

Abstract
Diffuse traumatic brain injury (DTBI) is associated with neuronal plasmalemmal disruption, leading to either necrosis or reactive change without cell death. This study examined whether enduring membrane perturbation consistently occurs, leading to cell death, or if there is the potential for transient perturbation followed by resealing/recovery. We also examined the relationship of these events to calpain-mediated spectrin proteolysis (CMSP). To assess plasmalemmal disruption, rats (n = 21) received intracerebroventricular infusion 2 h before DTBI of a normally excluded 10 kDa fluorophore-labeled dextran. To reveal plasmalemmal resealing or enduring disruption, rats were infused with another labeled dextran 2 h (n = 10) or 6 h (n = 11) after injury. Immunohistochemistry for the 150 kDa spectrin breakdown product evaluated the concomitant role of CMSP. Neocortical neurons were followed with confocal and electron microscopy. After DTBI at 4 and 8 h, 55% of all tracer-flooded neurons contained both dextrans, demonstrating enduring plasmalemmal leakage, with many demonstrating necrosis. At 4 h, 12.0% and at 8 h, 15.7% of the dual tracer-flooded neurons showed CMSP, yet, these demonstrated less advanced cellular change. At 4 h, 39.0% and at 8 h, 24.4% of all tracer-flooded neurons revealed only preinjury dextran uptake, consistent with membrane resealing, whereas 7.6 and 11.1%, respectively, showed CMSP. At 4 h, 35% and at 8 h, 33% of neurons demonstrated CMSP without dextran flooding. At 4 h, 5.5% and at 8 h, 20.9% of tracer-flooded neurons revealed only postinjury dextran uptake, consistent with delayed membrane perturbation, with 55.0 and 35.4%, respectively, showing CMSP. These studies illustrate that DTBI evokes evolving plasmalemmal changes that highlight mechanical and potential secondary events in membrane poration.
Introduction
Our understanding of diffuse traumatic brain injury (DTBI) has long moved on the assumption that the forces of injury evoke focal as well as diffuse neuronal death, contributing to morbidity and mortality. However, little work exists to support this premise. In animals and humans, contusions are associated with regional neuronal death, together with the finding of scattered neuronal death in the neocortex, hippocampus, and diencephalon (Cortez et al., 1989; Dietrich et al., 1994a,b; Colicos et al., 1996; Hicks et al., 1996; Saatman et al., 1996). In these cases, both apoptotic and necrotic cell death have been identified, with the assumption that the observed neuronal death proceeds from traumatically induced neuroexcitation, oxygen radical-mediated damage and/or secondary insults. Few have considered the potential that the forces of injury directly cause mechanically induced neuronal perturbation or death. Recently, after DTBI, we identified axotomy-mediated change in the neuronal somata associated with impaired, yet transient protein translation (Singleton et al., 2002). We also observed progressive necrosis in neurons adjacent to the axotomized neurons. Studies using extracellular tracers normally excluded by intact neuronal membranes revealed that these necrotic neurons suffered altered tracer permeability at the moment of injury most likely allowing for influx of damaging ions (Singleton and Povlishock, 2004). Other neurons in the same field, however, also took up these normally excluded tracers, yet reorganized them over time, manifesting no evidence of cell death. These observations led to the premise that the DTBI evoked direct mechanical poration of the neuronal cell membrane, which, if enduring, contributed to cell death. Conversely, it was posited that those neurons that contained tracers, yet remained viable, resealed their mechanically damaged membranes. In this communication, we tested this hypothesis by reexamining the potential for traumatically induced mechanical poration of neuronal membranes while evaluating whether continuing membrane perturbation leads to cell death. We also explored the potential that some neuronal membranes may reseal and recover. Fluorescently labeled tracers were administered both before and after injury to evaluate poration and its continuance. Parallel immunocytochemical investigations examined calpain-mediated spectrin proteolysis (CMSP) long associated with the damaging proteolytic death cascades activated in DTBI (Kampfl et al., 1996, 1997; Saatman et al., 1996; Newcomb et al., 1997; Pike et al., 1998; Buki et al., 1999; McCracken et al., 1999). In fact, the significance of CMSP is highlighted by experimental and clinical studies that have suggested that CSF CMSP breakdown products are surrogate markers of morbidity and mortality (Pike et al., 2001; Farkas et al., 2005). Confocal microscopy and concomitant ultrastructural analyses using antibodies targeting the fluorophore-tagged tracers confirmed that TBI caused direct mechanical poration in numerous neuronal somata scattered throughout the neocortex. Over 50% of the tracer-containing neurons were labeled with both tracers with many of these revealing necrotic cell death. Other flooded neurons contained only one tracer, consistent either with membrane resealing or delayed opening. Surprisingly, CMSP did not frequently colocalize with neurons, demonstrating tracer influx and/or necrosis. Collectively, these studies illustrate the complex and evolving plasmalemmal changes ongoing with DTBI.
Materials and Methods
In all, 21 adult male Sprague Dawley rats weighing 380–400 g were used for the experiments (n = 10 in 4 h survival group; n = 11 in 8 h survival group). Four animals served as tracer-infused, sham-injured controls to demonstrate that infusion into the lateral ventricle itself does not lead to neuronal cellular membrane disruption. Ten noninjected, injured animals were used as intracranial pressure (ICP) controls to obviate the concern that any tracer infusion-induced rise in ICP could constitute a confound.
Surgical preparation and injury induction.
For the induction of anesthesia, animals were exposed to 4% isoflurane in a mixture of 30% O2 and 70% N2O for 5 min, then intubated and ventilated with 1–2% isoflurane in the mixture of 30% O2 and 70% N2O. Anesthesia was maintained throughout the duration of the experiment until transcardial perfusion. Animals were placed on a feedback-controlled heating pad (Harvard Apparatus, Holliston, MA) to maintain body temperature at 37°C during surgery. A PE50 polyethylene tube (Becton Dickinson, Sparks, MD) was placed into the left femoral artery to monitor the mean arterial blood pressure (MABP) and blood gases. After cannulation animals were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). The skull between the coronal and lambdoidal sutures was exposed with a midline incision. A 2 mm burr hole was drilled in the right parietal bone. A 26 gauge needle connected to a pressure transducer and a microinfusion pump via a PE50 polyethylene tube was placed into the lateral ventricle 0.5 mm posterior and 1.3 mm lateral from bregma through the burr hole. During the needle placement, sterile saline was infused with a 3 μl/min rate within a closed fluid-pressure system, while intracranial pressure monitoring was obtained via a PowerLab system (ADInstruments, Colorado Springs, CO). With the advancement of the needle, the detection of a significant pressure drop indicated the moment that the needle had broached the ventricular wall (Amorini et al., 2003). This controlled and monitored needle placement assured consistent needle placement within the ventricular compartment without the potential confound of brain placement and its potentially damaging consequences on tracer infusion. After placing the needle into the ventricle, 75 μl of Alexa Fluor 488-labeled 10 kDa molecular weight dextran (Invitrogen, San Diego, CA) was infused into the ventricle at 2 μl/min rate using a CMA/100 microinjection pump (Carnegie Medicin, Stockholm, Sweden), with continuous intracranial pressure monitoring. The dextran was diluted in sterile saline in 26.67 mg/ml concentration; the final amount infused was 5 mg/kg. The needle was allowed to remain in the ventricle for 10 min after completion of the infusion, after which the needle was slowly removed. Gelfoam and bone wax were used to restore skull integrity before the induction of closed head injury. Next, the animal was prepared for injury as described in previous protocols (Foda and Marmarou, 1994; Marmarou et al., 1994). Briefly, a metallic disc-shaped helmet of 10 mm diameter was firmly glued between the bregma and lambdoid sutures of the skull. The animal was disconnected from the ventilator and placed in a prone position on a foam bed with the metallic helmet centered under the edge of a Plexiglas tube. The rat was prevented from falling by two belts secured to a foam bed of known spring constant (Type E bed; Foam to Size, Ashland, VA). Brass weights weighing 450 g were allowed to fall from a height of 2 m through the Plexiglas tube directly to the metallic disc fixed to the animal's skull, a setting that precluded cerebral contusion or subdural hemorrhage. Rebound impact was prevented by sliding the foam bed containing the animal away from the tube immediately after the initial impact. Each animal sustained injury 2 h after preinjury tracer administration. Noninjured controls were prepared for injury in the same manner but were not injured. Immediately after the injury, the animal was reconnected to the ventilator. The helmet was removed and the skull was studied for any sign of fracture, which, if found, disqualified the animal from further evaluation. The scalp wound was then sutured while the animal remained on artificial ventilation.
Delayed tracer infusion.
Before the second tracer infusion, animals were returned to the stereotaxic device, the skull was exposed, and another hole was drilled through the left parietal bone. Texas Red-labeled 10 kDa dextran (Invitrogen) was then infused at the same concentration as the preinjury dextran. Once again, this was accomplished via a needle placed in the left ventricle 0.5 mm posterior and 1.3 mm lateral to bregma with 2 μl/min infusion rate with continuous ICP monitoring. This tracer was infused either at 2 h after injury (n = 10) or 6 h after injury (n = 11). The tracer was then allowed to diffuse for additional 2 h before perfusion with fixative, resulting in the respective 4 and 8 h postinjury survival groups. Sham-injured animals were infused either 4 or 8 h after the first infusion. Non-injected-injured animals (n = 10) were prepared in the same manner for ICP monitoring and injury, but no tracers were infused in their ventricles. Rather, ICP was monitored through a needle placed in the ventricle at the same time points used in injected animals. Five minutes before transcardial perfusion, the animals were injected with 150 mg/kg euthasol. At either 4 or 8 h after injury or at either 6 or 10 h after the first infusion (sham injury), the animals underwent transcardial perfusion with saline and then 4% paraformaldehyde in Millonig's buffer.
Physiological assessment.
MABP was monitored via a femoral line in each animal for the duration of the surgical procedures. Blood gases were analyzed before the tracer infusions as well as before and after injury. As noted above, ICP was also monitored before, during, and after tracer infusion or throughout the duration of all procedures in noninjected animals. Body temperature was monitored via a rectal probe and maintained at 37°C using a feedback control heating pad. PowerLab was used to monitor all physiological parameters (MABP, ICP, temperature).
Tissue processing.
After perfusion, the brains were removed and stored in 4% paraformaldehyde in Millonig's buffer at 4°C for 24 h, after which they were coronally blocked at the optic chiasm and midbrain and sectioned in 0.1 m phosphate buffer with a vibratome (Leica, Banockburn, IL) at a thickness of 40 μm. Sections were collected serially in 24-well culture plates and stored in Millonig's buffer at 4°C until further processing.
Multiple fluorescent analysis.
Because dextrans infused to the brain were labeled with fluorophores (Alexa Fluor 488 for the preinjury dextran and Texas Red for postinjury dextran), no additional steps were needed to visualize the tracer or any tracer-containing neurons. In addition to the identification of tracer-containing neurons, antibody against the 150 kDa calpain-specific spectrin breakdown product was used to examine the link between any observed membrane perturbation and the ensuing induction of calpain-mediated spectrin proteolysis, previously linked to TBI-induced proteolytic cascades. The antibody Ab 38 was used for this approach, because it is well characterized, targeting the N-terminal fragment of the 150 kDa calpain-specific breakdown of αII-spectrin, a cytoskeletal protein known to be cleaved after traumatic brain injury (Siman et al., 1989; Roberts-Lewis et al., 1994). In this procedure, some sections were washed in PBS three times for 10 min and then subjected to temperature-controlled microwave antigen retrieval as described previously (Stone et al., 1999). After retrieval, sections were again rinsed in PBS three times for 10 min and treated for 60 min in 10% normal goat serum (NGS) with 0.2% Triton X-100 in PBS and 5% BSA. The tissue was then incubated in a 1:5000 dilution of Ab 38 (generous gift from Dr. R. Siman, University of Pennsylvania, Philadelphia, PA) in 1% NGS/PBS with 1% BSA overnight. The following day, sections were rinsed three times for 10 min 1% NGS/PBS and then incubated for 2 h in a 1:1500 dilution of Alexa Fluor 647-labeled anti-rabbit IgG (Invitrogen) in 1% NGS/PBS. After rinsing three times for 10 min in PBS and twice for 10 min in 0.1 m phosphate buffer, sections were mounted on nontreated slides and coverslipped (ProLong; Invitrogen).
Visualization of fluorophores at ultrastructural level.
Because the fluorescent dyes that were used are not detectable at electron microscopic level, for parallel assessments of the ultrastructural change, conversion of fluorophores to a stable, electron-dense product was performed. This was achieved through the use of an antibody against the fluorophores themselves. The same sections previously scanned with confocal microscope for tracer uptake or individually captured with a Spot RT camera attached to an epifluorescent Nikon (Tokyo, Japan) Eclipse E800 microscope were used to provide a visual fluorescent map to which the subsequently prepared chromagen-based sections could be directly compared at the LM and EM levels. After capture, the tissues were soaked off the slides, and brain regions demonstrating preinjury and/or postinjury tracer uptake were removed and processed to investigate any ultrastructural change related to membrane disruption by using antibodies targeted to Alexa Fluor 488. This method allowed the identification of the same cells that previously revealed evidence of the uptake of either one or both dextran tracers, allowing the ultrastructural assessment of any alteration associated with enduring membrane change and/or membrane resealing. In this approach, sections were postfixed with 0.1% glutaraldehyde in 4% paraformaldehyde, then rinsed five times for 10 min in Millonig's buffer and three times for 10 min in PBS. Endogenous peroxidase activity within the tissue was blocked with 0.5% H2O2 in PBS for 30 min. Sections were processed using the temperature-controlled microwave antigen retrieval approach described above. After microwave antigen retrieval, sections were preincubated for 60 min in 10% NGS with 0.2% Triton X-100 in PBS. The tissue was incubated overnight in a 1:5000 dilution of rabbit anti-Alexa Fluor 488 antibody (Invitrogen) in 1% NGS in PBS. Sections were then incubated for 1 h in biotinylated anti-rabbit antibody (IgG; Vector Laboratories, Burlingame, CA) diluted 1:2000 in 1% NGS in PBS and then for 1 h in a 1:200 dilution of an avidin–horseradish peroxidase complex (ABC Standard Elite kit; Vector Laboratories). The reaction product was visualized with 0.05% diaminobenzidine, 0.01% hydrogen peroxide, and 0.3% imidazole in 0.1 m phosphate buffer for 10–20 min.
Visualization of calpain-mediated spectrin proteolysis at the ultrastructural level.
Brain regions demonstrating evidence for tracer uptake with confocal microscopy were removed and processed to investigate CMSP using the previously described Ab 38 antibody. In this approach, after postfixation tissues were processed for EM analysis as described above, using Ab 38 as primary antibody in 1:8000 dilution and biotinylated anti-rabbit antibody in 1:2000 dilution.
Electron microscopy.
Using confocal and routine fluorescent images as described above, the once-fluorescent cells, which now contained a peroxidase reaction product, were prepared for EM. In this approach, the previously prepared fluorescent maps were used to identify specific sites of interest. These were dissected out of the tissue sections, and then osmicated, dehydrated, and embedded in epoxy resins on plastic slides with plastic coverslips. After resin curing, the plastic slides were studied with routine LM to identify the precise neurons of interest. Once identified, these sites were removed, mounted on plastic studs, and thick sectioned to the depth of the immunoreactive sites of interest. Serial 70 nm sections were cut and picked up on to Formvar-coated slotted grids. The grids then were stained in 5% uranyl acetate in 50% methanol for 2 min and 0.5% lead citrate for 1 min. Ultrastructural analysis was performed using a JEM 1230 electron microscope (JEOL-USA, Peabody, MA). Specifically, at 1000×, the grid field was scanned to identify the cell(s) of interest. Once identified, these cells were digitally acquired at 5000× as a montage. These digital montages were copied to a digital video disk and transferred to a Dell Dimension XPS Gen 4 computer (Dell Computer Company, Round Rock, TX), on which the images were further enlarged and assessed for any evidence of cellular/subcellular change.
Confocal fluorescent microscopy.
Zeiss (Oberkochen, Germany) 510 Meta and Leica TCS SP2 AOBS laser scanning confocal microscope systems were used to analyze double- and/or triple-labeled sections. Appropriate laser lines (argon 488 nm for Alexa Fluor 488, He-Ne 594 nm for Texas Red, and He-Ne 633 nm for Alexa Fluor 647) and emission filters (Zeiss) or emission bands (Leica) were used for exciting and detecting fluorescent signals.
Semiquantification of fluorescent images.
The TBI model used in the current study evokes diffuse injury throughout the neocortex, resulting in scattered neuronal injury with heterogeneous pathologies. These modest numbers of damaged neurons, particularly those revealing only one tracer, would violate a basic tenant of stereology and preclude further stereological analysis of the number of neurons with membrane perturbation at 4 or 8 h after injury. Densitometric evaluation of injured neurons showing different dextran uptake profiles could not be performed, considering the high and variable background level of the fluorophores associated with the intraventricular administration of the labeled dextrans, making the necessary standardization difficult. Based on these deficiencies, we conducted traditional cell counting using images captured by confocal microscopy with rigorous inclusion criteria to semiquantify neurons showing primary and/or secondary dextran uptake both at 4 and 8 h after injury. Sections captured at 40× magnification and containing at least three fluorescent labeled neurons were included (number of animals at 4 h, n = 8; at 8 h, n = 7; mean number of sections per animal, 7.8). The definition for positive labeled neurons was all neurons showing any dextran flooding. Neurons showing preinjury dextran uptake, postinjury dextran uptake, or both preinjury and postinjury dextran uptake were quantified as a percentage of the total number of neurons demonstrating any tracer uptake in each section and averaged for each animal. Similar methods were used to quantify calpain mediated spectrin proteolysis and its relationship to membrane disruption.
Statistical analysis.
Quantitative data are presented as mean ± SEM. One-way ANOVA was used to compare the physiological parameters (MABP, pO2, pCO2, body temperature, and ICP) between the different groups or between different time points. For further analysis, Duncan post hoc tests were performed, and a p value <0.05 was considered statistically significant. To detect a redistribution of tracer-filled neurons over time after injury, serial χ2 tests between proportions of neurons with specific membrane perturbations were performed. The three χ2 tests (resealed vs enduring, enduring vs delayed, resealed vs delayed) were evaluated at p < 0.0167 to correct for multiple analyses.
Results
Physiological assessments
Mean arterial blood pressure, pCO2, pO2, and pH were monitored and maintained at physiological levels in each animal for the duration of the experiments. There were no differences in these physiological parameters between injected and noninjected animals, demonstrating no systemic effects of the intracerebroventricular injections (data not shown).
ICP was also monitored to reveal the effects of preinjury and postinjury infusions on ICP as well as any ICP change related to diffuse traumatic brain injury. No elevation in ICP was found in the injected, noninjured sham animals. This indicated that the intraventricular infusion into the lateral ventricle did not increase ICP itself (data not shown). No significant differences in ICP over time were observed between injected-injured and non-injected-injured animals at any time point, indicating that intracerebroventricular infusion had no effect on ICP change related to DTBI. However, it is important to note that in this injury model, like the human condition, the traumatic episode itself was associated with ICP elevation. Specifically, ICP was elevated 2, 3, 6, and 7 h after injury compared with preinjury levels both in non-injected-injured and injected-injured animals (Fig. 1). In some cases, the ICP approached 20 mmHg, a value consistent with that seen in brain-injured humans (Miller et al., 1977; Marshall et al., 1979).

Bar charts representing the mean levels of intracranial pressure measured 1 and 2 h before injury as well as 2, 3, 6, and 7 h after injury (black column, noninfused injured, n = 10; gray column, infused injured, 4 h survival, n = 10; white column, infused injured, 8 h survival, n = 11). No elevation in ICP was found in injected-non-injured sham animals, indicating that controlled infusion into the lateral ventricle does not increase ICP (data not shown). No significant difference in ICP was observed between injected-injured and non-injected-injured animals at 4 or 8 h after injury, indicating that infusion into the lateral ventricle does not complicate any ICP change associated with closed head injury. Note, however, that in this injury model, the traumatic episode was associated with a significant elevation of ICP. The ICP was elevated at each time point measured postinjury, particularly at 2, 3, 6, and 7 h after injury. Data are shown as mean ± SEM. The asterisk indicates statistical significance (p < 0.001).
Evidence for altered neuronal membrane permeability after DTBI
Administration of high-molecular-weight dextrans normally excluded from the neuronal and glial cytoplasm by intact cell membranes was used to investigate altered membrane permeability related to diffuse brain injury. Based on our previous observation that the infusion of 10 kDa dextran into the lateral ventricle is a reliable approach for the detection of plasmalemmal alteration related to closed head injury (Singleton and Povlishock, 2004), Alexa Fluor 488-conjugated 10 kDa dextran was infused 2 h before injury. Additionally, Texas Red-conjugated 10 kDa dextran was used either 2 or 6 h after TBI to examine potential membrane resealing and/or enduring membrane disruption. The rationale here was that neurons containing both fluorophores sustained enduring membrane damage over the experimental time course, whereas the presence of one fluorophore alone would argue for either membrane resealing in the case of the preinjury-administered fluorophore or delayed opening in the case of the postinjury-administered fluorophore-labeled dextran. Sham-injured animals receiving fluorophore-labeled dextran infusions demonstrated interstitial or perivascular diffusion of the tracers throughout the neocortex without any evidence of cellular uptake. This indicated that controlled dextran infusion into the lateral ventricles did not lead to cellular uptake of the tracers (Fig. 2). This pattern of tracer distribution was consistent with that described with the use of comparable molecular weight tracers described by our laboratory and others (Singleton and Povlishock, 2004). In contrast, the injured animals revealed tracer-containing neurons indicative of membrane disruption scattered bilaterally throughout the rostrocaudal extent of the neocortex, primarily within layers IV and V (Fig. 3A). The relatively large number of dextran-flooded neurons in injured tissue indicated that the membrane disruption was not an isolated phenomenon but rather occurred throughout the extent of the neocortex scattered among other apparently unaltered neurons. In all cases, based on at both light and electron microscopy evaluation, there was no evidence of dextran-flooded glia. At 4 and 8 h after injury, respectively, 55.4% ± 7.4 and 54.7% ± 6.4 of neurons sustaining any membrane disruption and tracer flooding were double labeled both with the preinjury- and postinjury-infused dextrans (Alexa Fluor 488 and Texas Red conjugated). These findings suggest that neuronal cell membranes were opened initially after injury and maintained continued membrane disruption detected by the administration of the second tracer (Fig. 3B–D). With confocal microscopy, many of the neurons flooded with both dextrans demonstrated evidence of concomitant cell injury reflected in their irregular profiles and vacuolization, all of which were suggestive of necrotic alteration leading to cell death (Fig. 4A–C). In those neurons showing the most dramatic structural change, the dextrans typically were found within the nucleus. In contrast to these findings, a limited number of double-labeled neurons at both time points demonstrated little or no pathological damage. These neurons revealed homogenous tracer flooding without nuclear accumulation or vacuolization (Fig. 4D). Concomitant ultrastructural analyses confirmed and supplemented the morphological changes observed with confocal microscopy. With electron microscopy, many of the double-flooded neurons appeared damaged with evidence of necrosis. Such necrotic change was reflected in occasional nuclear pyknosis, dilation of the Golgi apparatus and the smooth endoplasmic reticulum (SER), together with occasional vesicular swelling, isolated mitochondrial change, increased nuclear and cytoplasmic electron density, and perisomatic glial swelling in the most severely necrotic neurons (Fig. 5E,F). However, as noted above, a limited number of double-flooded cells demonstrated only modest ultrastructural change, consistent with a less severe form of cellular perturbation (Fig. 5C–E).

This double-labeled confocal image from a sham-injured animal receiving fluorophore-labeled dextran infusions demonstrates the diffusion of the tracers throughout the interstices of the neocortex without any evidence of neuronal uptake of any of the administrated dextrans. Note that in addition to their passage through the interstices of the brain parenchyma, the labeled dextrans can be easily visualized in the perivascular regions, consistent with a control pattern of dextran distribution (arrows). Scale bar, 100 μm.
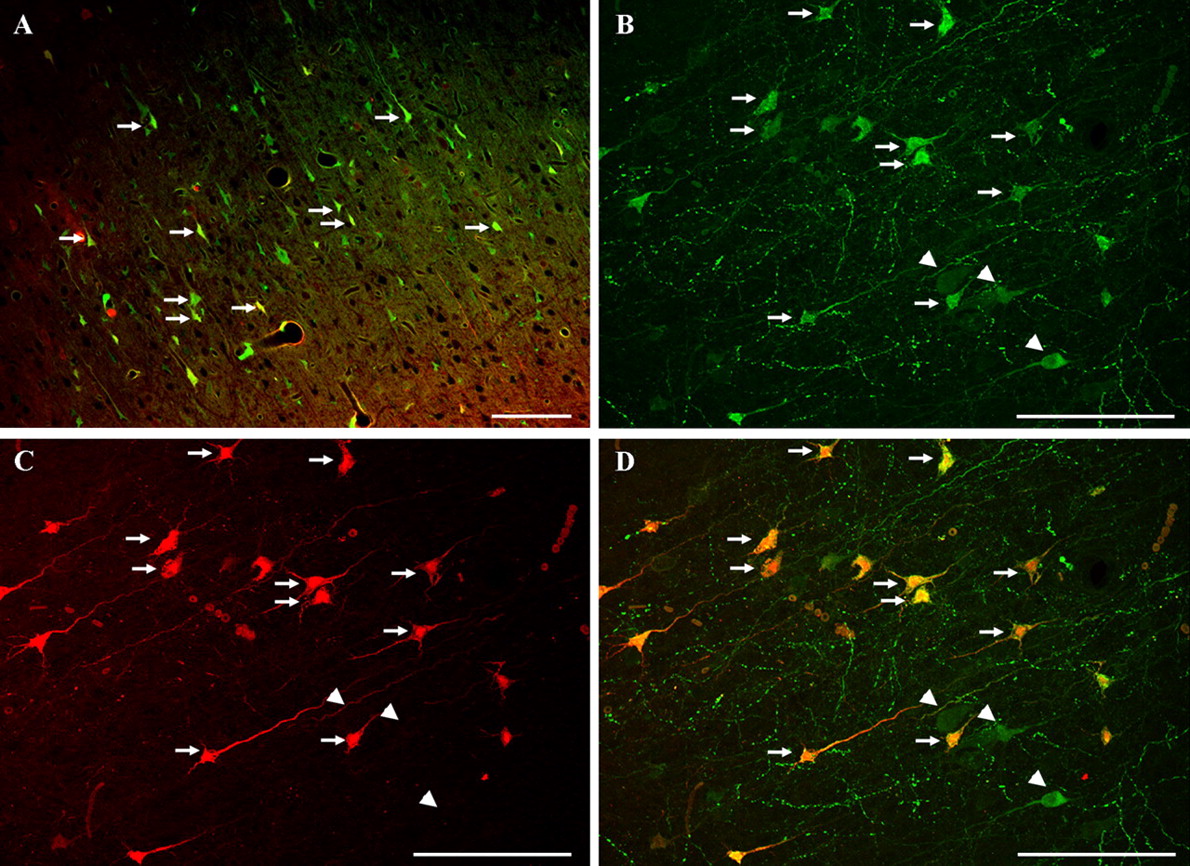
Confocal images of tracer-infused-injured animals. A, This double-labeled image demonstrates numerous cortical neurons (arrows) revealing intracellular tracer flooding with both the preinjury and postinjury infused dextrans. Scale bar, 100 μm. B–D, These confocal images, revealing the initially administered dextran flooding (B), the postinjury infused dextran flooding (C), and their overlay (D), demonstrate that the majority of the neurons sustaining membrane disruption and tracer uptake are double labeled both with preinjury- and postinjury-infused dextrans (arrows). Such double labeling was observed at both 4 and 8 h after injury. Note that some neurons flooding with the preinjury dextran alone can also be observed (arrowheads). Scale bar, 100 μm.

A–D, Double-labeled confocal images. In A–C, neurons flooding with both dextrans reveal evidence of concomitant cellular injury, reflected in their irregular, distorted profiles and vacuolization (arrows). In those cells showing the most severe damage, dextrans are also typically found within the nucleus (arrowhead). Note that other double-labeled neurons (D) demonstrate little or no pathological damage and that despite homogenous tracer uptake, no nuclear accumulation or vacuolization occurs (arrows). Scale bars, 100 μm.

Ultrastructural analysis of the dual tracer-containing neurons reveals various forms of pathologic change. A, The fluorescent image obtained via routine fluorescent microscopy. B, The anti-Alexa Fluor 488-immunostained image of the same region. C, D, An electron micrograph of the same neuron. Note that this neuron demonstrates moderately increased electron density, cytoplasmic and nuclear tracer flooding (arrows), and perinuclear organelle vacuolization (arrowheads) without overt mitochondrial damage (double arrowheads), best shown in the enlarged panel D. Scale bar, 2 μm. E, Three tracer-flooded neurons, again confirmed by routine fluorescent microscopy and followed via EM. The most severely damaged neuron (asterisk) demonstrates increased electron density, organelle vacuolization, and perisomatic glial ensheathment best illustrated in enlarged panel F. The two other cells (double and triple asterisks) demonstrate little or no pathological change. Note that the surrounding neuropil demonstrates little overt pathologic change consistent with the confocal observations. Scale bar, 5 μm
Within these injury foci, CMSP revealed a complex and unanticipated repertoire of responses in relation to the above-described pathologies. No CMSP-immunopositive neurons were observed in sham-injured animals. However, at both 4 and 8 h after injury, CMSP-immunoreactive neurons were observed throughout the neocortex (Fig. 6a). At 4 h after injury, 12.0% ± 8.1 of neurons demonstrating enduring membrane disruption, evidenced by their content of both tracers, were also found to be Ab 38 positive. By 8 h after injury, 15.7% ± 4.3 of the double-flooded neurons revealed evidence of CMSP. Furthermore, at 4 and 8 h respectively, 34.3% ± 3.1 and 35.4% ± 6.3 of all neurons analyzed revealed CMSP immunoreactivity without concomitant tracer flooding (Fig. 6B–D). Ultrastructural analysis of these Ab 38-positive neurons revealed only moderate subcellular damage (Fig. 7A–C). Typically, the CMSP reaction product was found scattered throughout the cytoplasm, with local concentrations around dilated mitochondria dispersed throughout the neuronal cell body. In contrast, neurons demonstrating severe ultrastructural alterations failed to show CMSP immunoreactivity (Fig. 7A,B).

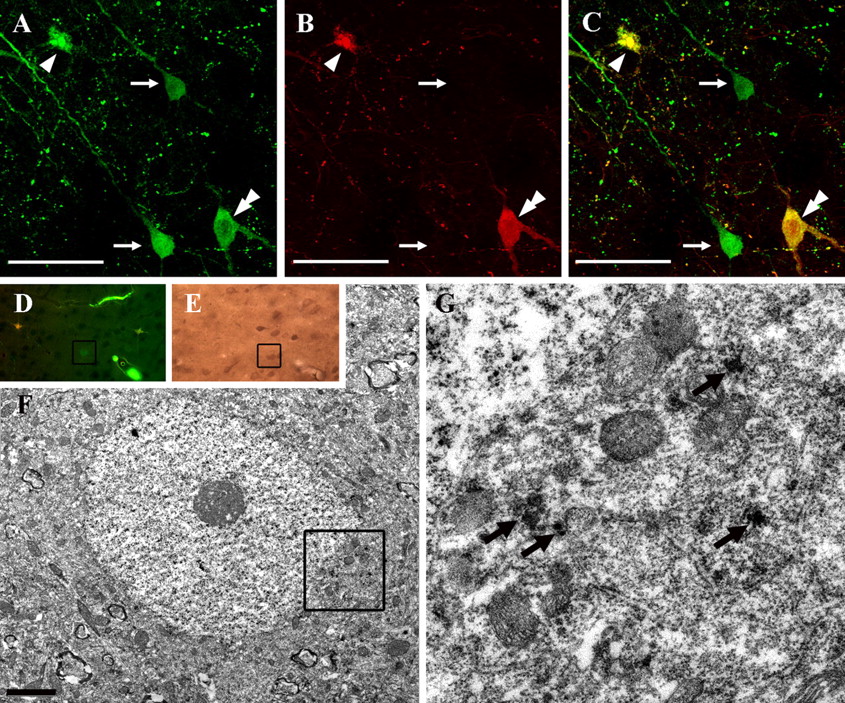
A, Triple-labeled confocal image demonstrating dextran flooded neurons as well as CMSP-immunopositive neurons. Note that at both 4 and 8 h after injury, CMSP-immunoreactive neurons were observed throughout the neocortex (arrows). Scale bar, 100 μm. B–E, Confocal images of preinjury dextran flooding (B), postinjury dextran flooding (C), CMSP immunopositivity (D), and their overlay (E) demonstrate neurons showing enduring membrane disruption reflected in their content of both tracers. Note that some neurons colocalize with CMSP immunopositivity (arrow), whereas other neurons demonstrate tracer flooding without CMSP (arrowheads). Also note that at these same time points, several CMSP-immunoreactive neurons also could be identified without concomitant tracer flooding (double arrowhead). Finally, note that in the same region, a neuron demonstrating only the initial tracer flooding (big arrow), as well as a neuron revealing only secondary tracer flooding (triple arrowhead), can also be seen. Scale bar, 50 μm.

A–C, A toluidine blue-stained 1-μm-thick section reacted for the visualization of CMSP is shown in A, whereas B and C reveal an EM of the same neurons. Note that in B, one CMSP-immunopositive neuron demonstrates moderate subcellular damage. Note that C, taken from that area blocked off in B, reveals the CMSP reaction product around swollen mitochondria (arrows) and dispersed throughout the cytoplasm (double arrowheads). A neuron demonstrating overt necrosis with no evidence of CMSP is also shown (arrowhead). Scale bar, 5 μm.
Evidence for membrane resealing after DTBI
In contrast to those neurons demonstrating enduring membrane alteration as evidenced by their content of both dextrans, at 4 h, 39.1% ± 6.4 and at 8 h, 24.3% ± 9.4 of cortical neurons, showing any evidence of tracer uptake, flooded with the preinjury infused dextran alone, thereby suggesting membrane resealing (Fig. 8A–C). With confocal microscopy, these neurons displayed normal cellular detail, an observation consistent with post-TBI membrane resealing and cell survival. Parallel ultrastructural analyses failed to reveal significant pathological change within these neurons (Fig. 8F,G). Furthermore, at 4 and 8 h respectively, 7.6% ± 5.5 and 11.1% ± 2.9 of these resealed neurons showed evidence of calpain-mediated spectrin proteolysis.

A–C, Confocal images of preinjury-infused dextrans (A), postinjury-infused dextrans (B), and their overlay (C) demonstrate some cortical neurons flooding with the preinjury-infused dextran alone without concomitant flooding with the postinjury-administrated tracer (arrows). Note that one double-flooded neuron demonstrating severe damage (arrowhead) and one double-labeled neuron demonstrating less severe pathology (double arrowhead) are also shown. Scale bar, 50 μm. D, A routine fluorescent image that reveals a single labeled cell. E, The same neuron as in D is visualized through the use of antibodies to the fluorophore and then carried to the EM level (F, G). Note that neurons flooding with the preinjury dextran alone do not show overt pathological damage. Immunoreactive products (anti-Alexa Fluor IR) are labeled with arrows in G, which is an enlargement of the area, blocked out in F. Scale bar, 2 μm.
Evidence for delayed membrane perturbation after DTBI
Despite the consistent finding of neurons containing the initially infused tracer without any delayed tracer flooding, at 4 h, 5.5% ± 2.7 and at 8 h, 20.9% ± 9.1 of all tracer-containing neurons were found to flood with the postinjury infused dextran alone (Fig. 9A–C). At 4 h, 55.1% ± 12.9 and at 8 h, 32.5% ± 11.8 of neurons demonstrating delayed membrane perturbation also showed CMSP.

A–C, Confocal images of preinjury-infused dextran (A), postinjury-infused dextran (B), and their overlay (C) demonstrate scattered neurons flooding with the postinjury-infused dextran alone (arrows), among with double-flooded neurons (arrowheads). Scale bar, 100 μm.
Importantly, all of the above-described populations of neurons could be found in the same neocortical loci, excluding the possibility that different regions exposed to different rates of dextran diffusion could have sustained different degrees of dextran flooding.
Redistribution of neuronal membrane perturbation after DTBI
As noted above, neurons scattered throughout the neocortex exhibited varying degrees of membrane perturbation and cellular pathology after DTBI. In the present study design, three categories of neurons with membrane perturbation were detected: (1) resealed neurons, (2) neurons with enduring membrane perturbation, (3) neurons with delayed membrane damage. Similarly, the experimental design separated each survival group (4 and 8 h after injury) into periods before and after the availability of the postinjury tracer. In this way, the relative proportions of neurons with resealed, enduring, and delayed membrane perturbations could be analyzed in relationship to time periods of tracer availability, to provide a model of membrane perturbation pathology after DTBI (Fig. 10).

Distribution of neurons with resealed, enduring, or delayed membrane perturbation within the population of neurons with DTBI-induced membrane perturbation. As membranes remain closed, open, or reseal in response to injury, tracer availability will determine the type of membrane perturbation categorically assigned to each neuron (table within figure legend). The preinjury administration of Alexa Fluor 488 and postinjury administration of Texas Red-conjugated dextrans permit the evaluation of neuronal membrane perturbation distribution at 4 and 8 h after injury. Across time points, the proportion of neurons with delayed membrane perturbation is significantly different from the proportion of resealed neurons and the proportion of enduring membrane perturbation (χ2, p < 0.016). The change in the proportion of neurons with resealed and enduring membrane perturbation over time was not significant (χ2, p = 0.19). The redistribution of the types of membrane perturbation between 4 and 8 h after injury may result from resealed neurons reopening to become neurons with enduring damage and/or additional neurons suffering delayed membrane perturbation. See Results for more details.
Of all the neurons that suffer membrane perturbation by 4 h after injury, 95% are damaged within the first 2 h. However, 40% of the neurons reseal by 2 h, leaving 60% with enduring or delayed membrane perturbations at 4 h after injury (Fig. 10). From the 8 h experimental group, 80% of all neurons with membrane perturbation suffer damage within the first 6 h, with 25% of the neurons resealing before the postinjury tracer administration at 6 h, and 75% of the population manifests enduring or delayed membrane perturbation (Fig. 10). Because the percentages of the population are normalized and derived from the same data set, statistical tests cannot be performed within a time point. Between time points, the proportion of neurons with delayed membrane perturbation differs significantly from the proportion of resealed neurons and the proportion of enduring membrane perturbation (χ2, p < 0.016). The change in the proportion of neurons with resealed and enduring membrane perturbation over time was not significant (χ2, p = 0.19). Together, these tests confirm a significant redistribution of the neuronal population with membrane perturbations between 4 and 8 h. These analyses were based on fractions of all neurons that demonstrate perturbation, but the total populations were not necessarily equal.
Discussion
Using tracer exclusion and immunohistochemical techniques, together with confocal and electron microscopy, the current study provides new evidence for the diversity of the neuronal cellular responses to DTBI, emphasizing the significance of neuronal membrane mechanoporation in DTBI pathobiology.
Membrane disruption and enduring membrane permeability
The present study is the first to demonstrate, in an in vivo model of DTBI, the potential for either enduring membrane perturbation progressing to cell death or neuronal membrane resealing compatible with recovery. These neuronal changes were both widespread and diffusely distributed, interspersed with apparently uninjured neurons, a finding consistent with the pathobiology of DTBI in humans (Povlishock and Katz, 2005).
These data emphasize that the mechanical forces initiate the observed pathology. The early, diffuse damage identified is not consistent with focal ischemia that would have precipitated a more delayed and highly focal neuronal response (Dietrich et al., 1987; Dereski et al., 1993; Dietrich, 1998; Mennel et al., 2000; Bramlett and Dietrich, 2004). Although in this study, DTBI evoked elevated ICP, this elevation never exceeded 20 mmHg, suggesting no significant alteration in cerebral perfusion pressure with the normal arterial blood pressure described in this study. This does not preclude, however, the potential that over time the elevated ICP evoked secondary neuronal damage (vide infra).
Using lactate dehydrogenase (LDH) release or high-molecular-weight tracer uptake to evaluate membrane damage in various TBI models, our laboratory and others have established that membrane disruption can occur at the moment of injury, suggesting that the traumatic event evokes direct mechanical poration of the neuronal cell membrane (Pettus et al., 1994; LaPlaca et al., 1997; Geddes and Cargill, 2001; Geddes et al., 2003; Prado and LaPlaca, 2004; Prado et al., 2004; Singleton and Povlishock, 2004). In these studies, it was suggested that the altered neuronal membrane permeability generated ionic disturbances to precipitate cell death. This premise, however, was neither directly addressed nor followed comprehensively via multiple endpoints (LaPlaca et al., 1997; Geddes et al., 2003; Prado et al., 2004). The current study significantly extends previous observations, demonstrating that membrane perturbation persists for, at least, several hours after injury in many injured neurons. Significance is also attached to the LM observation that these neurons revealed morphological features consistent with irreversible damage, including cellular distortion, vacuolization, and pyknotic nuclei. The parallel EM findings of increased electron density, together with the dilation of the Golgi and SER and increased numbers of dilated vesicles, were also consistent with a necrotic cascade triggered by the translocation of lysosomal cathepsins and other hydrolytic enzymes (White et al., 1993; Chan, 1996; Yamashima et al., 1998; Yamashima, 2004). However, we cannot rule out that this organelle and vesicular dilation are linked to an albeit abortive attempt at membrane repair mediated by the lysosomes and the synaptotagmin found on their surfaces (Andrews, 2005). These issues will require highly targeted investigations for their resolution. Although our finding of isolated neurons demonstrating enduring membrane permeability without severe ultrastructural damage at both 4 and 8 h after injury suggests that membrane perturbation may not always lead to cell death, we cannot preclude the possibility that with increased survival these same neurons would die.
Membrane resealing
In addition to enduring membrane alteration, other neurons displayed evidence of membrane resealing via their uptake of only the preinjury tracer. Previous studies in non-neuronal cells demonstrated that, after mechanical injury, membrane resealing can occur within seconds to minutes (McNeil and Ito, 1989; McNeil and Khakee, 1992; Yu and McNeil, 1992; Steinhardt et al., 1994; Terasaki et al., 1997; McNeil et al., 2000). These studies investigated cardiac muscle (Clarke et al., 1995), skeletal muscle (McNeil and Khakee, 1992), skin (McNeil and Ito, 1990), vascular endothelium (Yu and McNeil, 1992), or gastrointestinal cells (McNeil and Ito, 1989), which frequently experience mechanical stress/strain under normal conditions. To date, only limited studies have investigated membrane resealing in the CNS. In vitro or nonmammalian models have focused primarily on resealing after axonal transection (Yawo and Kuno, 1983, 1985; Fishman et al., 1990; Xie and Barrett, 1991; Godell et al., 1997; Shi and Pryor, 2000; Shi et al., 2001; Geddes et al., 2003). In contrast to non-neuronal cells, these studies suggested that neuronal cell membrane resealing takes minutes to hours rather than seconds described in non-neuronal cells. Our finding of neurons labeled with the preinjury tracer alone is consistent with membrane repair; however, the sampling period occurred over several hours after injury precluding the determination of whether this resealing was immediate or rather occurred at a later time point.
Delayed membrane perturbation
Our observation of neurons flooded with the postinjury infused dextran alone suggests the potential for delayed membrane disruption occurring several hours after injury. The precise mechanism(s) for this delayed opening is (are) unknown; however, the possibility exists that the sustained, elevated ICP occurring after injury could alter the neuronal cell membrane. This issue is of more than mere academic interest, because sustained elevated ICP is one of the major adverse prognostic factors for recovery after TBI and obviously this issue will merit continued evaluation (The Brain Trauma Foundation et al., 2000).
The coincidence of CMSP with altered membrane permeability
The present study also examined the potential role of calpain activation and its relation to membrane permeability and/or neuronal damage. Calpain activation and the subsequent CMSP have long been associated with the pathogenesis of different brain injuries and TBI cell death (Kampfl et al., 1996, 1997; Saatman et al., 1996; Newcomb et al., 1997; Pike et al., 1998, 2000, 2001; Buki et al., 1999; McCracken et al., 1999; Kupina et al., 2002, 2003). Calpain activation occurs at elevated intracellular calcium levels. Membrane perturbation can lead to excessive calcium influx from the extracellular space, resulting in calpain activation (Povlishock et al., 1997; Buki et al., 1999), that then causes direct subcellular damage or lysosome membrane rupture with the release of damaging enzymes such as cathepsins (Yamashima, 2004). The cysteine proteases, such as calpain and caspase, can also maintain increased membrane permeability, theoretically permitting additional pathology (Liu and Schnellmann, 2003; Liu et al., 2004). Furthermore, it has been demonstrated, in vitro, that CMSP correlates with LDH release and propidium iodide uptake, markers of increased membrane permeability after stretch injury (Pike et al., 2000). Other studies, however, have also suggested that calpain might have an important role in membrane resealing (Godell et al., 1997; Howard et al., 1999; Shi et al., 2000).
In the present study, the majority of cells demonstrating CMSP did not reveal tracer flooding either at 4 or 8 h after injury, suggesting that calpain activation occurs independent of cell membrane disruption. Furthermore, the majority of CMSP-positive neurons revealed only limited ultrastructural change, suggesting that calpain activation is not necessarily associated with cell death after DTBI, at least within the time frames assessed. Alternatively, the initiation of neuronal death may await the activation of a secondary calpain cascade, a premise supported by several studies (Saido et al., 1993; Yokota et al., 1995; Neumar et al., 2001; Saatman et al., 2003; Czogalla and Sikorski, 2005). Equally surprising was the finding that the majority of cells flooding with both dextrans and manifesting necrotic change did not show CMSP. Conceivably, the calpain-specific spectrin breakdown products in the necrotic neurons were cleaved before the assessment, yet, this argument is countered by the fact that spectrin fragments are highly stable in vivo and in vitro (Wang et al., 1989; Czogalla and Sikorski, 2005). Neurons triple labeled with both dextrans and Ab 38 were observed, yet they represented the smallest population of injured neurons. Collectively, these results suggest that the observed membrane disruption is not necessary or sufficient for calpain-mediated spectrin proteolysis. Together with other studies (Saatman et al., 1996; Brana et al., 1999), these findings emphasize the need for caution in interpreting the occurrence of CMSP and its overall implications for neuronal injury and death.
Redistribution of neuronal membrane perturbation after TBI
In addition to the above provocative information regarding membrane perturbation, resealing, and delayed opening, the shifts over time in the percentages of neuronal populations showing membrane damage provide compelling evidence for a redistribution of the membrane damaged population over time. Specifically, the fraction of neurons that reseal early postinjury may suffer secondary membrane reopening, converting these neurons to those linked with enduring membrane perturbation. Furthermore, with increased survival, proportionally more neurons suffer delayed membrane perturbation. Theoretically to achieve such a distribution from 4–8 h after injury, 25% of the resealed neurons would be required to reopen, with an additional 20% of the previously uninjured neurons now suffering delayed membrane perturbation. Together with our previous findings, these observations confirm that the plasmalemmal changes evoked by DTBI are complex, evolving, and potentially subject to modification by secondary mechanisms.
In sum, this study demonstrates the heterogeneity of injury-related membrane perturbation and the diversity of the neuronal responses to this perturbation, while also demonstrating the potential of delayed membrane repair after TBI. Furthermore, our findings indicate that calpain-mediated spectrin proteolysis can occur independent of membrane disruption and that membrane disruption itself does not necessarily lead to calpain activation. These findings highlight the complexity of DTBI, its dissimilarity to most CNS disorders that evoke homogenous, focal change, and its role in dynamic mechanical poration of the neuronal plasmalemma.
Footnotes
-
This work was supported by National Institutes of Health Grants R01 NS-045834, F32 HD-49343, and NS-47463 and by the Commonwealth Center for Traumatic Brain Injury. We thank Sue Walker, Lynn Davis, and Tom Coburn for their technical assistance. We also thank for Dr. Robert Hamm for his advice on statistical analysis and Dr. Scott Henderson for his help with confocal microscopy.
- Correspondence should be addressed to Dr. John T. Povlishock, Department of Anatomy and Neurobiology, Medical College of Virginia Campus of Virginia Commonwealth University, P.O. Box 980709, Richmond, VA 23298. Email: jtpovlis{at}vcu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}