distinct layers with 5.0 silk threads and wound clips, respectively, in all procedures. Sham surgeries for control groups were similarly performed without nerve injury. Fluorogold (FG) (2.5%; Fluorochrome, Denver, CO) was injected (eight times, 1 μl) in the sural skin territory of a subgroup of SNI-operated or control rats for retrograde tracing and identification of neurons of non-injured sural nerve. In addition, we used the unilateral arthritis complete Freund's adjuvant (CFA) model of inflammation consisting of a single injection of 50 μl of CFA (Sigma, St. Louis, MO) into the plantar surface of the hindpaw.
Animals were allowed to survive for 1, 3, 5, 7, 14, 21, or 28 d (n = 3-12 in treated and control groups at all time points and type of analysis).
β2 antibodies. Several anti-β2 subunit antibodies have been used in the present study. Antibodies were raised in rabbits against the human (GW83 and GW93) and the rat (GW2690) C-terminal cytoplasmic tail of β2 subunit using the peptides CVRRKKEQKLSTDDLKTEEEGKTDGEGNPDDGAK (GW83 and GW93) (Coward et al., 2001) and EGKTDGEGNAEDGAK (GW2690). β2-ec antibody, a generous gift from the laboratory of W. A. Catterall (University of Washington, Seattle, WA), was generated using a peptide corresponding to the extracellular residues 59-73 of the rat sodium channel β2 subunit (INLKLERFGDRVEFS) (Chen et al., 2002). None of the immunizing peptide sequences chosen have similarities with the sequences of the β1, β3, and β4 subunits. In addition to standard controls excluding primary antibody from reactions, characterization, and controls included incubation of blot membrane (GW93) or tissue sections (GW83) with β2 antibody preabsorbed for 1 h at room temperature (RT) with incremental concentrations (1-100 nm) of respective immunizing peptides. Western blot analyses were performed on proteins extracted from cell lines stably expressing the individual β subunits (isolated after transfection of human embryonic kidney HEK293 with control vector, pCIH-β1, pCIH-β2, and pCIH-β3) (Meadows et al., 2002) and from brain tissue of adult β2 subunit -/- mice and control wild-type mice.
Western blots. L4/L5 DRGs and nerves were homogenized and lysed in a solution of 2% SDS, 20% glycerol, and 100 mm Tris-HCl, pH 6.8, containing a protease inhibitor mixture (Complete; Roche Diagnostics, Rotkreuz, Switzerland). The homogenate was centrifuged at 10,000 × g for 20 min at 4°C, and protein concentration in supernatant was determined by the Bradford method (Protein Assay; Bio-Rad, München, Germany). The molecular weight of β2 subunit depends on the tissue (above 30 kDa in cell extracts/brain samples, at 25 kDa in DRG samples). This suggests different levels of glycosylation in the different tissues. To deal with this, an additional deglycosylation step was added for processing brain and cell extracts by incubating the lysate with 500 U of PNGase F enzyme in buffers supplied by the manufacturer (New England Biolabs, Frankfurt am Main, Germany) for 1.5 h at 37°C. Then, 1 μl of β-mercapto-ethanol and 1 μl of 1% bromophenol blue were added for each 40 μg of protein sample and incubated 3 min at 100°C for denaturation. A subcellular fractionation of cytosolic and plasma membrane proteins using ultracentrifugation was performed for a subset of samples: DRGs were lysed in a cold lysis buffer (10 mm Tris, pH 7.5, 300 mm sucrose, 1 mm EDTA, and protease inhibitor mixture) at 4°C, sonicated, and centrifuged at 6000 × g for 5 min. The supernatant was then centrifuged at 100,000 × g for 1 h. The pellet of membrane proteins was washed, centrifuged at 100,000 × g, and resuspended in lysis buffer.
Proteins were separated on 10% SDS-PAGE gel and transferred to polyvinylidene difluoride membrane with a Semidry Transfer System (Bio-Rad, München, Germany). Membranes were blocked at RT for 30 min in 5% milk in PBS containing 0.2% Tween-20, followed by incubation with β2 antibody (GW93, 1:1000; GW2690, 1:75; and β2-ec, 1:250) overnight at 4°C. The blots were then incubated for 90 min at RT with HRP-conjugated goat anti-rabbit secondary antibody (1:2000; Dako, Glostrup, Denmark), developed using enhanced chemiluminescence solution (ECL Plus; Amersham Biosciences, Little Chalfont, UK) for 5 min at RT, and exposed on films (BioMax MR Film; Eastman Kodak, Rochester, NY) for 30 s to 15 minutes. Blots were stripped and reprobed with extracellular signal-regulated kinase 1/2 (ERK1/ERK2) antibody diluted at 1:3000 (Cell Signaling Technology, Beverly, MA) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody at 1:1,000,000 (Abcam, Cambridge, UK). Constitutively expressed ERK1/ERK2 is not regulated by nerve injury and has served as a convenient protein loading control (Zhuang et al., 2005). To confirm that ERK1/ERK2 protein levels are unaltered by nerve injury, a control using GAPDH as an internal control on the same membrane was performed (Fig. 1 E). We find no difference in the relative amounts of ERK and GAPDH in control and DRGs from animals after SNI.
For analysis and quantification, films were scanned and the density of specific bands was measured and normalized with the corresponding loading control ERK2 band (Scion Image software; Scion, Frederick, MD).
Immunohistochemistry. Rats were terminally anesthetized with pentobarbital sodium (100 mg/kg, i.p) and transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. L4 and L5 DRGs and nerves (sural alone or tibial and common peroneal nerves) were dissected, postfixed at 4°C for additional 90 min in the same fixative solution, and transferred into 20% sucrose 0.1 m PB overnight. DRGs and nerves were mounted in O.C.T. compound (Tissue-Tek; Sakura Finetek, Zoeterwoude, The Netherlands). Tissue obtained from peripheral nerve injury and control groups were embedded in the same mold, frozen on dry ice, serially cryosectioned parallel to the direction of nerve fibers at 12 μm of thickness, and deposed on Superfrost plus-coated slides. Tyramide system amplification (TSA+ Fluorescence systems; NEN Life Science, Boston, MA) followed by direct immunofluorescence was used for coimmunodetection of β2 subunits with other markers (Amaya et al., 2000). Sections were incubated for 30 min at 37°C in phenylhydrazin (0.1% in PBS). After three washing steps with TNT buffer (0.15 m NaCl, 0.1 m Tris-HCl, pH 7.5, and 0.05% Tween 20), sections were blocked for 30 min at RT in TNB buffer (0.5% blocking reagent provided by the manufacturer, 0.15 m NaCl, 0.1 m Tris-HCl, pH 7.5, and 0.5% Tween 20) and then incubated overnight at 4°C with β2 GW83 antibody (1:1000). After washing, sections were incubated with biotin-conjugated anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA) at a dilution of 1:200 in TNB buffer for 2 h at RT, and then another incubation with streptavidin-HRP (provided in TSA+ kit) at 1:100 in TNB buffer was performed for 30 min at RT. Finally, tyramide was added (1:50 in amplification diluent buffer) to slides for 8 min at RT.

Spared nerve injury increases expression of the β2 subunit of VGSCs in the DRGs. A, Deglycosylated lysates of HEK293 cells transfected with β2, β1, or β3 subunit expression constructs or control expression vector only (ctrl). A single band is detected in β2-expressing cells only (GW93 antibody). B, Immunoblot with GW93 anti-β2 subunit antibody on deglycosylated brain tissue from β2 subunit null mutant (-/-) or wild-type (+/+) mice. The antibody was preincubated with a specific β2 peptide at increasing concentrations (0-100 nm) before Western blotting. C-E, Three antibodies specific for the β2 subunit all recognize a single band at 25 kDa in Western blot analyses of L4/L5 DRG tissue from SNI-operated and control rats. Enhanced β2 subunit immunoreactivity 1 week after SNI is observed using β2 subunit β2-ec (C), GW2690 (D), and GW93 (E) antibodies. To control for loading, the same blots were reprobed with an antibody recognizing both ERK1 (44 kDa) and ERK2 (42 kDa), which are constitutively expressed (bottom or middle panels in B-E) and confirmed with an GAPDH antibody (37 kDa) (E, bottom panel).
Direct fluorescent immunohistochemistry (IHC) was conducted thereafter. Sections were reblocked for 30 min in PBS 10% normal goat serum and 0.3% Triton X-100 and incubated overnight at 4°C with mouse anti-neurofilament-200 (NF-200) antibody (1:500; Sigma) or rabbit anti-activating transcription factor 3 (ATF3) antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA). Sections were washed three times in PBS and incubated for 90 min at RT with corresponding FITC-conjugated secondary antibody (Vector Laboratories). Direct fluorescent IHC was used for β2 subunit simple detection in DRGs and nerves using the same protocol with the exception that GW83 antibody was used at 1:100 dilution and sections were incubated at 4°C for 36 h. Slides were mounted with Vectashield mounting medium (Vector Laboratories).
Immunostained DRG sections were examined with an epifluorescent microscope (AxioPlan; Zeiss, Feldbach, Switzerland) under the appropriate filters, and images were recorded by digital camera using the same conditions (e.g., exposure and gain) for injured and control sections (AxioCam; Zeiss). Intensity and cell area were measured with analySIS software (Soft Imaging System, Münster, Germany), and the number of labeled neuronal profiles was counted in a blinded manner. For quantification, a slide from the first 10 sections was randomly selected, and then four sections were selected every fifth DRG section from the series of consecutive serially cut sections. Immunoreactive (IR) cell profiles were counted in each DRG section and expressed as the percentage of total neuronal cell profiles or for colocalization studies as the percentage of ATF3 neuronal profiles (for tibial and peroneal injured cells) and Fluorogold profiles (for sural non-injured cells). DRGs from three to four animals were analyzed in each treatment group. To construct profile size distribution histograms, four sections were similarly selected from each animal (n = 4), labeled neurons in a 10× optic field were identified, and the cross-sectional areas (in square micrometers) were measured (Decosterd et al., 2002).
Nerve sections were examined with a confocal laser scanning microscope (LSM 510 Meta Confocal Microscope; Zeiss). Images were taken through the central plane of the nerve at a thickness of 0.8 μm, and long parts of the nerves were scanned with the same parameters of image acquisition in controls and SNI sections. A montage of images along the nerve was made with Adobe Illustrator (Adobe Systems, San Jose, CA).
Real-time reverse transcription-PCR. Ipsilateral L4 and L5 DRGs were rapidly dissected and kept in RNA-later solution (Qiagen, Basel, Switzerland) to prevent degradation. Total RNA from each animal was isolated using RNeasy Mini kit (Qiagen), with a DNase step (RNase free DNase set; Qiagen) directly on column. Extracted RNA was eluted, and optical density was measured using 260 nm absorbance spectrophotometry and RNA quality controlled by gel electrophoresis. A total of 1.5 μg of RNA was reverse transcribed using Omniscript reverse transcriptase according to the protocol of the manufacturer (Qiagen).
Primer and probe sequences were designed using Beacon Designer 3.0 software (Premier Biosoft International, Palo Alto, CA) according to specification for SYBR-green (Vandesompele et al., 2002). Sequences for the forward primer and reverse primer are as follow: 5′-GATGTTCCTCCAGTTCCGAATG-3′ and 5′-GGGTTCCCCGAGAACTCTAC-3′ for β2; 5′-GGCTCATTAAATCAGTTATGGTTCCT-3′ and 5′-TCTCTGACCTCTTTCCCTTTGATTTC-3′ for Nav1.3; 5′-TCCCGGACAAATCAGAAACTG-3′ and 5′-GCCCCTGGTGACACTGTCA-3′ for Nav1.8; and 5′-CCCCCAATGTATCCGTTGTG-3′ and 5′-TAGCCCAGGATGCCCTTTAGT-3′ for GAPDH. GAPDH expression is not altered in our experimental conditions and was used as an endogenous control to normalize expression levels of sodium channel subunits. Real-time PCR reaction was performed in a final volume of 20 μl containing 50 ng of cDNA, 300 nm of both primers, 10 μl of 2× iQ SYBR-green mix containing nucleotides, iTaq DNA polymerase, SYBR-green, and fluorescein (Bio-Rad, Reinach, Switzerland) using the MyiQ Single Color real-time PCR Detection System (Bio-Rad, Reinach, Switzerland). The amplification protocol included 3 min at 95°C, followed by 45 cycles of 10 s at 95°C for denaturation and 45 s at 60°C for annealing and extension. Specificity of amplification was determined by DNA melting curve using the built-in feature in the software for automatic fluorescence data capturing during gradual temperature increments (0.5°C) from 55 to 95°C. Efficiency of amplification was determined by serial dilution of starting DNA, and standard curves were constructed from the respective mean critical threshold value for endogenous control and sodium channel targets. Individual control samples (sham control group, n = 4) and treated samples (SNI group, n = 4-6) were amplified in triplicate. The relative expression of the target gene is calculated based on real-time PCR efficiencies and the threshold value of the unknown sample versus the standard sample (Pfaffl, 2001).
Isotopic in situ hybridization. Isotopic in situ hybridization was performed as described previously (Karchewski et al., 1999). Two sets of 48 bp oligonucleotide probes were designed to be complementary and selective for regions 418-371 and 670-717 of the β2 subunit (GenBank accession number NM_012877), and one set was designed for Nav1.3 (GenBank accession number NM_013119). Probes were labeled at the 3′-end with 35S-dATP or 33P-dATP terminal deoxynucleotidyl transferase reaction. Cryosectioned fresh-frozen DRG tissue from SNI, SNL, and control groups mounted on slides were covered with hybridization buffer (50% formamide, 1× Denhardt's solution, 1% sarcosyl, 10% dextran sulfate, 0.02 m phosphate buffer, pH7.0, 4× SSC, 500 μg/ml heat-denaturated salmon sperm DNA, 200 mm DTT, and 107 cpm/ml labeled probe) and incubated for 18 h at 42°C. Slides were then washed four times for 15 min in 1× SSC at 55°C, cooled down to room temperature for 30 min in the last wash step, dipped in distilled water, and finally dehydrated in ethanol. Autoradiograms were generated by coating slides with NTB2 nuclear track emulsion (Eastman Kodak) and exposing in the dark at 4°C for 2-4 weeks. Sections were then developed for 3 min in Kodak D19 developer (Eastman Kodak), fixed for 6 min in Rapid Fixer (Eastman Kodak), washed, and mounted with glycerol. Tissue was viewed under dark field, and images were captured.
Behavioral assessment. Mechanical sensitivity was assessed in age-matched β2 subunit null mutant mice (n = 10; male/female ratio, 5:5) and wild-type C57BL/6 mice (n = 12; male/female ratio, 7:5). Gross animal appearance is similar between groups, and the experimenter was blinded to the mouse genotypes. Mice were acclimated to the testing environment for 2 weeks, two baselines (BL1 and BL2) were measured 6 and 4 d before SNI surgery, and then testing sessions were conducted at 3, 7, 14, 21, and 28 d after SNI surgery. The lateral side of the plantar surface of the paw (sural nerve territory) was stimulated by calibrated von Frey monofilaments (Stoelting, Wood Dale, IL) (Decosterd and Woolf, 2000; Suter et al., 2003). Frequency of paw withdrawal to 10 stimuli for each monofilament was recorded, and each filament was applied in a logarithmic ascending force order (0.008, 0.02, 0.04, 0.070, 0.16, 0.40, 0.60, 1.0, 1.4, and 2.0 g) (Mansikka et al., 2004). At 4 weeks, 50% withdrawal threshold was determined using the up and down method (Chaplan et al., 1994).
Statistics. Data are represented as mean ± SEM. Differences between groups were compared using Student's t test or ANOVA, followed by Bonferroni's or Fisher's PLSD post hoc analysis when appropriate. A two-way ANOVA for repeated measures was used to evaluate the overall effect of the genotype on mechanical sensitivity. von Frey series present logarithmic differences between hairs, and logarithmic transformed values were used for the analysis, enabling ANOVA tests (Decosterd et al., 2004). Statistical analyses were performed using JMP statistical software (version 5.01; SAS Institute, Cary, NC). A p value ≤0.05 was considered statistically significant.
Results
Antibody specificity
We confirmed the specificity of a number of anti-β2 subunit antibodies by Western blot, demonstrating the absence of cross-reactivity with β1 and β3 subunits of VGSCs (Fig. 1A) (Coward et al., 2001). Anti-β2 subunit antibodies GW83, GW93, and GW2690 were tested on membranes prepared from HEK293 cells transfected with the β1, β2, β3, or control plasmids. In extract from the β2 subunit cell line, a single band at apparent size above 30 kDa was reduced to a size slightly below 25 kDa when a deglycosylation step is added to the tissue processing (Fig. 1A), compatible with the expected β2 subunit mass of 21 kDa (Isom et al., 1994). Similarly, in brain samples, a 37 kDa band ran at 20-25 kDa after deglycosylation (Fig. 1B). This correlates with a previous biochemical characterization that suggests that >30% of the mass of the native rat brain β2 subunit is carbohydrate (Messner and Catterall, 1985). The slightly higher mass observed in Figure 1A might correspond to an incomplete deglycosylation during the processing of cell extracts.
There was no immunoreactivity in lanes containing brain tissue from β2 subunit -/- mice, and the single band visible from protein extracts of β2 subunit +/+ mice disappeared when GW93 antibody was preabsorbed with low concentrations (0.1 μm and above) of the corresponding blocking peptide (Fig. 1B). The same preabsorption experiments were included as a control for blots and immunohistochemistry with GW83 and GW93 antibodies on DRG tissue from control and SNI rats, and the signal was abolished (data not shown).
Peripheral nerve injury but not peripheral inflammation induces the β2 subunit in DRG neurons
Immunoblotting reveals a dramatic increase in β2 subunit expression in L4/L5 DRGs 1 week after SNI. Two different antibodies with cross-reactivity for the C-terminal tail of human and rat β2 subunit (GW93 and GW2690) and the β2-ec antibody targeting the extracellular domain of β2 subunit all yielded similar results (Fig. 1C-E). β2 subunit protein expression in the DRG was evaluated in three different models of peripheral nerve injury. Western blot analysis of β2 subunit expression in the L4/L5 DRGs using GW93 antibody reveals increased expression 1 week after SNI, total transection of the sciatic nerve, and after SNL (L5 DRGs) (Figs. 1E, 2A) (see Fig. 7A). Compared with SNI lesion (tibial and common peroneal nerves), a complete sciatic nerve axotomy will include an additional lesion to the sural as well as the tibial and common peroneal nerves, and the number of injured neurons is commensurately larger. However, the proportion of sural nerve neurons in L4/L5 DRGs is ≤10% of the total number of DRG neurons (Devor et al., 1985; Schmalbruch, 1986), which is at the limit of the sensitivity of Western blots; hence, there is no detectable difference in β2 subunit level between SNI and axotomy models.
The augmentation of β2 subunit level is significantly different from controls at 3, 5, and 7 d after SNI (p < 0.01; n = 4 for each time point groups) (Fig. 2B,C). β2 subunit levels at time points beyond day 3 are significantly different from the day 1 and day 3 time groups (p < 0.05), whereas no significant difference was observed between day 5 and day 7, suggesting a slow onset of upregulation (at least 3 d) that plateaus 5 d after nerve injury. The elevation in the β2 subunit persists throughout the 4 week time course of the experiment (Fig. 2D). Constitutive ERK levels were used as a protein loading control because these do not change after nerve injury (Fig. 1E). The difference in ERK1/ERK2 levels between the control and SNI lanes in Figure 2B is attributable to unbalanced protein loading and not regulation of ERK1/ERK2. To investigate whether β2 subunit upregulation after nerve injury is specific to this type of lesion, we measured β2 subunit expression levels after peripheral inflammation induced by intraplantar injection of CFA. Inflammation induced by CFA had no effect on β2 subunit expression at both 48 h and 1 week (Fig. 2 A).
An increase in β2 subunit is detected in membrane fractions from the DRGs of SNI rats, whereas signal is almost absent in these fractions of control rats (Fig. 3).
Immunohistochemistry using GW83 β2 antibody reveals a substantial increase in β2-IR neurons in the ipsilateral L4/L5 DRGs 1 week after SNI compared with only a few neurons that are labeled in control DRG sections (Fig. 4A). Cell profile quantification indicated that 27.3 ± 3.5% of total DRG neuronal profiles from SNI animals showed β2 immunoreactivity compared with 2.1 ± 0.7% of DRG neuronal profiles from control animals (p < 0.001; n = 4 in each group) (Fig. 4B).
To investigate whether the increase in the β2 subunit occurs in a particular subpopulation of DRG neurons, we colocalized β2-IR neurons with NF-200, a marker for large sensory neurons (myelinated A fibers) (Michael and Priestley, 1999). One week after SNI, β2 immunoreactivity was colocalized with NF-200-IR neurons, indicating that β2 subunit is induced in neurons with myelinated fibers. β2 immunoreactivity was also present in neurons that were not immunoreactive for NF-200, which possess unmyelinated axons (Fig. 4C). In addition, we examined the cross-sectional areas of β2-IR cell profiles 1 week after SNI. A total of 56.2% of the 575 β2-IR profiles counted were small neurons (<600 μm2), 23.3% corresponded to medium-sized sensory neurons (600-1200 μm2), and 20.5% were large neurons (>1200 μm2) (Fig. 4D). Relative to the total number of DRG neuron profiles, 25.5, 27.8, and 37.1% of small-, medium-, large-sized DRG neurons were β2-IR positive. The number of β2-positive neurons in control DRG sections was too low to establish a reliable distribution profile, but occasional small, medium, and large cells were observed.

Peripheral nerve injury induces a rapid and persistent increase in β2 subunit immunoreactivity in the DRG. A, Western blot analyses of the L4/L5 DRGs 1 week after an SNI, total axotomy of the sciatic nerve (Axo), or 48 h and 1 week (1W) after intraplantar CFA induced inflammation (longer exposure). B, D, Representative Western blots from at least four independent experiments at 1, 3, 5, and 7 d after SNI and 1, 2, 3, and 4 weeks after SNI. C, Quantitation of β2 subunit protein levels in the L4/L5 DRGs at the indicated days after SNI. Fold represents comparative increase over control level after normalizing to total ERK2 (n = 4 for each time point; *p < 0.01). ctrl, Control.
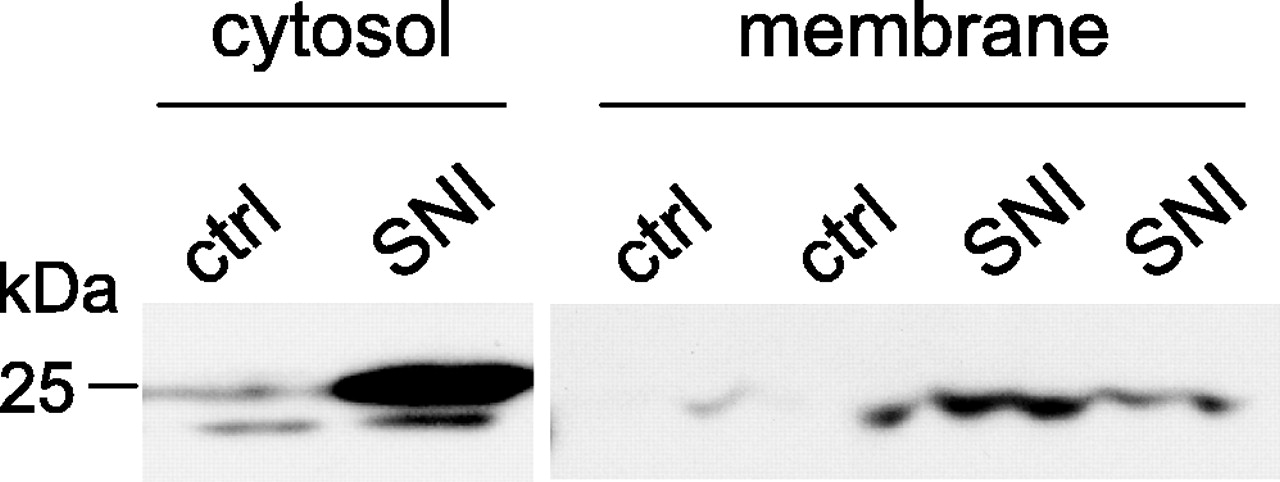
Redistribution of the β2 subunit to the plasma membrane. Western blot analyses of cytosolic (left) and membrane (right) fractions show that SNI causes an increase in the amount of β2 subunit at the plasma membrane, whereas signal in this fraction is almost absent in controls (ctrl) (L4 and L5 DRGs; n = 4 in each lane conditions).
Differential regulation of VGSC subunits in injured and non-injured neurons after peripheral nerve injury
We used real-time reverse transcription-PCR to compare VGSC subunit mRNA expression in L4/L5 DRG at different time points after SNI (Fig. 5A). VGSC transcript levels were normalized to GAPDH, a housekeeping gene that is unaltered by nerve injury, and expressed as the ratio of VGSC transcript level in DRGs from SNI rats to the VSGC transcript level in DRGs from controls. β2 subunit mRNA levels in L4/L5 DRGs did not change over the 7 d after SNI (p > 0.05; n = 4-6 in each group). As expected from other studies (Black et al., 1999; Sleeper et al., 2000), Nav1.8 mRNA significantly decreased 7 d after SNI compared with control (p < 0.05), and the level of Nav1.3 mRNA significantly increased at 7 d after SNI (p < 0.05). The unchanged level of β2 subunit mRNA after SNI was confirmed by isotopic in situ hybridization studies using two separate sets of radiolabeled probes and performed 1 week after SNI or 2 weeks after SNL (Fig. 5B). Absence of a change in β2 subunit mRNA after nerve injury was also confirmed by Northern blot analysis (data not shown). In control conditions, β2 subunit mRNA transcript expression is more prevalent in DRG neurons than β2 subunit protein expression (compare Figs. 4A,5B). Although a low sensitivity of detection by the immunohistochemistry assay cannot be excluded, the presence of the transcript in neurons in control naive DRGs, with high expression after nerve injury, support a posttranscriptional regulation of β2 subunit produced by the nerve injury.
Isotopic in situ hybridization demonstrated an upregulation of Nav1.3 mRNA after both SNL and SNI (Fig. 5C). After SNL, Nav1.3 mRNA upregulation occurs exclusively in the L5 DRGs, in which neurons are axotomized by the L5 spinal nerve injury procedure but not in the non-axotomized L4 DRG neurons (Fig. 5C).

β2 subunit augmentation and distribution in primary sensory neuron populations after SNI. A, Representative photomicrograph of fluorescent immunohistochemistry for β2 subunit in L4 DRGs 1 week after SNI compared with control rats. B, Percentage of total neuronal profile that express β2 subunit in control DRGs and DRGs 1 week after SNI. *p < 0.001; n = 4 in each group. C, Double immunohistochemistry for β2 subunit (red) and NF-200 (green) shows a distribution of β2 immunoreactivity in both large cells (colocalization with NF-200 appears yellow on the merged image) and small cells (absence of colocalization, red). D, Profile area frequency histogram constructed from 575 cell profiles shows an increase of β2 subunit-IR neurons 1 week after SNI in small-sized (<600 μm2), medium-sized (600-1200 μm2), and large-sized (> 1200 μm2) neurons. Scale bars, 100 μm.

Regulation of VGSC subunit mRNA expression after nerve injury. A, Relative quantification by real-time reverse transcription-PCR amplification of mRNA for β2 subunit and for α subunits Nav1.3 and Nav1.8 at indicated times. The SNI/control ratio refers to the ratio of mRNA levels in L4/L5 DRG tissue of SNI-injured and sham-operated animals (triplicate analysis of 4-6 rats for each time point; *p < 0.05). B, C, Representative dark-field photomicrographs of isotopic in situ hybridization in L4 DRGs from control animals or 1 week after SNI, as well as L4 (intact) DRGs and L5 (injured) DRGs 2 weeks after SNL. B, In situ hybridization for β2 subunit mRNA. C, In situ hybridization for Nav1.3 α subunit mRNA. Scale bar, 350 μm.
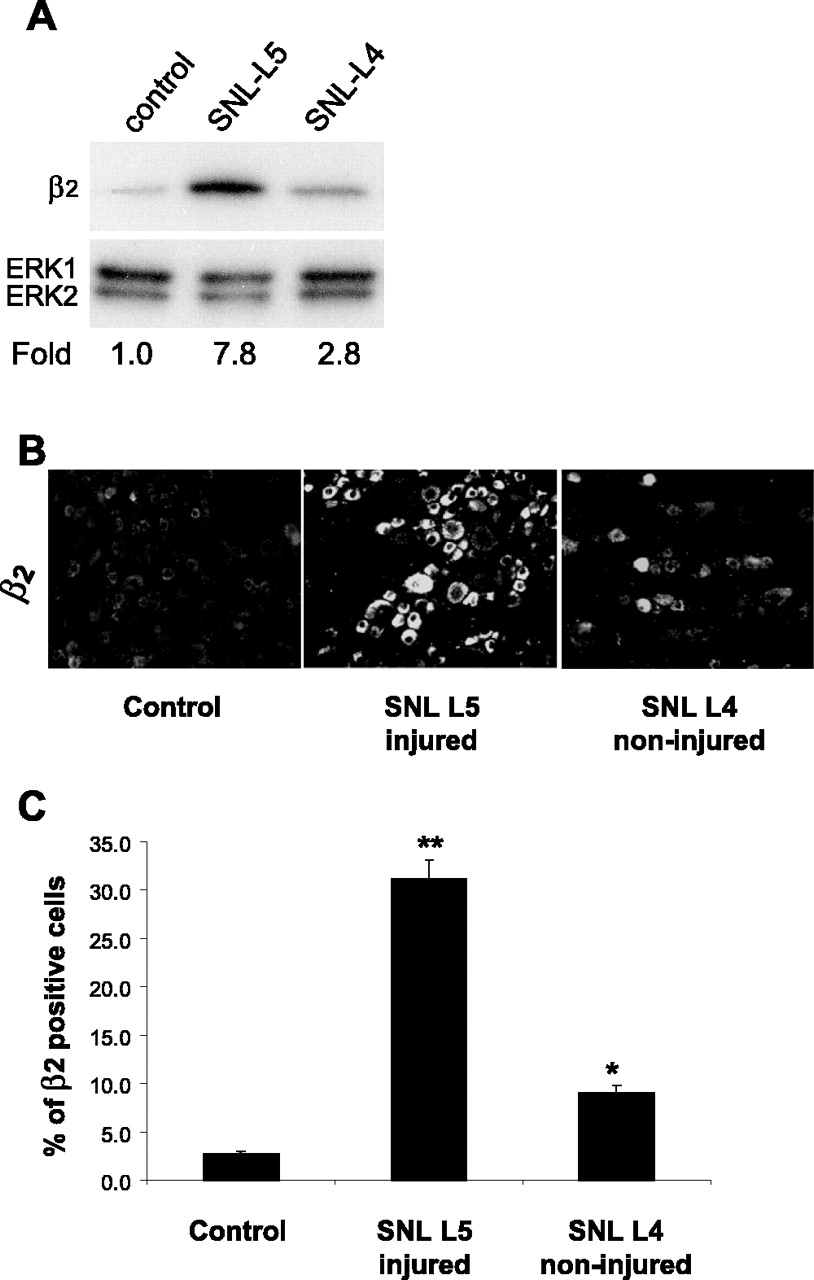
After SNI, injured neurons from the transected tibial and common peroneal nerves and non-injured neurons from the sural nerve comingle in the L4 and L5 DRGs. To discriminate injured and non-injured neurons in the same DRGs, we used a strategy combining retrograde tracing of the intact sural nerve and immunohistochemical detection of injured neurons with antibody to ATF3 (a marker of axonal injury) (Benn et al., 2002; Decosterd et al., 2002; Obata et al., 2004). As expected, 1 week after SNI, significant ATF3 immunoreactivity was present but did not overlap with Fluorogold (Fig. 6). β2 immunoreactivity was predominantly observed in ATF3-IR neurons: 85.9 ± 2.9% of injured ATF3-IR neurons expressed the β2 subunit, and 83.6 ± 2.5% β2-IR neurons were ATF3 positive. Only 7.6 ± 1.4% of retrogradely traced non-injured sural neurons were β2-IR, a much smaller subset than seen in injured ATF3-positive neurons (p < 0.0001; n = 4 in each group). In control sections, β2 immunoreactivity in sural FG-positive cells was very rare (one or less FG/β2-positive cell on each DRG section). To evaluate that both injured and non-injured neurons upregulate the β2 subunit after peripheral nerve injury, we investigated the β2 subunit expression in the L4 and L5 DRGs 1 week after SNL (Fig. 7) and found that the β2 subunit is increased after SNL in both L4 (intact) and L5 (injured) DRGs by Western blot analysis (Fig. 7A) and by immunohistochemistry (Fig. 7B,C). Similar to findings in the SNI model, the increase in β2 subunit-IR profiles is much more pronounced in the injured neurons of the L5 DRGs, but it also augmented, although to a lesser extent, in the non-injured L4 DRG neurons (n = 3; p < 0.001 and 0.01, respectively) (Fig. 7B,C).

Characterization of β2 immunoreactivity in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Colocalization of immunoreactivity for β2 with immunoreactivity for ATF3 and retrograde labeling with FG. The majority of β2 immunoreactivity (red) is seen in ATF3-IR neurons (green), corresponding to injured cell population (green). To a limited extent, β2 immunoreactivity is also seen in non-injured neurons of the sural nerve (white arrow) retrogradely traced with FG (bright gray). Scale bars, 100 μm.
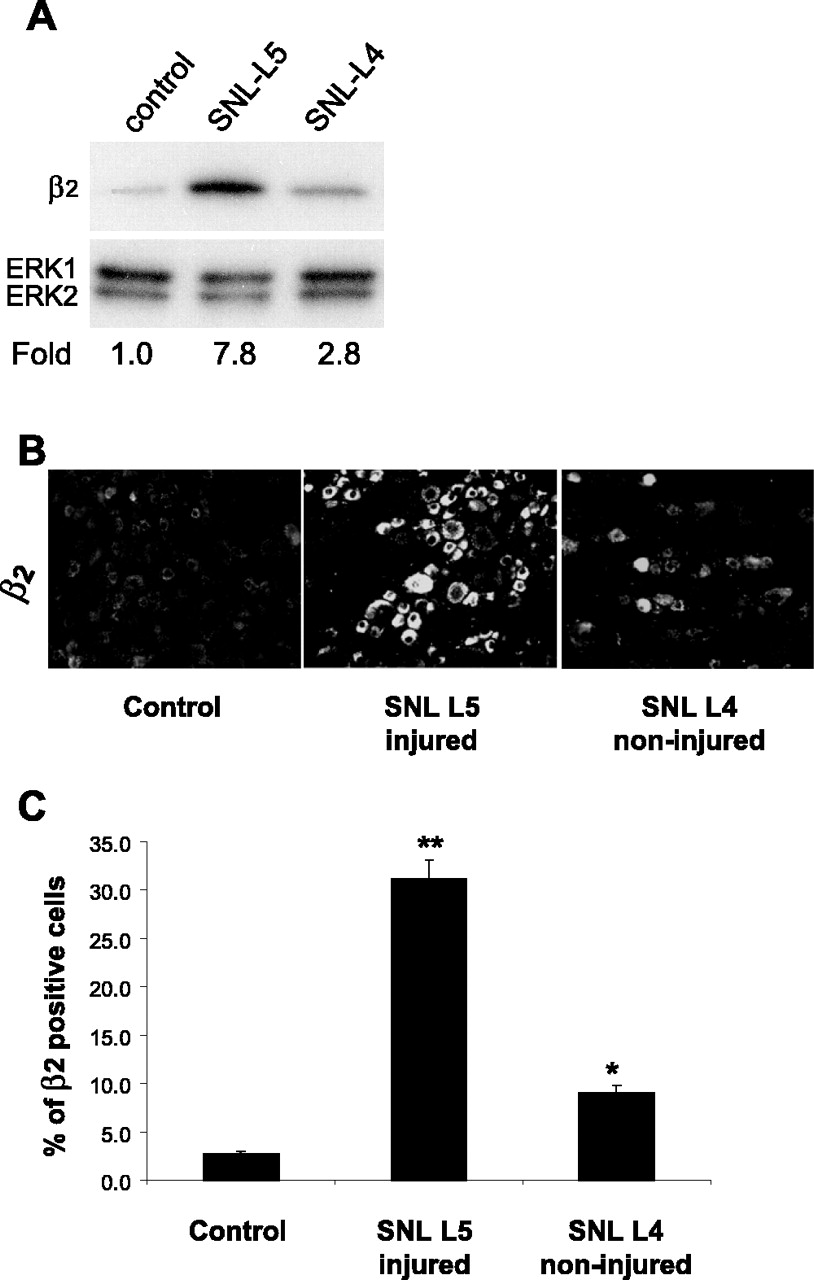
Characterization of β2 immunoreactivity in injured and non-injured neurons in the L5 SNL model of neuropathic pain. A, β2 immunoreactivity in L5 and L4 DRGs 1 week after SNL and in control. B, Immunohistochemistry for β2 subunit in sections of L4 and L5 DRGs 1 week after SNL and in a section of a control DRG. C, Percentage of neuronal profiles in injured (L5) and intact (L4) DRGs that express β2 subunit compared with control contralateral DRG (*p < 0.01; **p < 0.001; n = 3). Fold represents comparative increase over control level after normalizing to ERK2 loading control.
β2 subunit expression is present in injured nerves and clusters in the neuroma
Common peroneal and tibial nerves were separated from the sural nerve proximal to the ligation, and a nerve portion of a length of 4 mm including the neuroma tip was dissected. Western analysis revealed a twofold to threefold increase of β2 subunit in the injured tibial and common peroneal nerves (n = 4 for each time point) (Fig. 8A). However, no change in the level of β2 subunit protein was detected in sural nerve in the 4 weeks after SNI (1 week after SNI, Fig. 8A; 2-4 weeks after SNI, data not shown). Regulation of β2 subunit expression in these nerves was further investigated by immunohistochemistry of nerve sections. Confocal images (n = 4-5 in each group) reveal a large increase in intensity of staining for β2 subunit in the injured tibial and peroneal nerves relative to control nerves and a smaller increase in staining in the sural nerve after SNI (Fig. 8B,C). Particularly intense β2 subunit immunoreactivity was present at the transected axon tips within the neuroma (Fig. 8B).
Mechanical allodynia-like behavior induced by SNI is attenuated in β2 subunit null mutant mice
Responsiveness to mechanical stimuli in uninjured β2 subunit -/- and wild-type +/+ mice was assessed by measuring the frequency of response to repeated application of a standard set of calibrated von Frey hairs. No response was elicited at the very lowest von Frey hairs tested (0.008 and 0.02 g) and >70% response at 2 g, the highest (Fig. 9A). No difference was found in responsiveness in the two sets of mice (Fig. 9A). The rate of response increased significantly relative to the non-injured control mice after SNI in both the wild type +/+ and β2 subunit -/- mice for all forces <1 g, with a recruitment of responses at previously subthreshold stimuli and greater responses to suprathreshold stimuli at 3, 7, 14, 21, and 28 d (p < 0.01; -/-, n = 10; +/+, n = 12) (Fig. 9B). No statistically significant difference between the two groups was found 3 d after SNI (data not shown). However, at day 7 and all other times tested up to 28 d, the response in -/- mice after SNI was significantly attenuated compared with +/+ mice (p < 0.001) (Fig. 9B). Four weeks after SNI, the 50% mechanical threshold of the hindpaw measured ipsilateral to the nerve injury was higher in β2 subunit -/- mice compared with the threshold in wild-type mice (p = 0.02) (Fig. 9C), with no difference in the contralateral paws (p > 0.05).

β2 subunit is increased in peripheral nerves after SNI and is transported along the injured nerve to the neuroma. Western blot (A) and immunohistochemistry (B, C) analyses for β2 subunit in injured and non-injured nerves 1 week (W) after SNI. A, Representative blots, each from at least three independent experiments from control and SNI groups (n = 4-6 in each group). The signal is augmented in the injured tibial and common peroneal nerves after SNI at all time points compared with nerves from control rats. B, C, Nerves were probed with anti-β2 subunit antibody, and confocal images of representative sections show β2 staining in injured common peroneal and tibial nerves 1 week after SNI (B) and in the spared sural nerve after SNI (C). High-magnification photomicrographs are of portions of nerves at a distance ∼2.5 mm away from the distal extremity of the neuroma. Fold represents comparative increase over control level after normalizing to ERK2 loading control.
Discussion
Using specific antibodies, we found a striking induction of VGSC β2 subunit immunoreactivity after peripheral nerve injury. The increase in protein levels is most dramatic in the cell bodies, fibers, and nerve stump of injured neurons, but β2 subunit expression was also increased in adjacent intact neurons. That this increase has functional significance was indicated by our observation that mechanical allodynia-like behavior after nerve injury was attenuated in β2 subunit null mutant mice.
The β2 subunit remained elevated in DRG neurons for at least 4 weeks after peripheral nerve injury, and this appears to be independent of transcriptional regulation because no increase in β2 subunit mRNA transcript levels was detected, as with previous studies (Black et al., 1996; Shah et al., 2000; Takahashi et al., 2003). Nerve injury can induce expression of target genes by activating transcription factors such as ATF3, cAMP response element-binding protein, or c-jun, but protein synthesis in the nervous system can be also modulated without involving new transcription of mRNA by a modification of translational control. Such a mechanism is based on our data, likely to be initiated by nerve injury. It may include increases in translational rate as well as modulation by small micro-RNAs (Gebauer and Hentze, 2004; Klann and Dever, 2004). There is a precedent for posttranscriptional regulation in DRG neuron: the augmentation of TRPV1 protein after inflammation without a parallel change in TRPV1 mRNA levels (Ji et al., 2002). This is dependant on activation of the p38 mitogen-activated protein kinase pathway, and, after nerve injury, p38 is also activated in DRG neurons (Obata et al., 2004). p38 could potentially stimulate the eukaryotic initiation factor 4E, an mRNA cap-binding protein essential for the initiation and rate of translation, and this or similar changes could augment β2 subunit protein levels without change in transcript number (Scheper and Proud, 2002). A change in protein turnover rate could also augment β2 subunit levels by decreasing protein degradation by the lysosomal proteolytic pathway.
Both the β2 subunit and the Nav1.3 α subunit are increased in injured neurons. This parallels the emergence of a rapidly repriming TTX-sensitive sodium current in injured neurons that may contribute to a spontaneous firing of DRG neurons or firing at unusually higher frequencies (Black et al., 1999; Cummins et al., 2001; Takahashi et al., 2003). In contrast, TTX-resistant sodium currents and Nav1.8 and Nav1.9 channel expression decrease in injured neurons (Sleeper et al., 2000; Decosterd et al., 2002). TTX-resistant channels persist in intact adjacent neurons (Decosterd et al., 2002), and Nav1.8 is redistributed in the spared nerve so that this channel may contribute to pain hypersensitivity (Gold et al., 2003). Our data suggest that the upregulation and redistribution of β2 subunit to the plasma membrane after nerve injury may contribute to an increase in the translocation of the sodium channel complex to the plasma membrane.
Neuropathic pain is associated with development of ectopic discharge in A- and C-fibers in both injured and non-injured nerves at the neuroma and in the cell somata that is associated with a subthreshold oscillation of the membrane potential of DRG neurons (Gold, 2000; Liu et al., 2000; Wu et al., 2001; Amir et al., 2005). VGSCs mediate intrinsic neuronal excitability and play a major role in the initiation and propagation of action potentials, repetitive firing, and amplification of synaptic potentials, as well as in generating subthreshold membrane potential oscillations (Klink and Alonso, 1993; Franceschetti et al., 1995; Catterall, 2000). Increased expression of the β2 subunit after nerve injury may contribute to the altered sodium channel activity that is so characteristic of neuropathic pain. The β2 subunit can regulate the number of VGSCs at the plasma membrane by controlling trafficking from the intracellular to the membrane compartment and alter VGSC kinetics (Schmidt and Catterall, 1986; Chen et al., 2002). The subunit is also a cell adhesion molecule that may mediate interactions with extracellular matrix and other signaling molecules. We discuss these properties of the β2 subunit below and how each might be involved in VGSC function after nerve injury.

Attenuation of mechanical allodynia-like behavior after SNI in β2 subunit -/- mice. A, B, Paw withdrawal frequencies to mechanical stimuli using a series of calibrated monofilaments with logarithmic increment. Frequency of response (paw withdrawal) to each monofilament in wild-type (+/+) and β2 subunit knock-out mice (-/-) are shown before (A; two baselines, BL1 and BL2) and after SNI (B; 7, 14, 21, and 28 d). Baseline curves did not differ significantly between genotypes, whereas the response curves for the +/+ and -/- mice at day 7 and after are significantly different, with the -/- mice showing an attenuated response compared with the +/+ mice (p < 0.001; n = 12 and 10, respectively). C, The 50% withdrawal threshold 4 weeks after SNI. The mechanical threshold is higher in β2 subunit -/- mice than wild-type mice ipsilateral but not contralateral to the nerve injury (*p = 0.02; n = 12 and 10, respectively).
The β2 subunit has a well established effect on VGSC cell surface expression (Isom et al., 1995). Many α subunits exist in a free intracellular pool, which serves as a source of precursors for subunit assembly by disulfide linkage with β2 subunits. The appearance of VGSCs at the cell surface correlates with β2 association (Catterall et al., 1986; Schmidt and Catterall, 1987; Isom et al., 1995; Isom, 2001). Cell expression studies have shown that association with β2 regulates VGSC cell surface density and localization in developing neurons (Schmidt and Catterall, 1986, 1987; Kaplan et al., 2001; Chen et al., 2002). Several observations in β2 subunit -/- mice are consistent with a reduction in cell surface expression of VGSCs. Neurons from these animals bind less 3H-saxitoxin and have lower sodium current density than neurons from wild-type mice (Chen et al., 2002). These results suggest that the β2 subunit is involved in the translocation of VGSC complex to and its immobilization in the cell membrane. After peripheral nerve injury, increased levels of β2 subunit in the DRG cell body, along axons, and in the nerve stump may contribute to a local generation of ectopic activity by increasing VGSC density in the plasma membrane in both injured and non-injured neurons. This mechanism would be attenuated in β2 subunit null mutant mice, which may explain why their mechanical hypersensitivity is less than that in wild-type mice.
The effect in vitro of the β2 subunit on sodium channel properties is dependant on both cell type and the other subunit types present. When coexpressed in Xenopus oocytes with Nav1.1 or Nav1.2, β2 promotes fast gating and a negative shift in voltage dependence of inactivation (Isom et al., 1995; Smith and Goldin, 1998). Conversely, β2 causes a positive shift in voltage dependence of inactivation when expressed in HEK293 cells (Qu et al., 2001). When complexed with Nav1.3 in Chinese hamster ovary cells or HEK293 cells, β2 does not affect gating (Cummins et al., 2001; Meadows et al., 2002). The β2 subunit-mediated effect on the TTX-resistant Nav1.8 sodium channel in Xenopus oocytes is only a small depolarizing shift in the voltage dependence of inactivation. However, β2 overrides the hyperpolarizing shift in activation that is observed when β1 is coexpressed with Nav1.8 (Vijayaragavan et al., 2004). Studies in β2 null mutant mice reveal only subtle and small change in the voltage dependence of inactivation of VGSCs (Chen et al., 2002). It is possible therefore that the β2 subunit has only limited effects on VGSC electrophysiological properties and that β2 subunit prime function is to regulate the membrane density of the VGSC complex, whereas the β3 subunit, which is also upregulated in injured neurons, modifies sodium channel kinetics (Morgan et al., 2000; Takahashi et al., 2003). Nevertheless, the small changes observed in channel properties in vitro may be critical for a fine-tuning of the more complex membrane excitability of sensory neurons in vivo.
The β2 subunit is also a cell adhesion molecule with sequence homology to the neural cell adhesion molecule contactin, a member of the Ig superfamily. Contactin, which associates with VGSC α subunits via β1, is upregulated in axotomized neurons and accumulates together with Nav1.3 at injured axon tips in the neuroma (Shah et al., 2004). β2 and contactin may share a similar mechanism for increasing hyperexcitability in neuropathic pain (Isom et al., 1995; Kazarinova-Noyes et al., 2001). β2 interacts with the extracellular matrix proteins tenascin-R and tenascin-C (Xiao et al., 1999), recruits ankyrin to the plasma membrane at points of cell-cell contact, and transduces signals between adjacent cells or adjacent axons and the cytoskeleton (Malhotra et al., 2000). After nerve injury, cell adhesion molecules of the Ig super-family may play a role in neuronal regeneration and the reformation of nodes of Ranvier (Kaplan et al., 2001; Kazarinova-Noyes et al., 2001; Isom, 2002). An increase in β2 subunit in the active growth cone of sensory axons might participate, therefore, in the regeneration response to nerve injury (Davis et al., 2004).
In conclusion, we find a rapid and persistent transcription-independent increase in the β2 VGSC subunit after peripheral nerve injury. Upregulation of the β2 subunit in injured and non-injured sensory afferents may regulate both the translocation and immobilization of sodium channels in the cell membrane and modify their kinetics, leading to membrane hyperexcitability and ectopic activity, and thereby, contribute to neuropathic pain.
Footnotes
This work was supported by Swiss National Science Foundation Grant 3232-066354/3200-066355 (I.D.), the Pierre Mercier Science Foundation (I.D.), the Swiss Society of Anesthesiology (I.D.), National Institutes of Health Grants NS040698 (R.-R.J.) and NS039518 (C.J.W.), and National Multiple Sclerosis Society Grant RG2882 (L.I.). We thank Egbert Welker for his critical reading of this manuscript, Michael Costigan, Romano Regazzi, and Severine Cheviet for scientific advice, Nadège Roduit for technical assistance, Eric Bernardi and Susan Lewis for editorial assistance, and Jean-Yves Chatton and Yannick Krempp for the use and advice at the Cellular Imaging Facility at the University of Lausanne.
Correspondence should be addressed to Isabelle Decosterd, Anesthesiology Pain Research Group, Department of Anesthesiology, Lausanne University Hospital and Department of Cell Biology and Morphology, Lausanne University, Bugnon 9, CH-1005 Lausanne, Switzerland. E-mail: isabelle.decosterd{at}chuv.ch.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510970-11$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}