

Abstract
Hepatic encephalopathy (HE) is a major neurological complication in patients with severe liver failure. Elevated levels of ammonia have been strongly implicated as a factor in HE, and astrocytes appear to be the primary target of its neurotoxicity. Mechanisms mediating key aspects of ammonia-induced astrocyte dysfunction such as cell swelling and inhibition of glutamate uptake are not clear. We demonstrated previously that cultured astrocytes exposed to ammonia increase free radical production. We now show that treatment with antioxidants significantly prevents ammonia-induced astrocyte swelling as well as glutamate uptake inhibition. Because one consequence of oxidative stress is the phosphorylation of mitogen-activated protein kinases (MAPKs), we investigated whether phosphorylation of MAPKs may mediate astrocyte dysfunction. Primary cultured astrocytes exposed to 5 mm NH4Cl for different time periods (1–72 h) significantly increased phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2), p38MAPK, and c-Jun N-terminal kinase (JNK) 1/2/3, which was inhibited by appropriate MAPK inhibitors 1, 4-diamino-2, 3-dicyano-1, 4-bis (2-aminophenylthio) butadiene (UO126; for ERK1/2), trans-1-(4-hydroxyclyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxypyrimidin-4-yl)imidazole (SB 239063; for p38MAPK), and anthra[1,9-cd]pyrazol-6(2H)-one (SP600125; for JNK1/2/3), as well as by antioxidants. Kinase inhibitors partially or completely prevented astrocyte swelling. Although SB239063 and SP600125 significantly reversed glutamate uptake inhibition and ammonia-induced decline in glutamate-aspartate transporter protein levels, UO126 did not, indicating a differential effect of these kinases in ammonia-induced astrocyte swelling and glutamate transport impairment. These studies strongly suggest the involvement of oxidative stress and phosphorylation of MAPKs in the mechanism of ammonia-induced astrocyte dysfunction associated with ammonia neurotoxicity.
Introduction
Liver failure is the fourth leading cause of death in the United States and is an important cause of morbidity and mortality among the poor (Lockwood, 1992). Approximately 50–75% of these individuals have varying degrees of neurological symptoms resulting in a condition known as hepatic encephalopathy (HE). HE can present acutely [fulminant hepatic failure (FHF)] resulting from massive liver necrosis after viral hepatitis, acetaminophen toxicity, or exposure to other hepatotoxins. It presents with the abrupt onset of delirium, seizures, and coma and has an extremely poor prognosis with an 80–90% mortality rate (Capocaccia and Angelico, 1991). The only effective treatment for this condition is an emergency liver transplantation (Bismuth et al., 1996). Chronic HE (portal-systemic encephalopathy) is characterized by change in personality, altered mood, diminished intellectual capacity, abnormal muscle tone and tremor (asterixis), stupor, and coma (Plum and Hindfelt, 1976).
Although the pathogenetic factors responsible for HE are still not clearly known, elevated levels of brain ammonia resulting from failure of the liver to detoxify it has been strongly implicated as an important element in this condition (for review, see Albrecht and Jones, 1999; Hazell and Butterworth, 1999). Agents and experimental procedures that increase blood or brain ammonia reproduce clinical and pathological changes observed in HE. Likewise, patients with hereditary hyperammonemia have similar clinical and pathological changes as found in HE. Exposure of cultured astrocytes to ammonia mimics many of the changes observed in HE in vivo. However, mechanisms by which ammonia exerts its neurotoxicity still remain poorly understood. Views have ranged from electrophysiological disturbance (Raabe, 1989), neurotransmitter dysfunction (Butterworth, 2001), excitotoxicity (Marcaida et al., 1992), and more recently oxidative stress (Norenberg, 2003).
Brain edema with increased intracranial pressure and brain herniation is the major clinical complication associated with acute HE (Vaquero et al., 2003). Although the basis for such edema remains to be determined, it is clear that swelling of astrocytes represents a major component of the edema (Martinez, 1968; Norenberg, 1977, 2001; Traber et al., 1989). Mechanisms underlying such astrocyte swelling in acute HE are still not well understood, although elevated brain ammonia levels are likely involved. Increased brain water has been described in dogs with urease-induced hyperammonemia (Levin et al., 1989), as well as in rats after ammonium acetate infusions (Takahashi et al., 1991). Treatment of cerebral cortical slices with ammonia in concentrations equivalent to those reported in brain in experimental FHF resulted in significant swelling and concomitant reductions of the inulin space (Ganz et al., 1989). Similarly, swelling of cerebral cortical astrocytes was observed after ammonia infusion in primates (Voorhies et al., 1983), as well as in cultured astrocytes (Norenberg et al., 1991; Zwingmann et al., 2000).
Another aspect of HE is a decline in glutamate transport by astrocytes, which may result in elevated extracellular glutamate levels and lead to abnormalities in glutamatergic neurotransmission. Thus, whereas total brain glutamate levels are decreased in various models of HE (Hindfelt et al., 1977; Bosman et al., 1990; Dejong et al., 1992), extracellular glutamate levels are consistently increased in rat models of acute liver failure (Moroni et al., 1983; Tossman et al., 1987; de Knegt et al., 1994). Ammonia has been shown to suppress high affinity glutamate uptake (Bender and Norenberg, 1996). Likewise, downregulation of the glutamate transporter GLT-1 [excitatory amino acid transporter 2 (EAAT-2)] has been shown in hyperammonemic rats, in rats with portacaval anastomosis (model of chronic HE), as well as in thioacetamide-induced acute liver failure (Knecht et al., 1997; Norenberg et al., 1997). A decrease in glutamate-aspartate transporter (GLAST) (EAAT-1) mRNA levels was found in cultured astrocytes exposed to a pathophysiological concentration of ammonia (Zhou and Norenberg, 1999).
The mechanism(s) by which ammonia induces astrocyte swelling and glutamate uptake inhibition is unclear. Oxidative stress has been hypothesized to play an important role in the pathogenesis of ammonia neurotoxicity (Norenberg, 2003). One important consequence of oxidative stress is the phosphorylation of mitogen-activated protein kinases (MAPKs), including p38MAPK, c-Jun N-terminal kinase (JNK), and the extracellular signal-regulated kinase (ERK) (Kyriakis and Avruch, 1996; Aikawa et al., 1997; Mielke and Herdegen, 2000; Ono and Han, 2000). Such MAPK phosphorylation has also been shown in cultured astrocytes in response to oxidant signaling (Luo and Roth, 2000; Mizuhashi et al., 2000; S. H. Chen et al., 2001; Lennon et al., 2002).
In this study, we investigated whether MAPK phosphorylation occurs in ammonia-treated astrocytes and whether such phosphorylation mediates key aspects of ammonia-induced astrocyte dysfunction. Our findings indicate that antioxidants reduce ammonia-induced astrocyte swelling and glutamate uptake inhibition; that MAPK phosphorylation occurs after ammonia treatment, a process which is inhibited by antioxidants; and that inhibition of MAPKs attenuates aspects of ammonia-induced astrocyte dysfunction. These studies indicate an important role for oxidative stress and MAPK activation in the mechanism of ammonia neurotoxicity.
Materials and Methods
Astrocyte cultures.
Primary cultures of cortical astrocytes were prepared as described previously (Ducis et al., 1990). Briefly, brains of 1- to 2-d-old rat pups were seeded on 35 mm culture dishes in DMEM containing penicillin, streptomycin, and 15% fetal bovine serum and incubated at 37°C with 5% CO2 and 95% air. The culture media were changed twice weekly. Cultures consisted of 95–99% astrocytes as determined by glial fibrillary acidic protein (GFAP) immunohistochemistry. All cultures used in the experiments were 25–30 d old. Procedures followed guidelines established by National Institute of Health Guide for the Care and use of Laboratory Animals and were approved by the local animal care committee (Institutional Animal Care and Use Committee).
Cell volume determination.
Cell volume was estimated by measuring the intracellular water space by the method of Kletzein et al. (1975), as modified by Kimelberg (1987) and Bender and Norenberg (1998). Briefly, 1 mm 3-O-methylglucose (3-OMG) and 0.5 μCi/ml [3H]-3-OMG were added to the culture 6 h before the volume assay. At the end of the incubation period, culture medium was aspirated, and an aliquot was saved for radioactivity determination. Cells were rapidly washed six times with ice-cold buffer containing 229 mm sucrose, 1 mm Tris-nitrate, 0.5 mm calcium nitrate, and 0.1 mm phloretin, pH 7.4. Cells were harvested into 0.5 ml of 1N sodium hydroxide. Radioactivity in the cell extracts and medium were determined, and an aliquot of the cell extract was used for protein estimation with the Bio-Rad bicinchoninic acid kit. Values were normalized to protein level, and cell volume was expressed as microliters/milligram protein.
Western blots.
Astrocytes were solubilized in lysis buffer: 125 mm Tris-HCl 6.8; 4% SDS, phosphatase inhibitors, and protease inhibitor mixture (Roche Products, Indianapolis, IN). Lysates were centrifuged at 10,000 rpm for 6 min, and protein levels were measured by the BCA method. Equal amounts of protein (10 μg of total protein per lane) were subjected to SDS-PAGE using 12% gels (Tris-HCl) and then electrophoretically transferred to a nitrocellulose membrane. Blots were blocked with 5% nonfat dry milk in TBS-T (TBS; 20 mm Tris-HCl, 150 mm NaCl, pH 7.4, and 0.05% Tween 20) for 2 h at room temperature and then incubated with respective primary antibodies at 4°C overnight. Membranes were washed with TBS-T and incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. After washing, membranes were visualized using enhanced chemiluminescence reagents (ECL-plus; Amersham Biosciences, Piscataway, NJ). Primary antibodies to detect only phosphorylated forms of ERK1/2, p38MAPK, and JNK1/2/3 as well as total ERK1/2, p38MAPK, and JNK1/2/3 were purchased from Cell Signaling Technology (Beverly, MA). Anti-GLAST antibody was purchased from Chemicon (Temecula, CA). Anti-α-tubulin antibody was obtained from Oncogene (San Diego, CA). All primary antibodies were used at 1:1000 dilutions. Anti-rabbit, anti-mouse (Vector Laboratories, Burlingame, CA), and anti-guinea pig (Santa Cruz Biotechnology, Santa Cruz, CA) HRP-conjugated secondary antibodies were used at 1:1000. Optical density of the bands was measured with the Chemi-Imager digital imaging system (Alpha Innotech, San Leandro, CA), and results were quantified with Sigma (St. Louis, MO) Scan Pro as a proportion of the signal of a house-keeping protein band (α-tubulin). Controls, which included omission of the primary antibodies, did not show any band.
Measurement of glutamate uptake.
l-Glutamate uptake studies were done as described by Drejer et al. (1982) and Bender et al. (1989). Assays were done in DMEM at 37°C in 5% CO2/95% air. Uptake experiments were initiated by adding 50 μm and 7.4 MBq/L d-[3H] aspartate (a nonmetabolizable analog of l-[3H] glutamic acid) to the media for 1 min. The uptake reaction was terminated by washing cells rapidly three times in ice-cold DMEM. Cells were then harvested into 500 μl of 1N NaOH solution and mixed thoroughly by vortexing. Separate aliquots were taken for protein determination and liquid scintillation spectroscopy. Uptake rates were calculated from the measured radioactivity of the cells and the specific activity of the media, and the results were expressed as nmol/min/mg protein.
Statistical analysis.
Each group consisted of four to five culture dishes per experiment for each time point studied for the cell swelling and glutamate uptake experiments. At least two to four plates were used for Western blot analysis. Experiments were performed from four to seven separate seedings. The extent of cell swelling and glutamate uptake rates obtained were normalized to protein values and subjected to ANOVA followed by Tukey's post hoc comparisons. Intensity unit values obtained from optical density of the bands in Western blots were also subjected to ANOVA followed by Tukey's post hoc comparison test and were expressed as percentage change over control. At each time point, the experimental cultures were compared with their respective control.
Results
Antioxidants and nitric oxide synthase inhibition attenuate ammonia-induced astrocyte swelling
Astrocyte swelling in cultures, after treatment with 5 mm ammonia, is first observed at 12 h (Rama Rao et al., 2003), but it peaks at 48 h. All studies for assessing swelling used the 48 h time point. To investigate the role of oxidative stress in ammonia-induced astrocyte swelling, primary astrocyte cultures were treated for 2 d with a pathophysiological concentration of ammonia (NH4Cl; 5 mm); such value is commonly found in brains of animals with acute liver failure (Swain et al., 1992). Antioxidant treatment was done in the presence or absence of different concentrations of individual antioxidants, including superoxide dismutase (SOD; 10, 25, and 100 U/ml), catalase (250, 500, and 1000 U/ml), α-tocopherol (vitamin E; 50, 100, and 250 μm), or N-tert-butyl-α-phenylnitrone (PBN; 100, 250, and 500 μm), a spin trapping free radical scavenger as well as l-NAME (250 and 500 μm), a nitric oxide (NO) synthase (NOS) inhibitor. Control plates were treated with the same volume of respective solvents (vehicle). Ammonia (5 mm) increased cell swelling by 41% compared with controls (p < 0.05) (Fig. 1). Treatment with antioxidants and N-nitro-l-arginine methyl ester (l-NAME) significantly reduced ammonia-induced swelling. l-NAME at 250 μm decreased ammonia-induced swelling by 76% (p < 0.05) (Fig. 1). Lesser reduction of swelling was observed with 100 μm vitamin E (52%; p < 0.05), 25 U/ml SOD (48%; p < 0.05), and 250 U/ml catalase (35%; p < 0.05). Although PBN at 250 μm diminished cell swelling by 27%, this decrease was not significant (Fig. 1). PBN at 100 or 500 μm also did not inhibit ammonia-induced astrocyte swelling. Cultures treated with the same volume of respective solvents or with antioxidants alone did not change cell volume.

Reduction of ammonia-induced astrocyte swelling by antioxidants. Cultured astrocytes exposed to 5 mm NH4Cl significantly increased cell swelling (2 d). Similarly, NO donors SNAP (25 μm) and SIN-1 (500 μm) also significantly increased astrocyte swelling. Treatment with SOD (25 U/ml), catalase (250 U/ml), vitamin E (100 μm), and the NOS inhibitor l-NAME (250 μm) significantly diminished ammonia-induced astrocyte swelling. PBN (500 μm) had no significant effect on swelling. ANOVA, n = 4. ∗p < 0.05 versus control; †p < 0.05 versus NH4Cl. Error bars represent mean ± SEM.
Because the magnitude of protection by l-NAME in diminishing astrocyte swelling was more than other antioxidants, we tested whether NO donors cause astrocyte swelling. Astrocytes exposed to S-nitroso-N-acetyl penicillamine (SNAP; 25 μm) or 3-morpholinosydnoimine HCl (SIN-1; 500 μm) for 48 h increased astrocyte swelling by 21.4 ± 3.6 and 30.7 ± 4.5%, respectively, when compared with controls (both, p < 0.05) (Fig. 1).
Antioxidants and NOS inhibition diminish ammonia-induced glutamate uptake inhibition
Inhibition of glutamate uptake by 5 mm ammonia occurs at 24 h, the earliest time point investigated (Bender and Norenberg, 1996), but it peaks at 72 h. To determine whether antioxidants prevent ammonia-induced glutamate uptake inhibition, cultures were treated with different antioxidants, SOD (10, 25, 50, and 100 U/ml), catalase (100, 250, 500, and 1000 U/ml), vitamin E (50, 100, and 250 μm), PBN (100, 250, and 500 μm), as well as l-NAME (100, 250, and 500 μm) with or without ammonia for 3 d. Control plates were treated with the same volume of respective solvents. The rate of uptake was performed at 1 min. Astrocytes exposed to ammonia significantly decreased d-aspartate uptake by 46% compared with controls (p < 0.05) (Fig. 2). This inhibition was significantly reversed by treatment with 25 U/ml SOD, 250 μm l-NAME, and 100 μm vitamin E (36, 54, and 47%, respectively; p < 0.05) (Fig. 2). Catalase or PBN did not significantly reverse the ammonia-induced glutamate uptake inhibition. Similar to the observation with astrocyte swelling, l-NAME was effective in abrogating ammonia-induced glutamate uptake inhibition. To determine whether SNAP and SIN-1 can also inhibit glutamate uptake, we examined their effect in cultured astrocytes. Results showed a 24.9 ± 4.2 and 36 ± 2.7% inhibition of glutamate uptake with SNAP and SIN-1, respectively, when compared with controls (both, p < 0.05) (Fig. 2).
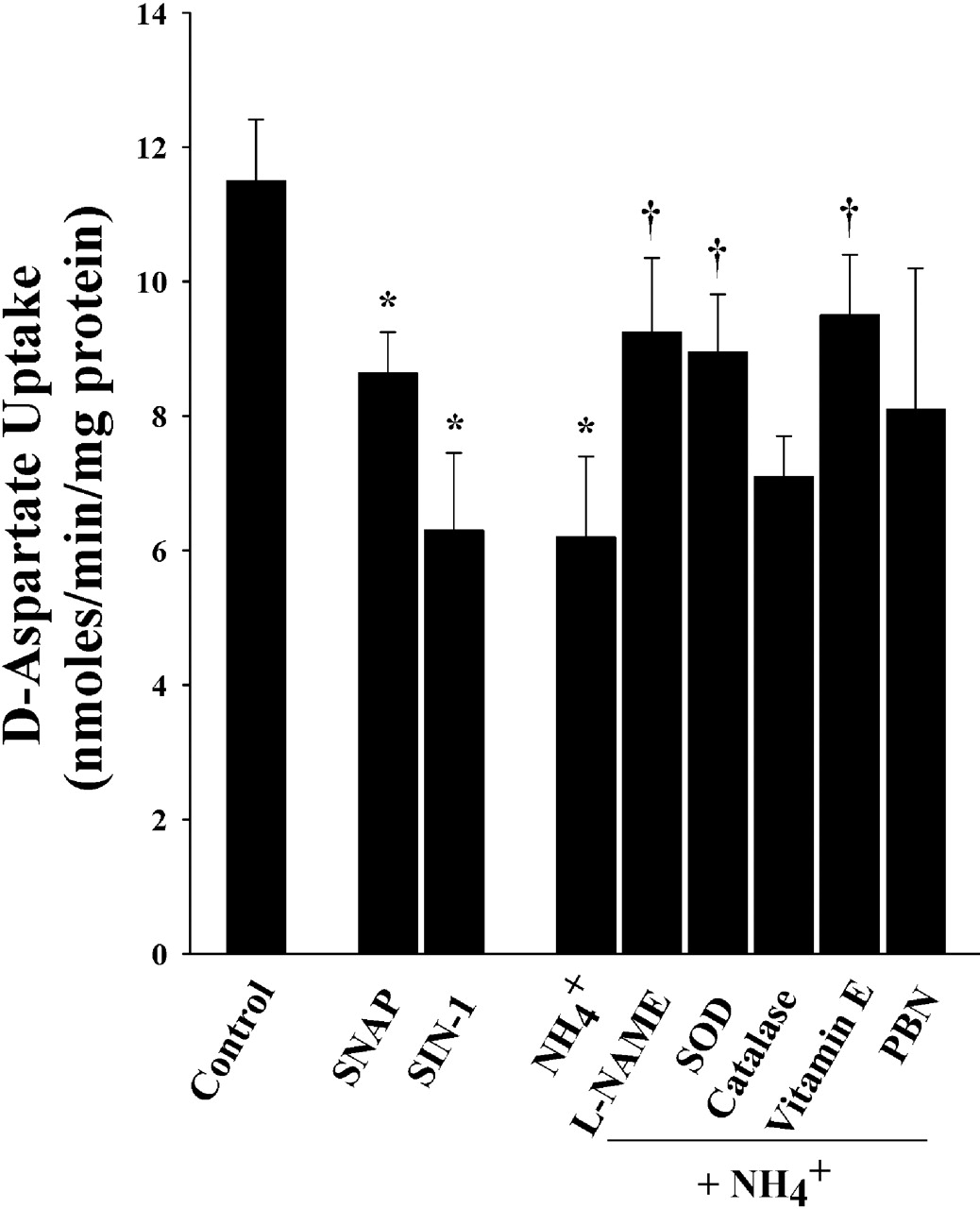
Effect of antioxidants on ammonia-induced glutamate uptake inhibition in cultured astrocytes. A 45% decrease in glutamate uptake was observed when cells were exposed to 5 mm NH4Cl (3 d). NO donors, SNAP (25 μm), and SIN-1 (500 μm) also decreased glutamate uptake significantly. Treatment with SOD (25 U/ml), vitamin E (100 μm), and l-NAME partially prevented the ammonia-induced glutamate uptake inhibition. PBN (500 μm) had no significant effect on glutamate uptake inhibition. ANOVA, n = 11. ∗p < 0.05 versus control; †p < 0.05 versus ammonia. Error bars represent mean ± SEM.
Ammonia increases phosphorylation of MAPKs in astrocytes
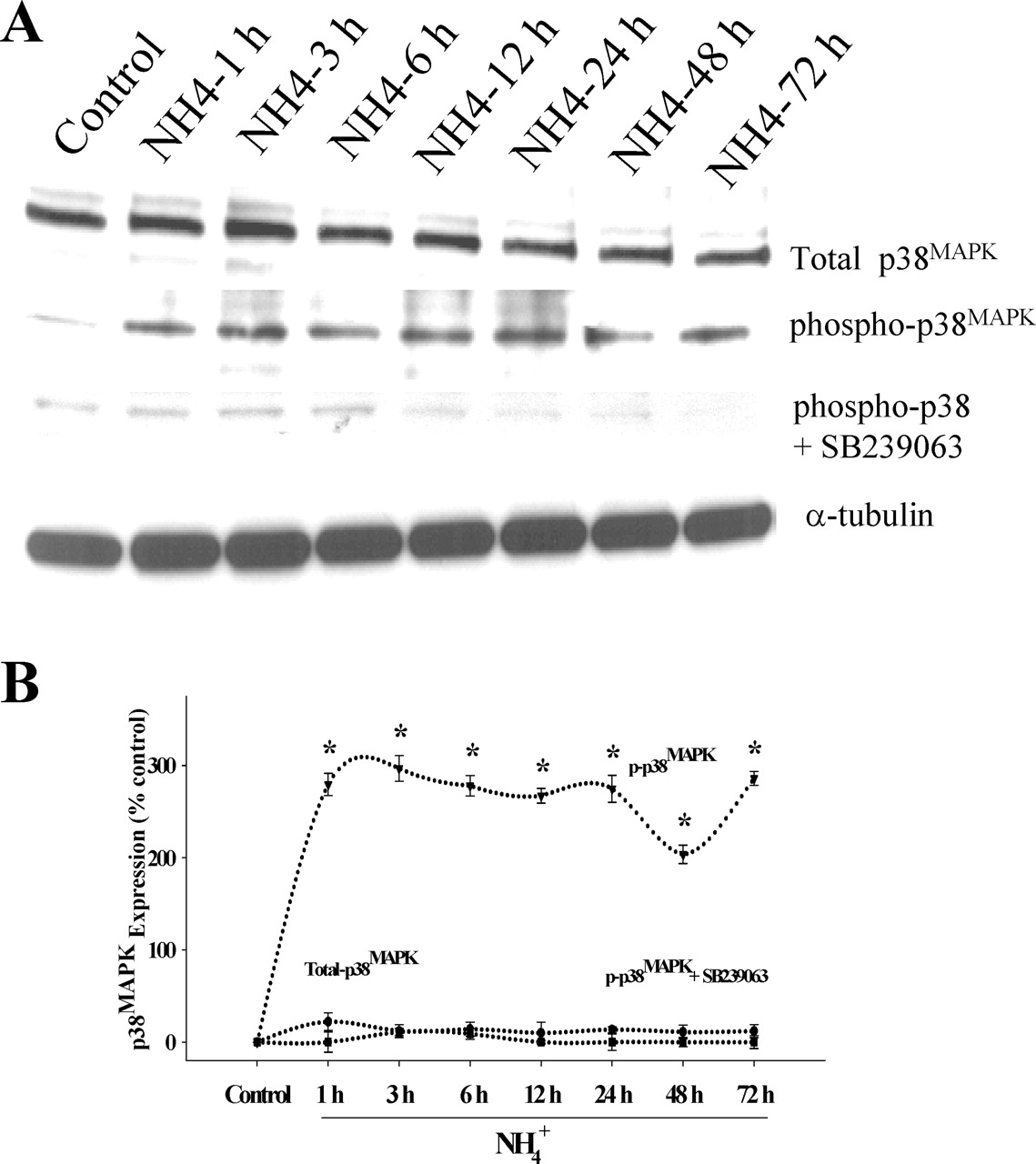
Because oxidative stress has been shown to activate MAPKs, including p38MAPK, JNK1/2/3, and ERK1/2, we examined whether MAPK phosphorylation also occurs after ammonia treatment. Cultured astrocytes were exposed to 5 mm NH4Cl for different time periods (1, 3, 6, 12, 24, 48, and 72 h). Changes in total and phosphorylated ERK1/2, JNK1/2/3, and p38MAPK were determined by Western blots. Ammonia significantly increased the phosphorylation of ERK1/2 at all time points. Peak phosphorylation was observed at 6 h, after which there was a decline at 12–24 h with a subsequent increase at 48–72 h (Fig. 3). Increased phosphorylation of p38MAPK was observed at 1 h after ammonia treatment, which generally remained elevated at this level at all time points (p < 0.05 vs control) (Fig. 4). In contrast, the peak increase in phosphorylation of JNK1/2/3 was observed at 3 h, followed by a subsequent decline (Fig. 5). Total ERK1/2, p38MAPK, and JNK1/2/3 expression did not change at any time point during ammonia treatment. These results demonstrate a differential temporal pattern of phosphorylation of MAPKs in cultures exposed to ammonia.

Time course of ERK activation by ammonia in cultured astrocytes. A, Western blots reveal no significant change in total ERK1/2 level when cultures were exposed to 5 mm NH4Cl. A sustained increase in the level of phospho-ERK1/2 was observed after ammonia treatment. UO126, an ERK1/2 inhibitor, prevented the NH4Cl-induced ERK1/2 phosphorylation. B, C, Quantitation of NH4Cl-induced phospho-ERK1 (B) and phospho-ERK2 (C) levels in astrocytes with and without the ERK1/2 inhibitor. Phospho-ERK1/2 levels are normalized against α-tubulin. ANOVA, n = 4. ∗p < 0.05 versus control; †p < 0.05 versus phospho-ERK. UO, UO126. Error bars represent mean ± SEM.

Time course of phospho-p38MAPK phosphorylation by ammonia in cultured astrocytes. A, Western blot analysis reveals no significant change in total p38MAPK level when cultures were exposed to 5 mm NH4Cl. A significant increase in the level of phospho-p38MAPK was identified when cultures were exposed to NH4Cl. SB239063, a p38MAPK inhibitor, prevented the ammonia-induced p38MAPK phosphorylation. B, Quantitation of ammonia-induced phospho-p38MAPK levels in the astrocytes. Phospho-p38MAPK levels are normalized against α-tubulin. ANOVA, n = 4.∗p < 0.05 versus control; †p < 0.05 versus phospho-p38MAPK. Error bars represent mean ± SEM.

A, Time course of JNK1/2/3 phosphorylation by ammonia in cultured astrocytes. Western blot analysis reveals no significant change in total JNK1/2/3 level when cultures were exposed to 5 mm NH4Cl. A significant increase in the levels of phospho-JNK1/2/3 was observed after ammonia treatment. SP600125, a JNK inhibitor, partially prevented the ammonia-induced JNK1 phosphorylation but not JNK2 or JNK3. B–D, Quantitation of ammonia-induced changes in phospho-JNK1 (B), phospho-JNK2 (C), and phospho-JNK3 (D). Phospho-JNK1/2/3 levels are normalized against α-tubulin. ANOVA, n = 5. ∗p < 0.05 versus control; †p < 0.05 versus NH4Cl. Error bars represent mean ± SEM.
Antioxidants and NOS inhibition attenuate ammonia-induced MAPK phosphorylation
To determine whether oxidative stress contributes to ammonia-induced MAPK phosphorylation in cultured astrocytes, we examined the effect of various antioxidants, including SOD (25 and 50 U/ml), catalase (250, 500, and 1000 U/ml), vitamin E (100 and 250 μm), PBN (50, 100, and 250 μm), and the NOS inhibitor l-NAME (100, 250, and 500 μm) on ERK1/2, JNK1/2/3, and p38MAPK phosphorylation. Treatment with SOD and l-NAME, but not the other antioxidants, significantly attenuated ammonia-induced ERK1/2 phosphorylation at the dose of (25 U/ml and 250 μm, respectively). All antioxidants, SOD (20 U/ml), catalase (250 U/ml), vitamin E (100 μm), as well as l-NAME (250 μm) significantly decreased phosphorylation of both JNK1/2/3 and p38MAPK (Fig. 6). Astrocytes treated with PBN had no effect on ammonia-induced ERK1/2, JNK1/2/3, and p38MAPK phosphorylation.

Antioxidants prevent ammonia-induced MAPK phosphorylation. Cultured astrocytes exposed to 5 mm NH4Cl significantly increased phosphorylation of ERK1/2 (6 h), JNK1/2/3 (3 h), and p38MAPK (1 h). Treatment with SOD (25 U/ml) and l-NAME (250 μm) (but not other antioxidants) significantly attenuated ammonia-induced ERK1 phosphorylation. SOD also inhibited the ERK2 phosphorylation. All antioxidants, as well as l-NAME, significantly decreased the phosphorylation level of both JNK1/2/3 and p38MAPK. ANOVA, n = 3. ∗p < 0.05 versus control; †p < 0.05 versus NH4Cl. Vit-E, Vitamin E. Error bars represent mean ± SEM.
MAPK inhibitors decrease ammonia-induced phosphorylation of MAPK and astrocyte swelling
We then examined whether MAPK phosphorylation contributes to ammonia-induced astrocyte swelling. Experiments to test the ability of various agents to inhibit phosphorylation of MAPKs in astrocytes were conducted with MAPK inhibitors. Cultures were treated with the following: 1–100 μm 1, 4-diamino-2, 3-dicyano-1, 4-bis (2-aminophenylthio) butadiene (UO126), an inhibitor of MAP kinase kinase 1/2 (MEK1/2), the upstream kinase that activates ERK1/2; trans-1-(4-hydroxyclyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxypyrimidin-4-yl)imidazole (SB 239063; 1-100 μm, an inhibitor of p38MAPK), and anthra[1,9-cd]pyrazol-6(2H)-one (SP600125; 1-100 μm, an inhibitor of JNK1/2/3), with and without ammonia. When compared with cells treated with ammonia, UO126 (10 μm) prevented the phosphorylation of both ERK1/2 (Fig. 3). Similarly, cells treated with SB239063 (10 μm) prevented ammonia-induced p38MAPK phosphorylation (Fig. 4). Although both JNK1 and JNK2 were phosphorylated in response to ammonia, the JNK inhibitor SP600125 (1 μm) only partially diminished the phosphorylation of JNK1, and it did not prevent phosphorylation of JNK2 or JNK3 (Fig. 5). Doses >1 μm either had no additional effect on JNK phosphorylation (5 μm) or resulted in astrocyte death (100 μm). Cultures treated with vehicle had no effect on MAPK phosphorylation. Doses that prevented MAPK phosphorylation from these experiments were chosen for subsequent studies (10 μm UO126; 10 μm SB239063; 1 μm SP600125), unless noted otherwise.
Cultures were treated with the above-mentioned MAPK inhibitors with and without 5 mm NH4Cl. Astrocytes treated with 5 mm NH4Cl for 48 h increased cell volume (45% over control; p < 0.05 vs control). Treatment of cultures with UO126 (10 μm) and SB239063 (10 μm) completely prevented ammonia-induced astrocyte swelling (p < 0.05 vs NH4Cl). In contrast, the JNK inhibitor SP600125 (1 μm) only partially prevented ammonia-induced astrocyte swelling (56%; p < 0.01 compared with NH4Cl alone). SP600125 (5 μm) did not inhibit swelling (Fig. 7).

MAPK inhibitors prevent ammonia-induced astrocyte swelling. Cultured astrocytes exposed to 5 mm NH4Cl significantly increased cell swelling (2 d). Cotreatment with SB239063 and UO126, p38MAPK and ERK inhibitors, respectively, attenuated ammonia-induced astrocyte swelling. SP600125, a JNK inhibitor, partially diminished astrocyte swelling. ANOVA, n = 4. ∗p < 0.05 versus control; †p < 0.05 versus ammonia. Posttreatment of MAPK inhibitors immediately after the peak phosphorylation of MAPKs by ammonia (UO126, 6 h; SB239063, 1 h; SP600125, 3 h) did not reduce cell swelling. UO, UO126; SB, SB239063; SP, SP600125. Error bars represent mean ± SEM.
To determine the significance of the early phosphorylation of MAPKs in ammonia-induced swelling, we added MAPK inhibitors immediately after the occurrence of the peak phosphorylation (ERK1/2 at 6 h, p38MAPK at 1 h, and JNK 1/2/3 at 3 h). The results showed that delayed addition of these inhibitors did not reduce astrocyte swelling. Furthermore, a lower dose of ammonia (2 mm) that did not cause significant astrocyte swelling also did not result in phosphorylation of MAPKs at early time points (1, 3, and 6 h; data not shown).
UO126, in addition to inhibiting ERK1/2, also inhibits p90 ribosomal S6 kinase (RSK) (Zhang et al., 2002). To rule out the effect of UO126 on RSK, we used rapamycin, an inhibitor of RSK. Rapamycin (1–50 nm) did not prevent ammonia-induced astrocyte swelling (data not shown), indicating that the effect of UO126 was not through inhibition of RSK.
MAPK inhibitors diminish ammonia-induced glutamate uptake inhibition
To examine whether MAPK inhibitors reverse ammonia-induced glutamate uptake inhibition, astrocyte cultures were treated with the above MAPK inhibitors, with or without NH4Cl (5 mm), for 3 d. Cultures treated with NH4Cl significantly inhibited d-aspartate uptake (46%, compared with control; p < 0.05). Treatment with SB239063 (10 μm) and SP600125 (1 μm) significantly reduced ammonia-induced d-aspartate uptake inhibition (88 and 79%, respectively; p < 0.05 compared with NH4Cl), whereas the MEK1/2 inhibitor UO126 did not (Fig. 8). Cultures treated with higher doses of UO126 (20, 50, or 100 μm), as well as vehicle, also had no effect on ammonia-induced glutamate uptake inhibition (data not shown). As with astrocyte swelling, we observed that addition of MAPK inhibitors immediately after the occurrence of the peak phosphorylation did not improve glutamate uptake inhibition. Likewise, a lower dose of ammonia (2 mm) did not cause glutamate uptake inhibition (data not shown).

MAPK inhibitors reduce ammonia-induced glutamate uptake inhibition in cultured astrocytes. A 45% decrease in glutamate uptake was observed when exposed to 5 mm NH4Cl (3 d). Cotreatment with MAPK inhibitors SB239063 (p38MAPK) and SP600125 (JNK1/2/3), but not UO126 (ERK1/2), significantly diminished ammonia-induced glutamate uptake inhibition. ANOVA, n = 5. ∗p < 0.05 versus control; †p < 0.05 versus ammonia. Posttreatment of MAPK inhibitors, as in Figure 7, did not reduce ammonia-induced glutamate uptake inhibition. UO, UO126; SB, SB239063; SP, SP600125. Error bars represent mean ± SEM.
MAPK inhibitors reverse ammonia-induced decrease in GLAST protein levels
We also determined whether the protective action of MAPK blockers on glutamate uptake inhibition was caused by an effect on the glutamate transporter. Western blot analysis showed a significant decrease in GLAST protein in cultures treated with 5 mm NH4Cl alone (38.8%, compared with control; p < 0.05). Although the decrease in GLAST is in agreement with our previous report showing a decrease in GLAST mRNA in cultured astrocytes exposed to ammonia (Zhou and Norenberg, 1999), the decline may also be attributable to, in part, increased protein degradation. Treatment with SB239063 and SP600125 significantly reversed the ammonia-induced decline in GLAST level (72 and 63%, respectively; p < 0.05, compared with NH4Cl), whereas UO126 had no inhibitory effect (Fig. 9).

A, A representative Western blot showing a 38% decrease in GLAST protein levels in astrocyte cultures exposed to 5 mm NH4Cl (3 d). Treatment with MAPK inhibitors SB239063 (p38MAPK) and SP600125 (JNK1/2/3), but not UO126 (ERK1/2), significantly abrogated the ammonia-induced decline in GLAST. B, Quantitation of GLAST levels normalized to α-tubulin. ANOVA, n = 3. ∗p < 0.05 versus control; †p < 0.05 versus ammonia. UO, UO126; SB, SB239063; SP, SP600125. Error bars represent mean ± SEM.
Discussion
This study demonstrates that key astroglial defects associated with HE such as cell swelling and glutamate uptake impairment can be attenuated by the use of antioxidants and NOS inhibition. Our studies also show that ammonia significantly enhances the phosphorylation of MAPKs, and that MAPK inhibitors reduce the severity of ammonia-induced astrocyte swelling and glutamate uptake inhibition. These findings lend support for a key role of oxidative stress and phosphorylation of MAPKs in the mechanism of ammonia neurotoxicity.
Oxidative/nitrosative stress in ammonia-induced astrocyte swelling
Oxidative/nitrosative stress is an evolving concept in the pathogenesis of HE and ammonia neurotoxicity. There is evidence of lipid peroxidation in the brain of hyperammonemic mice (Kosenko et al., 1999). Ammonia increases superoxide production and decreases the activity of the antioxidant enzymes glutathione peroxidase, SOD, and catalase (Kosenko et al., 1997, 1999). Additionally, increased brain nitric oxide production is reported in portacaval-shunted rats after ammonia infusion (Master et al., 1999). Ammonia also increases free radical production, including nitric oxide, in cultured astrocytes (Murthy et al., 2001). Together, these studies strongly suggest the involvement of oxidative/nitrosative stress in ammonia neurotoxicity.
Oxidative stress has been proposed as an important factor in the development of cell swelling. Free radicals have been shown to cause cell swelling in brain slices (Chan et al., 1982, 1989; Brahma et al., 2000), as well as in cultured astrocytes (Chan et al., 1989; Cubells et al., 1994; Staub et al., 1994; Sharma, 1996; C. J. Chen et al., 2000). In keeping with these findings, our study shows that SOD, catalase, and vitamin E all significantly blocked astrocyte swelling caused by ammonia. l-NAME also significantly reduced ammonia-induced astrocyte swelling, consistent with findings reported by Zielinska et al. (2003), who showed that l-NAME prevented ammonia-induced swelling in brain slices.
Because l-NAME robustly attenuated cell swelling, it is likely that NO plays a major role in the ammonia-induced astrocyte swelling. In support of a role for NO in HE, NOS activity and gene expression have been shown to be elevated in experimental models of HE (Rao et al., 1997), and there is increased brain NO production in portacaval-shunted rats given ammonia infusions (Master et al., 1999). In addition, we found that the NO donors, SNAP and SIN-1, increased astrocyte swelling. Additional support is provided by Schliess et al. (2002), who showed protein tyrosine nitration after ammonia treatment in vivo and in vitro. Collectively, these studies support a key role of oxidative and nitrosative stress in ammonia-induced astrocyte swelling.
Antioxidants and NOS inhibition attenuate ammonia-induced inhibition of glutamate uptake
Another important aspect of HE and hyperammonemia includes alterations in glutamatergic neurotransmission (Butterworth, 2001), likely because of failure of astrocytes to take up extracellular glutamate. In support of this, it has been shown that cultured astrocytes exposed to pathophysiological concentration of ammonia suppresses the high affinity uptake of glutamate in cultured astrocytes (Bender and Norenberg, 1996) and decreases GLAST mRNA levels in rat brain with acute liver failure (Knecht et al., 1997). Oxidative stress has been shown to inhibit glutamate uptake in cultured astrocytes (Volterra et al., 1994a,b; Y. Chen et al., 2000) and in rat brain synaptosomes (Berman and Hastings, 1997). Allen et al. (2001) showed that methylmercury-mediated inhibition of 3H-d-aspartate transport was reversed by the antioxidant catalase in cultured astrocytes. Volterra et al. (1994a) reported that xanthine plus xanthine oxidase-induced inhibition of glutamate transport was prevented by addition of SOD. Our findings are consistent with these observations in that the antioxidants SOD and vitamin E significantly attenuated the ammonia-induced inhibition of astrocytic glutamate uptake.
Cultures exposed to SNAP and SIN-1 significantly decreased glutamate uptake. Furthermore, l-NAME robustly prevented glutamate uptake inhibition, indicating an important role of NO in the mechanism of ammonia-induced astrocyte dysfunction. These results suggest that ammonia-induced inhibition of glutamate uptake, like astrocyte swelling, is mediated by oxidative/nitrosative stress.
MAPK phosphorylation by ammonia
The above-mentioned results suggest that oxidative stress after ammonia exposure may be involved in the mechanism of ammonia-induced cell swelling and inhibition of glutamate uptake. However, the mechanism(s) by which oxidative stress results in these astroglial changes is still unknown. One important molecular consequence of oxidative stress is the activation of intracellular signaling cascades. Numerous studies indicate that reactive oxygen species (ROS) activate protein kinases (Remacle et al., 1995; Sen and Packer, 1996; Suzuki et al., 1997; Czaja et al., 2003). In particular, p38MAPK, JNK, and ERK1/2 are phosphorylated by exposure to exogenous H2O2, and this phenomenon can be inhibited by treatment with antioxidants (Abe et al., 1996; Guyton et al., 1996; Lo and Cruz, 1996). MAPK signaling cascades have also been shown to be activated in cultured astrocytes in response to oxidant signaling (Luo and Roth, 2000; Mizuhashi et al., 2000; S. H. Chen et al., 2001; Lennon et al., 2002).
Our study shows that ammonia increased the phosphorylation of ERK1/2, p38MAPK, and JNK. Similar findings were also observed by Schliess et al. (2002), who reported that cultured astrocytes exposed to ammonia increased the phosphorylation of ERK1 and p38MAPK at 1 d. In addition, we now show that ammonia increases the phosphorylation of JNK1/2/3. ERK1/2 and p38MAPK were phosphorylated in a biphasic manner, although in contrast, JNK was only phosphorylated at earlier time points. The reason for the different time course of MAPK phosphorylation by ammonia is unknown. It is possible that the ERK1/2, p38MAPK, and JNK1/2/3 might be differentially phosphorylated by upstream MAPK kinases in response to ROS generation (Fukunaga et al., 2000; Nègre-Aminou et al., 2002; Ding et al., 2002). For example, the ERK1/2 may be activated by the Ras/Raf pathway upstream of MEK1/2, whereas the JNK and p38MAPK may be activated by Rac, an upstream kinase of MEK4/7 and MEK3/6 (Z. Chen et al., 2001). Therefore, differently activated Ras and Rac pathways by ammonia-induced oxidative stress may be responsible for the differential pattern of ERK1/2, p38MAPK, and JNK phosphorylation. One possible explanation for the biphasic phosphorylation of ERK1/2 is that the first peak of ERK1/2 phosphorylation might be attributable to ROS generation (Canals et al., 2003; Clausen et al., 2004). The second peak might be attributable to the inactivation of mitogen-activated protein kinase phosphatase, which might occur in the second phase of ERK1/2 phosphorylation (Berlett and Stadtman, 1997; Camps et al., 1998; Muda et al., 1998). Although the pattern of activation of these kinases is different and its significance is unclear, as mentioned below, the early phosphorylation of MAPKs by ammonia appear to be critical for the ammonia-induced astrocyte dysfunction.
Antioxidants and NOS inhibition prevent ammonia-induced MAPK phosphorylation
Treatment with SOD but not vitamin E or catalase, significantly diminished the ammonia-induced phosphorylation of ERK1/2. Additionally, l-NAME significantly attenuated the cell swelling. All antioxidants tested in our study, including vitamin E and catalase, as well as l-NAME significantly diminished both JNK and p38MAPK phosphorylation. It is unclear why SOD but not vitamin E prevented ERK1/2 phosphorylation, because both antioxidants can scavenge superoxide. Although vitamin E can scavenge both superoxide and NO, SOD and l-NAME are more potent in scavenging superoxide and NO, respectively (Osakada et al., 2003). It is possible that high levels of ammonia-induced superoxide and NO may generate peroxynitrite and that the effect of peroxynitrite may not be diminished by vitamin E. Thus, whereas ammonia increases phosphorylation of all these kinases, the free radical species involved in activating these various kinases may be different.
It should be noted that SOD is a large molecular weight protein, which is not freely permeable across the plasma membrane. Nevertheless, in our study, SOD diminished the astrocyte dysfunction produced by ammonia likely by quenching the superoxide radicals, which diffused out of the cell (Terada, 1996; Tolias et al., 1999; Murthy et al., 2001).
MAPK inhibitors diminish astrocyte swelling
Our results strongly implicate ERK1/2 and p38MAPK as important kinases involved in ammonia-induced swelling, because such swelling was completely inhibited by UO126 (inhibitor of MEK1/2, the upstream kinase specific for ERK1/2) and SB239063 (inhibitor of p38MAPK). JNK inhibition by SP600125 only partially prevented ammonia-induced astrocyte swelling. However, SP600125 only inhibited the phosphorylation of JNK1 but not of JNK2/3. Whether the additional inhibition of JNK 2/3 is also required to prevent astrocyte swelling is not known. Importantly, delayed addition of these inhibitors (after the peak phosphorylation of MAPKs) did not reduce astrocyte swelling, suggesting that early phosphorylation events induced by ammonia, and not the prolonged phosphorylation, is critical for astrocyte swelling.
Phosphorylation of MAPKs is known to occur after hypo-osmotic swelling in cultured astrocytes (Crépel et al., 1998), human intestine 407 cells (Tilly et al., 1996), H4IIE rat hepatoma cells (Schliess et al., 1995), and isolated rat hepatocytes (Noe et al., 1996; Kim et al., 2000). These studies suggest that cell swelling may act as a stress signal to induce the phosphorylation of MAPKs. Our observations, in contrast, indicate that MAPK phosphorylation occurs before ammonia-induced swelling and that blockade of MAPKs diminishes astrocyte swelling. It appears that the role of kinases in the mediation of osmotic swelling and ammonia-induced swelling are different. However, it is important to note the difference in temporal sequence of swelling in these two conditions, with hypo-osmotic challenge resulting in almost immediate swelling, whereas with ammonia, the swelling is only first detected at 12 h. The difference in rates of swelling may contribute to the pattern of MAPK phosphorylation.
Although it is possible that cell swelling induced by ammonia, rather than ammonia per se, may be the cause of MAPK phosphorylation, the increase in p-JNK occurred before the onset of swelling, and thus the possibility that swelling is responsible for JNK phosphorylation can be discounted. As for ERK and p38MAPK, whereas their peak phosphorylation occurred before the onset of swelling, the activation of these two kinases persisted during the period of swelling. Thus, the possibility that swelling may have contributed to ERK and p38MAPK activation cannot be completely ruled out.
MAPK inhibitors reduce glutamate uptake inhibition by ammonia
Our results showed that ammonia significantly diminished glutamate uptake inhibition, which was prevented by inhibitors of p38MAPK and JNK1/2/3 probably by preventing a decline in GLAST levels. Inhibition of ERK1/2 did not affect glutamate uptake inhibition. Abe and Misawa (2003) reported that β-amyloid-induced promotion of extracellular glutamate clearance (intracellular uptake) was enhanced in cultured astrocytes by the ERK1/2 inhibitor UO126. In both our study and theirs, an increase in ERK1/2 phosphorylation was detected after ammonia and β-amyloid treatment, respectively. However, inhibiting ERK1/2 in our study had no effect on glutamate uptake inhibition, suggesting that the role of ERK1/2 in glutamate transport is likely stress specific. Although p38MAPK is involved in both swelling and glutamate uptake inhibition, ERK1/2 phosphorylation appears to mediate astrocyte swelling but not glutamate uptake inhibition. As with swelling, delayed treatment with inhibitors did not improve glutamate uptake inhibition.
Mechanisms underlying the differential effect of kinases on astrocyte dysfunction are not clear. It is known that activation of the Ras-Raf pathway leads to activation of MEK1/2 and subsequently of ERK1/2. But activation of Ras also activates MEK3 (Efimova et al., 2002), which then activates p38MAPK. In such a case, blockade of only ERK1/2 by UO126 may not be sufficient because of p38MAPK activation by the Ras-MEK3 pathway.
Inhibition of JNK almost completely prevented glutamate uptake inhibition but only partially prevented cell swelling. Whether the partial inhibition of swelling is attributable to partial inhibition of JNK is not clear, because SP600125 prevented phosphorylation of JNK1 but not JNK2/3. Nevertheless, blockade of JNK1 by SP600125 prevented glutamate uptake inhibition by ammonia, indicating an important role of JNK1 but not JNK2/3 in glutamate uptake inhibition. These results suggest that although ammonia activates all of these MAPKs, the kinases differentially contribute to astrocytic dysfunctions.
Our study links the early generation of ROS with MAPK phosphorylation and the subsequent development of astrocyte dysfunction. Although there is a time gap between kinase phosphorylation and astrocyte dysfunction, the reason for this gap is not clear. A likely explanation is that the MAPKs activate downstream targets, including kinases and transcription factors (Z. Chen et al., 2001), to eventually bring about the observed astroglial abnormalities.
In summary, our study shows that ammonia-induced astrocyte swelling and inhibition of glutamate uptake is mediated through oxidative and/or nitrosative stress-induced phosphorylation of MAPKs. Antioxidants and inhibition of MAPKs blocked or attenuated these major aspects of ammonia neurotoxicity. Treatment aimed at diminishing oxidative stress or modulating MAPK phosphorylation might represent useful therapeutic approaches in HE and other hyperammonemic states.
Footnotes
-
This work was supported by a Merit Review from the Department of Veterans Affairs and by National Institutes of Health Grant DK063311. A.R.J. is supported by the American Association for the Study of Liver Disease/American Liver Foundation Grant. We thank Dr. Chang Kim and Alina Fernandez-Revuelta for the preparation of cell cultures. We also thank Jan Kelly for assistance with Western blot quantitation.
-
↵†Deceased Dec. 1, 2003.
- Correspondence should be addressed to Dr. Michael D. Norenberg, Department of Pathology (D-33), University of Miami School of Medicine, P.O. Box 016960, Miami, FL 33101. Email: mnorenbe{at}med.miami.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}