

Abstract
Synaptic transmission between hippocampal mossy fibers (MFs) and CA3 pyramidal cells exhibits remarkable use-dependent plasticity. The underlying presynaptic mechanisms, however, remain poorly understood. Here, we have used fluorescent Ca2+ indicators Fluo-4, Fluo-5F, and Oregon Green BAPTA-1 to investigate Ca2+ dynamics in individual giant MF boutons (MFBs) in area CA3 traced from the somata of granule cells held in whole-cell mode. In an individual MFB, a single action potential induces a brief peak of free Ca2+ (estimated in the range of 8–9 μm) followed by an elevation to ∼320 nm, which slowly decays to its resting level of ∼110 nm. Changes in the somatic membrane potential influence presynaptic Ca2+ entry at proximal MFBs in the hilus. This influence decays with distance along the axon, with a length constant of ∼200 μm. In giant MFBs in CA3, progressive saturation of endogenous Ca2+ buffers during repetitive spiking amplifies rapid Ca2+ peaks and the residual Ca2+ severalfold, suggesting a causal link to synaptic facilitation. We find that internal Ca2+ stores contribute to maintaining the low resting Ca2+ providing ∼22% of the buffering/extrusion capacity of giant MFBs. Rapid Ca2+ release from stores represents up to 20% of the presynaptic Ca2+ transient evoked by a brief train of action potentials. The results identify the main components of presynaptic Ca2+ dynamics at this important cortical synapse.
Introduction
Mechanisms of Ca2+-dependent neurotransmitter release are central to understanding synaptic function. Studies in giant calyceal terminals in the brainstem have provided critical insights into the underlying molecular machinery (Schneggenburger et al., 2002; von Gersdorff and Borst, 2002; Meinrenken et al., 2003). Similar advances with regard to cortical synapses have been made possible in large part by single-synapse imaging in the neocortex (Koester and Sakmann, 2000; Koester and Johnston, 2005) and whole-terminal recordings from giant mossy fiber (MF) boutons (MFBs) in the hippocampus (Geiger and Jonas, 2000; Engel and Jonas, 2005). Synapses formed by giant MFBs on CA3 pyramidal cells have also been intensely studied because they exhibit unusual short-term facilitation (Griffith, 1990) and an NMDA receptor-independent form of long-term potentiation (Zalutsky and Nicoll, 1990).
Several candidate mechanisms of short-term facilitation at these synapses have been proposed. Recordings from giant MFBs show that action potentials (APs) broaden during repetitive spiking, thus boosting presynaptic Ca2+ influx (Geiger and Jonas, 2000; Bischofberger et al., 2002). However, at moderate frequencies (10–30 Hz), this enhancement, in terms of either Ca2+ current amplitude or transferred charge, amounts to only <10% during the first 10 spikes (Geiger and Jonas, 2000). A 10% increase in presynaptic Ca2+ entry is expected to augment postsynaptic responses in CA3 pyramidal cells less than to the second power (Geiger and Jonas, 2000; Blatow et al., 2003; Mori-Kawakami et al., 2003) or by <21%. AP broadening alone thus cannot fully account for the profound synaptic facilitation routinely observed at similar frequencies.
Another candidate mechanism involves progressive saturation of endogenous Ca2+ buffers (Rozov et al., 2001). In young rodents, increasing the MF buffering capacity with Ca2+ chelators EGTA (high concentration) or BAPTA reduces short-term facilitation or paired-pulse ratios of postsynaptic responses (Regehr et al., 1994; Blatow et al., 2003; Mori-Kawakami et al., 2003). However, the effect is small in mature animals (Mori-Kawakami et al., 2003) and the residual presynaptic Ca2+ facilitates faster than do synaptic responses (Regehr et al., 1994). Furthermore, single-cell measurements of the main MF endogenous Ca2+ buffer calbindin-28K suggest that buffer saturation cannot explain strong facilitation of presynaptic Ca2+ (Muller et al., 2005).
An additional mechanism of short-term enhancement relies on presynaptic kainate receptors (Contractor et al., 2001; Lauri et al., 2001; Schmitz et al., 2001; Kamiya et al., 2002), which have been implicated in triggering Ca2+ release from Ca2+ stores (Liang et al., 2002; Lauri et al., 2003). However, AP-driven Ca2+ responses recorded in multiple MFs appear insensitive to Ca2+ store blockade (Carter et al., 2002; Breustedt and Schmitz, 2004).
In summary, the machinery of presynaptic Ca2+ signaling at MF synapses remains poorly understood. Because optical recordings from tissue-loaded MFs do not report resting Ca2+ levels and may incorporate signals from unidentified cellular structures, testing at the level of single synapses is preferable (Jackson and Redman, 2003; Ruiz et al., 2003). In addition, integrity of the presynaptic cell is important for quantitative evaluation of presynaptic Ca2+ signaling. Here, we combine two-photon microscopy with an improved slicing method, electrophysiology, and kinetic modeling to evaluate the role of somatic voltage, Ca2+ buffering, and Ca2+ stores in shaping Ca2+ dynamics in giant MFBs in area CA3 supplied by axons of intact granule cells.
Materials and Methods
Preparation and electrophysiology.
Acute 300 μm hippocampal slices from 3- to 4-week-old male Sprague Dawley rats were transferred to a recording submersion-type chamber (Scientific Systems Design, Montclair, NJ), superfused with the following (in mm): 124 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, and 10 glucose, and bubbled with 95:5 of O2/CO2. The slice cutting (close to the parasagittal plane) is detailed in Results. Internal solution included the following (in mm): 150 K methanesulfonate, 5 KCl, 10 HEPES, 3 MgATP2, and 0.4 GTP, and also included fluorophores as indicated. Unless specified otherwise, granule cells were held at −80 mV. Orthodromic action potentials or escape currents recorded in current- or voltage-clamp mode, respectively, were evoked by 2 ms command voltage pulses (single, 5, or 50, at 20 Hz, as indicated). Alternatively, 100 μs electrical stimuli were applied to stratum lucidum with a bipolar electrode. Recording sweeps (normally 500 ms long) were collected at a 5 kHz rate in 30 s or 1 min intervals using LabView (National Instruments, Austin, TX). Routinely, experiments were performed in the presence of 5 μm CGP-52432, 50 μm APV, and 100 μm picrotoxin at room temperature. Receptor antagonists were purchased from Tocris Cookson (Bristol, UK), and fluorescent probes were purchased from Invitrogen (San Diego, CA). All animal-handling procedures followed current United Kingdom regulations.
Two-photon excitation fluorescence imaging.
Imaging experiments were performed using a multiphoton installation comprising a Radiance 2000 imaging system (Zeiss, Oberkochen, Germany) optically linked to a femtosecond laser MaiTai (SpectraPhysics, Mountain View, CA) and integrated with a single-cell electrophysiology setup (Rusakov and Fine, 2003). Granule cells were held in whole-cell mode and loaded with two fluorophores, a morphological tracer Alexa Fluor 594 (20 μm) and a Ca2+ indicator [Fluo-4, Fluo-5F, or Oregon Green BAPTA-1 (OGB-1), as specified]. In granule cells with intact axons (see Results) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), 15–20 min were initially allowed for indicator equilibration before switching the system into fluorescence mode to trace the axon into the stratum lucidum. Fluorophores were excited in two-photon mode at 810 nm, with the laser power optimized for emission detection at different depths in the slice.
The axon was followed from the soma into stratum lucidum using frame mode scanning (256 × 256 pixels, 500 Hz; the number of taken frames was kept small to minimize phototoxic damage) and the system was focused on a giant MFB identified by its distinct morphology, at the maximum optical resolution (∼0.2 μm; digital capture, 70 nm per pixel). Recording started when the baseline fluorescence in both channels was stable (∼1 h later) (see Results) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material); the experiment lasted for up to 3–4 h and dye equilibration was routinely controlled post hoc, by comparing the resting fluorescence of Alexa Fluor 594 at the end of the experiment with that recorded 1–1.5 h earlier. We conducted two additional tests to verify that Ca2+ indicators are equilibrated along the axon and are not extruded from the cells appreciably with time. In the first test, we carefully pulled out the patch pipette after initial dye equilibration. The seal was confirmed by the fact that we obtained outside-out patches as a result and that cells could respond to extracellular stimulation by generating action potential-dependent Ca2+ transients. The subsequent recordings indicated no detectable loss of the resting fluorescence F over 100–200 min (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In the second test, we measured the baseline fluorescence in the axonal regions at different distances from the soma and found no spatial gradient (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The results of both tests argued against any significant loss of fluorescence indicators from the axon.
Fluorescence responses were recorded in line-scan mode at 500 Hz (500 or 1000 ms sweeps; intersweep interval, 30 s or 1 min) and stored for off-line analysis. The Ca2+-dependent fluorescence response ΔF/F (integrated over the visible MFB width) was routinely calculated as follows: (Fpost − Fpre)/(Fpre − F0). The values of Fpre and Fpost stand for the line-scan fluorescence averaged over, respectively, 100 ms before the first spike and either 50 ms in the case of single-response amplitude measurements or 250 ms in the case of five-response amplitude measurements (20 Hz train of five APs) after the first spike onset. F0 denotes the background fluorescence measured outside any cell structures filled with the indicator. Because special care was taken to avoid escape of the indicator from the pipette, and because the site of imaging was hundreds of micrometers away from the pipette tip, F0 was likely to represent the photomultiplier tube dark current. Image analyses were performed on stacks of stored line-scan images using a set of custom NIH Image macros. False color tables and averaged images were used for illustration purposes but the original (gray level) pixel brightness values in each line-scan image were used for the quantitative analysis. In most experiments, we reconstructed the axon trajectory using a collage of high-resolution Kalman-filtered z-stacks 15–20 μm deep. In total, we obtained full reconstruction of 43 axons, with an average distance between the recorded MFB and the soma of 686 ± 38 μm. Throughout the experiments, we observed no failures of spike-driven Ca2+ signals propagating along the main axonal trunk including giant MFBs. This, however, does not rule out the possibility that propagation could fail at higher spiking frequencies and/or in thin axon collaterals.
The two-photon excitation probability profile is proportional to the squared illumination light intensity I2p2 (Zipfel and Webb, 2001):  where the canonical coordinates u = 4ksin2(α/2)z and v = ksin(α)r represent the axial distance z; the radial distance r; the numerical aperture of the objective, NA = sin(α) = 0.9; and wave number k = n(2π/λ) (n = 1.33 is the medium refraction index and λ = 810 nm is the wavelength); J0 denotes zero-order Bessel’s function of the first kind. This theoretical function, however, represents the lower-limit estimate of the excitation profile: in reality, optical aberrations and imperfect alignment of the experimental optical system are likely to increase the spread of excitation. Similar considerations apply to the emission path. We therefore obtained an estimate of the excitation–emission profile by recording the point-spread function (PSF) of the system using 0.17 μm fluorescent beads (PS-Speck Microscope Point Source kit; Invitrogen) as illustrated below.
where the canonical coordinates u = 4ksin2(α/2)z and v = ksin(α)r represent the axial distance z; the radial distance r; the numerical aperture of the objective, NA = sin(α) = 0.9; and wave number k = n(2π/λ) (n = 1.33 is the medium refraction index and λ = 810 nm is the wavelength); J0 denotes zero-order Bessel’s function of the first kind. This theoretical function, however, represents the lower-limit estimate of the excitation profile: in reality, optical aberrations and imperfect alignment of the experimental optical system are likely to increase the spread of excitation. Similar considerations apply to the emission path. We therefore obtained an estimate of the excitation–emission profile by recording the point-spread function (PSF) of the system using 0.17 μm fluorescent beads (PS-Speck Microscope Point Source kit; Invitrogen) as illustrated below.
Estimating the resting concentration of presynaptic Ca2+.
Classically, the resting Ca2+ level ([Ca2+]rest) can be estimated from the following relationship:  where Kd is the Ca2+ indicator dissociation constant, and Fmax, F, and Fmin represent maximum, resting, and Ca2+-independent fluorescence of the indicator.
where Kd is the Ca2+ indicator dissociation constant, and Fmax, F, and Fmin represent maximum, resting, and Ca2+-independent fluorescence of the indicator.
However, the high dynamic range of Ca2+ indicators such as Fluo-4 implies that the resting fluorescence F could be comparable with the detection threshold of the system. In this case, the useful fluorescence signal could blend with the background noise in a nonlinear manner, which makes it difficult to obtain reliable ratios ΔF/F or Fmax/F. This difficulty could be addressed by relating ΔF to the Ca2+-independent fluorescence R recorded in the “red” (e.g., Alexa) channel (Oertner et al., 2002). We have modified this approach by diverting part of the Alexa Fluor 594 emission signal into the “green” Fluo-4 channel using a 560 nm dichroic mirror and a 700 nm short-pass filter. The use of the cross talk Alexa Fluor 594 signal, denoted here as Rct, was advantageous because this (1) elevated emission in the Fluo-4 channel well above the detection threshold of the system (see above) and (2) allowed both the Ca2+-dependent signal ΔF and Rct to be recorded through the same channel. (This method, however, is unlikely to be advantageous when the useful resting green fluorescence is substantially above the background noise.) The average ratio 〈Rct/R〉 was measured in separate experiments, by comparing emission in the two corresponding channels while imaging giant MFBs loaded with Alexa Fluor 594 only (see Results). In these terms, the recorded resting and maximum fluorescence (F and Fmax, respectively) will include a Ca2+-indepedent fluorescence fraction Rct + Fmin. Equation 2 can be therefore recast as follows:  In the case of the high dynamic range indicator Fluo-4, Fmin/Fmax ≈ 0 (Maravall et al., 2000; Oertner et al., 2002), implying that estimation of [Ca2+]rest simply requires experimental measurements of Rct and R, according to Equation 3. In the case of the lower dynamic range indicator OGB-1 (Kd ≈ 200 nm), this equation also includes the known value of Fmax/Fmin ≈ 6 (Maravall et al., 2000; Oertner et al., 2002; Jackson and Redman, 2003).
In the case of the high dynamic range indicator Fluo-4, Fmin/Fmax ≈ 0 (Maravall et al., 2000; Oertner et al., 2002), implying that estimation of [Ca2+]rest simply requires experimental measurements of Rct and R, according to Equation 3. In the case of the lower dynamic range indicator OGB-1 (Kd ≈ 200 nm), this equation also includes the known value of Fmax/Fmin ≈ 6 (Maravall et al., 2000; Oertner et al., 2002; Jackson and Redman, 2003).
Nonstationary presynaptic Ca2+ kinetics.
To translate the recorded fluorescence into the underlying Ca2+ kinetics, we used a single-compartment, multicomponent kinetic model (Helmchen et al., 1997; Neher, 1998; Jackson and Redman, 2003) in its nonstationary form, which requires no steady-state approximations (Sabatini and Regehr, 1998; Rusakov et al., 2005). Multicompartmental modeling was deemed unfeasible in the present study because (1) the Ca2+ fluorescence time course was indistinguishable between subregions inside a single bouton (the PSF of the system was too large to distinguish between such subregions) (see Fig. 4) and (2) Ca2+ exchange between multiple space partitions has little effect on the volume-average Ca2+ concentration transient (Sabatini and Regehr, 1998). Parameters of the model were constrained and cross-validated by (1) an independent estimate of [Ca2+]rest (see above), (2) an independent estimate of the endogenous buffer (calbindin-28K) concentration (Jackson and Redman, 2003; Muller et al., 2005), and (3) comparing the effects of different configurations of Ca2+ indicators/buffers on the recorded fluorescence signal versus the predicted kinetics.
The finite-difference kinetic scheme explicitly included the rate of Ca2+ entry, jCa, the binding–unbinding reactions with the endogenous buffer (calbindin-28K) B and Ca2+ indicator I, and the Ca2+ removal rate P, according to the following equation:  where brackets denote concentrations, CaI and CaB stand for the Ca-bound indicator and buffer, respectively, and the kinetic constants (top indices denote buffer B or indicator I; bottom indices indicate on and off constants) are summarized in Table 1. The mass conservation rules are as follows:
where brackets denote concentrations, CaI and CaB stand for the Ca-bound indicator and buffer, respectively, and the kinetic constants (top indices denote buffer B or indicator I; bottom indices indicate on and off constants) are summarized in Table 1. The mass conservation rules are as follows:  where index “tot” denotes total amount. The AP-evoked Ca2+ influx rate jCa followed the Gaussian (Sabatini and Regehr, 1998; Rusakov et al., 2004) as follows:
where index “tot” denotes total amount. The AP-evoked Ca2+ influx rate jCa followed the Gaussian (Sabatini and Regehr, 1998; Rusakov et al., 2004) as follows:  in line with experimental observations (Meinrenken et al., 2003), with the MFB spike half-width σ ≈ 0.5 ms (Bischofberger et al., 2002), onset at t0, and the time integral of Δ[Ca2+]tot reflecting total Ca2+ entry. Removal of Ca2+, which is a complex process involving diffusion, pumping out and/or sequestration was approximated by a first-order reaction (Jackson and Redman, 2003) (however, for discussion, see Matveev et al., 2004; Holcman et al., 2005) at the rate P([Ca2+] − [Ca2+]rest) (Eq. 4). The model, therefore, operated with only two adjustable (free) parameters: Δ[Ca2+]tot and P. However, varying either Δ[Ca2+]tot or P had virtually independent effects on the calculated amplitude (ΔF/F) and decay of fluorescent responses, respectively (see Results) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Each of the two parameters could be therefore constrained by a straightforward fitting procedure that would match the calculated and the experimental fluorescence, as discussed previously (Rusakov et al., 2005).
in line with experimental observations (Meinrenken et al., 2003), with the MFB spike half-width σ ≈ 0.5 ms (Bischofberger et al., 2002), onset at t0, and the time integral of Δ[Ca2+]tot reflecting total Ca2+ entry. Removal of Ca2+, which is a complex process involving diffusion, pumping out and/or sequestration was approximated by a first-order reaction (Jackson and Redman, 2003) (however, for discussion, see Matveev et al., 2004; Holcman et al., 2005) at the rate P([Ca2+] − [Ca2+]rest) (Eq. 4). The model, therefore, operated with only two adjustable (free) parameters: Δ[Ca2+]tot and P. However, varying either Δ[Ca2+]tot or P had virtually independent effects on the calculated amplitude (ΔF/F) and decay of fluorescent responses, respectively (see Results) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Each of the two parameters could be therefore constrained by a straightforward fitting procedure that would match the calculated and the experimental fluorescence, as discussed previously (Rusakov et al., 2005).
Adopted Ca2+ buffering constants in Ca2+ kinetics measurements (see Fig. 4)
Assessing calbindin-28K washout from the axon.
Gradual washout of endogenous buffers in whole-cell mode could alter presynaptic Ca2+ dynamics in the recorded cell (Rozov et al., 2001; Blatow et al., 2003). A previous study has estimated that washout of the endogenous Ca2+ buffer calbindin-28K from the granule cell soma occurs with a time constant of ∼9.7 min (Muller et al., 2005). To assess its washout rate at different regions of the axon, we used a multicompartmental diffusion model based on a detailed quantitative study of granule cell morphology (Claiborne et al., 1990). The modeled cell included the 19-μm-long/10-μm-wide elliptical soma and a 2000-μm-long/0.4-μm-thick axon. The average dendritic tree of granule cells (two primary dendrites, each ∼2.5 μm in diameter, giving rise to an arbor spreading over ∼300 μm with the total dendritic length of 3221 μm) (Claiborne et al., 1990) was represented by a single 300-μm-long trunk of the matching cross section (9.6 μm2) and total volume (2900 μm3). The patch pipette (tip diameter, 1.8 μm) provided a concentration clamp source linked to a somatic compartment. The initial calbindin concentration was 160 μm, and at t = 0, the concentration clamp was imposed through the pipette. Diffusion simulations were performed with the compartment size of 5 μm (464 compartments in total) using numerical algorithms described previously (Scimemi et al., 2004). The single unknown parameter, the effective diffusivity of calbindin was adjusted to obtain the best fit between the experimental data on somatic washout (Muller et al., 2005) and the computed calbindin concentration time course in the soma (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Under these conditions, washout of calbindin from the axonal regions where giant MFBs occur (the average distance from the soma, 686 ± 38 μm; n = 43 fully reconstructed axons) is predicted in the range of <5% within 2–3 h after break-in (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). This is consistent with the stable amplitude of recorded ΔF/F during that period (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Average data are shown as mean ± SEM; a t test was used for statistics.
Results
Tracing granule cell axons from the soma into the stratum lucidum
Obtaining connected granule cell–CA3 pyramidal cell pairs has been difficult in acute slices because of the intrinsically low probability of finding such connections through random sampling (Amaral et al., 1990). In addition, granule cell somata occur 200–300 μm outside the main MF projection plane (Tamamaki and Nojyo, 1991; Acsady et al., 1998) (Fig. 1A) and therefore are likely to be cut off during preparation of slices that preserve MFs in the stratum lucidum. To reduce the chances of cutting granule cell axons close to the soma, hippocampal slices were sectioned at an angle (close to the parasagittal direction) to the plane of the main MF projections (Fig. 1A,B). We then loaded granule cells, in whole-cell mode, with two fluorescence indicators (see Materials and Methods) and monitored APs evoked antidromically in the soma by extracellular stimulation of the stratum lucidum. Although virtually all monitored cells generated APs, only a small proportion (<10%) showed an AP latency exceeding 3–4 ms in response to the minimum strength stimuli sufficient to generate an AP (Fig. 1C). On subsequent visual inspection in a microscope, only these cells projected their axon into area CA3 (Fig. 1D) (Alexa Fluor 594 channel); the axons of cells with a short AP latency were truncated at <200 μm from the soma. These observations suggested a simple electrophysiological test to facilitate selection of cells with long axons before imaging. In the microscope, giant MFBs were identified in area CA3, >300 μm from the soma, by their distinctive large size (5–8 μm) and thin filopodial protrusions (Fig. 1D,E, image panels; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).

Two-photon excitation imaging of giant MFBs projected by intact granule cells to stratum lucidum. A, Three-dimensional (3D) representation of the granule cell axon trajectory (blue connected spheres) reconstructed from a pair of orthogonal projections reported previously [Acsady et al. (1998), their Fig. 2B]. Disconnected blues spheres, Dendritic ends; gray-shaded dots, two orthogonal projections. Dashed lines in the right panel, Preferred slicing position showing the cutting planes normal to the plane of image. B, Positioning of the cutting blade with respect to the hippocampus, a 3D view (the rat brain silhouette is shown for reference); note the oblique cutting angle. C, Characteristic traces of antidromic action potentials (escape currents) evoked by stimulation of stratum lucidum (inset diagram) in cut and uncut axons identified by microscopy. The black traces correspond to the minimum stimulus intensity sufficient to generate a spike (the intensity depended on the juxtaposition of the electrode and the axon and varied between slices), and the gray traces correspond to further increases in the stimulus strength. Note a dramatic difference in the poststimulus latency between cut and intact axons. D, An example of the reconstructed granule cell axon; Alexa Fluor 594 channel, whole-cell mode (see Materials and Methods); the dashed frames and image panels illustrate four giant MFBs (many thin filopodia are masked by the z-stack fusion image). E, A characteristic giant MFB (for the full axon reconstruction, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material); the arrows here and in subsequent figures represent the line scan position. F, Line-scan recording (Fluo-4 channel; 500 Hz) following five APs at 20 Hz; average of five sweeps. G, Line-scan fluorescence time course; the dashed lines and gray segments represent integration windows for single-pulse and five-pulse ΔF/F measurements.
We used 2 ms somatic depolarizing command voltage pulses to evoke orthodromic APs. These were rapidly followed by fluorescence transients in giant MFBs (Fig. 1F,G). After a period of equilibration lasting 1 h or more depending on the distance from the soma, both Ca2+-dependent and Ca2+-independent fluorescence in giant MFBs remained stable for several hours (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, proximal MF boutons tended to increase their baseline fluorescence, as well as their evoked Ca2+ responses, by the end of 30–40 min recording sessions in similar conditions (Ruiz et al., 2003); this is consistent with partial washout of endogenous buffers at short distances from the soma (Blatow et al., 2003; Muller et al., 2005), as discussed in Materials and Methods (supplemental Fig. 6, available at www.jneurosci.org as supplemental material).
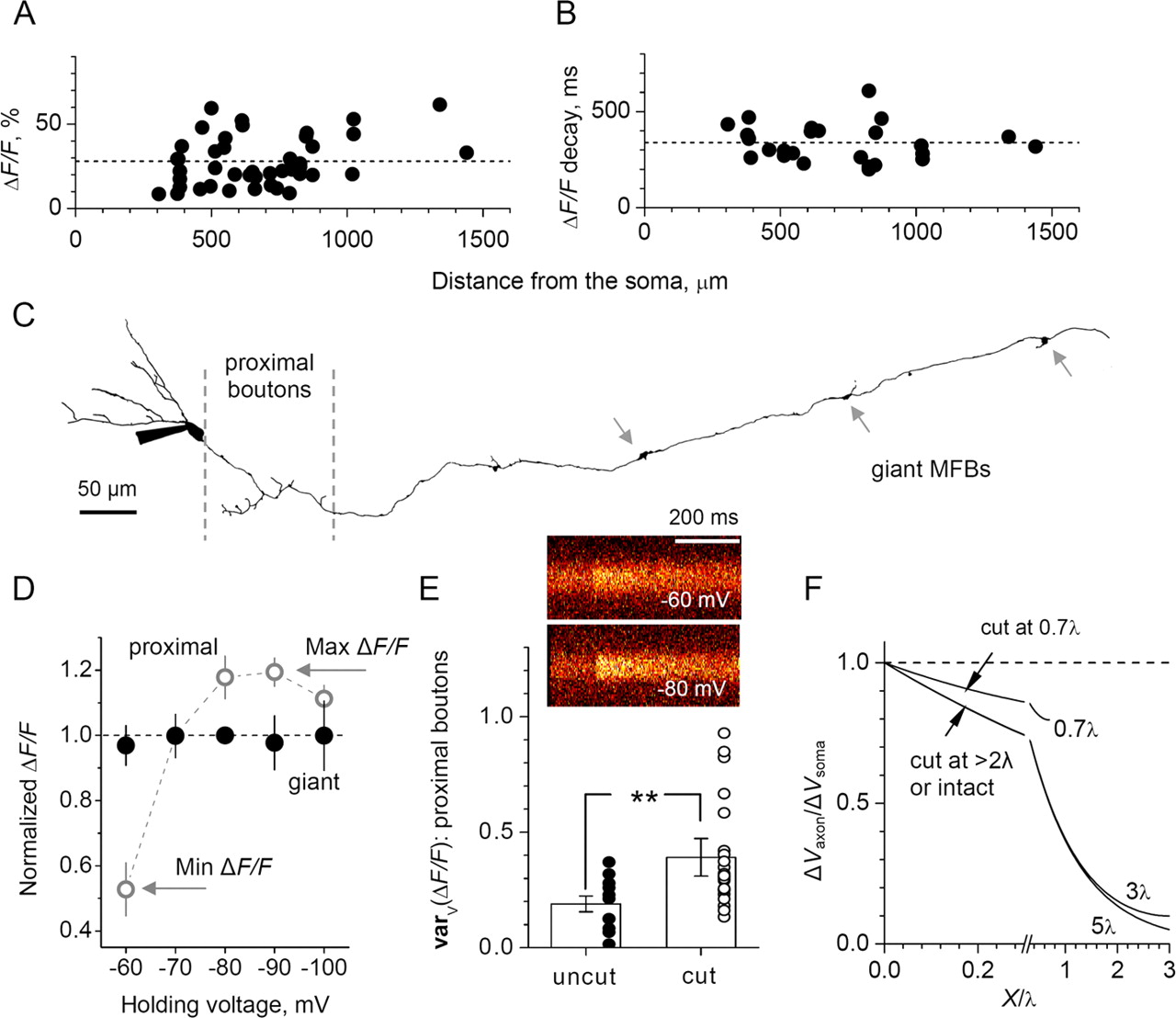
Neither the amplitude nor the decay time of the AP-evoked fluorescence transients varied systematically with the distance from the soma in n = 43 giant MFBs with fully reconstructed axons (Fig. 2A,B). In approximately one-third of all experiments, we compared Ca2+ responses evoked under voltage clamp (Vh = −80 mV) or current clamp (current adjusted to correspond to Vm = −80 mV). Because no consistent differences were found, the data from these experiments were pooled.

Differential electrotonic control of presynaptic Ca2+ signaling in hilar and CA3 MF boutons. A, B, Amplitude (A) and decay time constant (B) of the Ca2+-dependent fluorescence signal induced in individual giant MFBs by a single orthodromic AP. The values are plotted against the distance from the soma (average, 686 ± 38 μm; n = 43). The dashed lines indicate average values: ΔF/F = 28 ± 2% (n = 43); decay constant, 339 ± 19 ms (n = 26; decay constant estimates in the remaining MFBs were unreliable because of the low signal-to-noise ratio: the cells were discarded if the outcome of single-exponent fitting changed by >20% depending on the sampling time widow). C, An example of the reconstructed granule cell axon (Alexa Fluor 594 channel; whole-cell mode). The dashed lines indicate location of proximal boutons; the arrows indicate giant MFBs. D, ΔF/F amplitude (relative values) versus somatic holding voltage; filled circles, giant MFBs (n = 8); open circles, data from proximal en passant boutons (Ruiz et al., 2003) (n = 15). The arrows depict the average maximum and minimum Ca2+-dependent signals, Max ΔF/F and Min ΔF/F, respectively. The error bars in D and E represent SEM. E, Image panels, A single-cell line-scan example depicting Max ΔF/F and Min ΔF/F signals in the same giant MFBs at −80 mV and −60 mV, respectively. Plot, The average normalized Min–Max range of the fluorescence response varV(ΔF/F) = (Max ΔF/F − Min ΔF/F)/Max ΔF/F in proximal boutons of intact axons (19 ± 3%; n = 11; filled circles) and cut axons (39 ± 8%; n = 22; open circles; ∗∗p < 0.01). F, The “local-to-clamped” voltage ratio V/V0 plotted against the distance to the voltage-clamped soma along the axon (equivalent cable), according to Equation 7; distances are measured in electrotonic length units X/λ; the plotted lines are marked with the corresponding axon length value, L. For the axons cut at L > 2λ, the curves are indistinguishable in the somatic vicinity (L < λ). The arrows illustrate that cutting a long axon at ∼0.7 λ improves local voltage-clamp control approximately twofold.
Electrotonic control of Ca2+ signaling in MFs
We have shown previously (Ruiz et al., 2003) that varying the somatic holding voltage modulates the AP-evoked presynaptic Ca2+ transient up to 40% in MF axonal boutons in the hilus (Fig. 2C). We found, however, that Ca2+ signals at giant MFBs were unaffected by changing the holding voltage (Fig. 2D) (n = 8). To determine the extent of the somatic electrotonic influence in our conditions, we repeated these experiments in proximal axonal boutons (Fig. 2C). Changing the somatic voltage between −110 and −60 mV resulted in a Ca2+ signal variation, cast in relative terms as varV(ΔF/F) = (Max ΔF/F − Min ΔF/F)/Max ΔF/F (Fig. 2D), of 19 ± 3% (n = 11) (Fig. 2E). Surprisingly, this influence was only one-half of that detected in the previous study where varV(ΔF/F) was ∼40% (Ruiz et al., 2003).
This discrepancy, however, has a simple explanation: the previous study dealt with axons cut, on average, at 100–150 μm from the soma, whereas the present experiments were performed on cells with intact axons. According to cable theory, cutting and sealing the axon increases its electrotonic space constant, thus facilitating remote voltage control. [Although open-ended axons would have a different effect on the length constant, they are unlikely to be compatible with cell survival; in addition, sealed axonal ends, with no detectable escape of fluorescence, were routinely observed in a microscope and the intracellular level of Alexa Fluor 594 was unchanged throughout the experiment (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).] Indeed, when we recorded from proximal boutons in cells that had their axons cut off 100–150 μm from the soma (n = 7), the values of varV(ΔF/F) were indistinguishable from the previous results (Ruiz et al., 2003); combining the current and previous data in the cut axons gave an average varV(ΔF/F) of 39 ± 8% (n = 22) (Fig. 2E).
We used the observed twofold difference in varV(ΔF/F) between cut and uncut axons to estimate the electrotonic space constant of the axon. In an equivalent cable approximation, the steady-state voltage V at a distance x from a point clamped at V0 is given by the following relationship (for discussion, see Jackson, 1992):  where X and L represent distance x and the axon length l, respectively, cast in terms of the space constant λ: X = x/λ and L = l/λ (Fig. 2F). This relationship predicts that, to reduce the extent of somatic voltage control approximately twofold, one has to cut the long axon at L ∼ 0.7λ (Fig. 2F, arrows). Because the actual MF axons were cut at L ≈ 100–150 μm in these experiments, the estimated MF axon space constant was λ = L/0.7 = 150–200 μm. This value is consistent with electrotonic isolation of Ca2+ transients in the giant MFBs (Fig. 2D), also indicating that MF synapses in the hilus could be affected by the granule cell resting somatic voltage.
where X and L represent distance x and the axon length l, respectively, cast in terms of the space constant λ: X = x/λ and L = l/λ (Fig. 2F). This relationship predicts that, to reduce the extent of somatic voltage control approximately twofold, one has to cut the long axon at L ∼ 0.7λ (Fig. 2F, arrows). Because the actual MF axons were cut at L ≈ 100–150 μm in these experiments, the estimated MF axon space constant was λ = L/0.7 = 150–200 μm. This value is consistent with electrotonic isolation of Ca2+ transients in the giant MFBs (Fig. 2D), also indicating that MF synapses in the hilus could be affected by the granule cell resting somatic voltage.
Resting Ca2+ concentration at giant MFBs
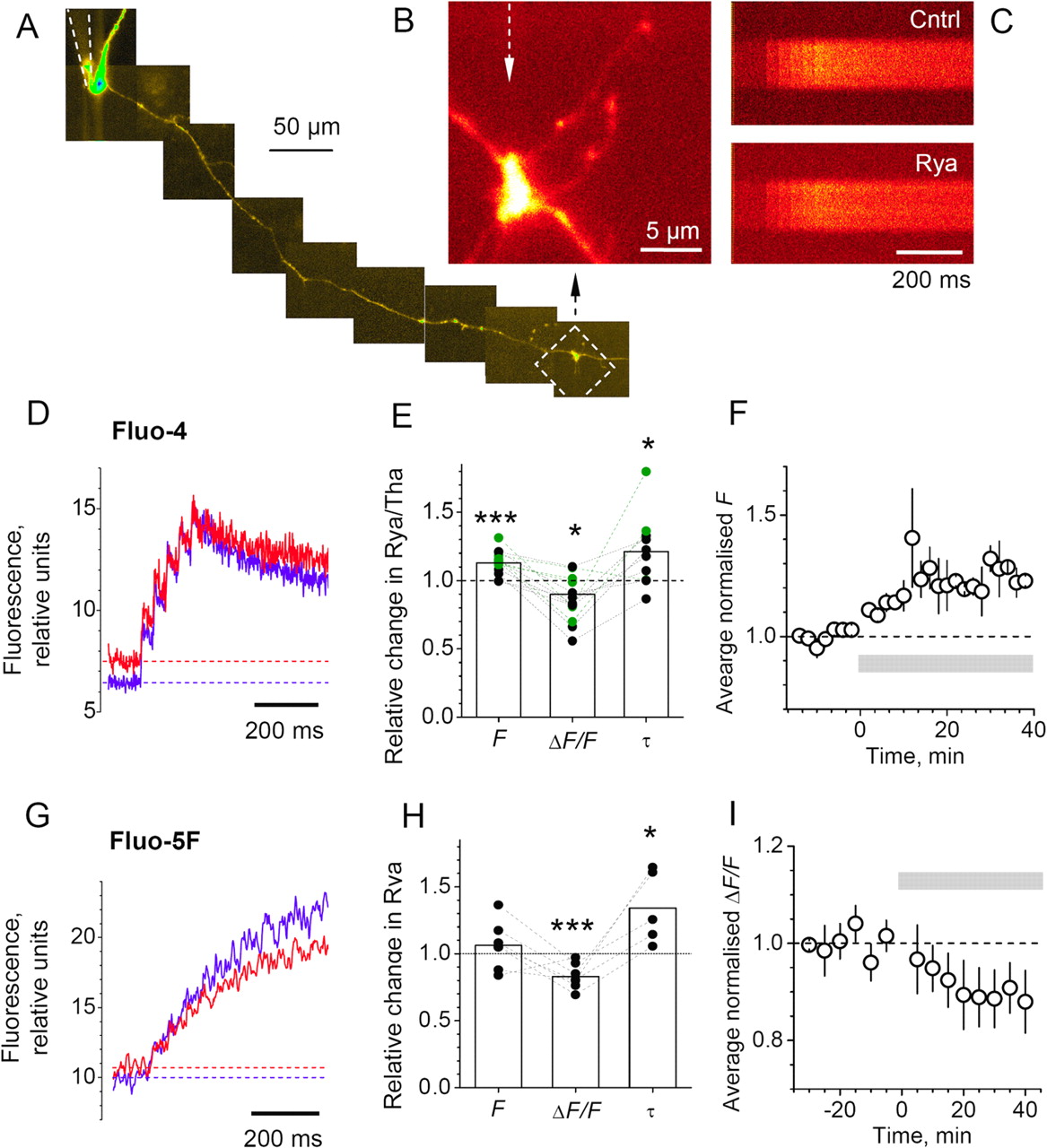
To estimate [Ca2+]rest from Equation 3, we first measured the cross-talk fraction of Alexa Fluor 594 fluorescence in the Fluo-4 (Ca2+ signal) emission channel, Rct/R. Granule cells were loaded with 20 μm Alexa Fluor 594, and MFBs were imaged in both channels (Fig. 3A,B). Comparing direct versus cross-talk signals of Alexa Fluor 594 indicated an average Rct/R ratio of 0.035 ± 0.002 (n = 24) (Fig. 3C). We then loaded cells with 200 μm Fluo-4 (in addition to 20 μm Alexa) and, after indicator equilibration (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), recorded Ca2+ fluorescence in giant MFBs in response to 50 APs at 20 Hz (Fig. 3D). The resulting fluorescence plateau (Fig. 3D) suggested that the indicator was close to saturation (Jackson and Redman, 2003; Rusakov et al., 2005). To confirm that this is indeed the case, we compared 20 and 50 Hz trains of APs in seven giant MFBs and found no significant difference in the maximum fluorescence signal Fmax (difference, 7 ± 5%; n = 7); similar experiments with a lower-affinity indicator Fluo-5F (Kd ∼ 1 μm; trains of 75–150 APs) gave a qualitatively identical result (difference, 5 ± 3%; n = 7) (Fig. 3E; supplemental Fig. 7, available at www.jneurosci.org as supplemental material). The saturating trains of APs in Fluo-4 therefore gave average Fmax/F and R/F ratios for Fluo-4 of 2.63 ± 0.31 and 6.8 ± 1.2, respectively (n = 14). The estimates of [Ca2+]rest in each individual giant MFB were obtained by substituting individual Fmax/F and R/F values in Equation 3; this gave an average [Ca2+]rest of 116 ± 20 nm (n = 14) (Fig. 3F).

Estimation of the resting Ca2+ at giant MFBs using a cross-talk reference. A, An example of a giant MFB filled with 20 μm Alexa Fluor 594 only (no Ca2+ indicator); average of five frame scans. B, Line scans of the bouton shown in A (arrows) imaged in the Alexa Fluor 594 and Fluo-4 (cross-talk) channels, as indicated; arrow, cross-talk fluorescence signal of Alexa Fluor 594, Rct. C, A summary of experiments shown in A and B. The Alexa Fluor 594 cross-talk signal recorded in the Fluo-4 channel (ordinate) is plotted against the Alexa Fluor 594 direct fluorescence signal (abscissa). The dotted line represents the best-fit linear regression, as indicated; background fluorescence is subtracted. D, A giant MFBs filled with 200 μm Fluo-4 and 20 μm Alexa Fluor 594 (left panel, Alexa Fluor 594 channel) and the corresponding line-scan ΔF/F signal (right panel and trace, Fluo-4 channel; recorded at 500 Hz) in response to 50 APs at 20 Hz; the relationship between Fmax, F, and Rct is illustrated. E, Comparison of the plateau Fmax levels obtained in response to 20 and 50 Hz trains of APs generated in the same giant MFB in experiments using Fluo-4 (200 μm; Kd ∼ 350 nm; n = 7) and Fluo-5F (200 μm; Kd ∼ 1 μm; n = 7), as indicated. F, The average Fmax/F ratios (left panel) and estimated [Ca2+]rest (right panel) in giant MFBs obtained using fluorescent indicators Fluo-4 (Fmax/F = 2.62 ± 0.31; [Ca2+]rest = 116 ± 20 nm; n = 15) and OGB-1 (Fmax/F = 2.15 ± 0.14; [Ca2+]rest = 103 ± 14 nm; n = 8), as indicated. Error bars indicate SEM.
An alternative estimate of [Ca2+]rest was obtained using another high-affinity Ca2+ indicator, Oregon Green BAPTA-1 (200 μm; Kd = 205 nm) for which Fmax/Fmin ∼ 6 (Maravall et al., 2000; Jackson and Redman, 2003). With a similar protocol, these experiments gave an average Fmax/F ratio of 2.15 ± 0.14, predicting [Ca2+]rest at 103 ± 14 nm (n = 8) (Fig. 3F). This was in good correspondence with the estimate based on Fluo-4.
Ca2+ kinetics at giant MFBs
To quantify the kinetics of presynaptic Ca2+ transients triggered by APs, we routinely evoked five spikes at 20 Hz, a firing pattern compatible with that of individual granule cells in vivo (Henze et al., 2002). The fluorescence response was analyzed using a single-compartment model of Ca2+ dynamics: this approach was relevant because the characteristic point-spread function of the imaging system was comparable with or larger than the characteristic cross section of giant MFBs (Fig. 4A–C) (see Materials and Methods). To account for the nonstationary kinetics during and shortly after rapid Ca2+ entry, we simulated the binding–unbinding reactions using an explicit finite-difference scheme (Sabatini and Regehr, 1998; Matveev et al., 2004; Rusakov et al., 2005).

Action potential-evoked Ca2+ transients in giant MFBs in the CA3 region. A, The two-photon excitation PSF evaluated for the optical settings used. Left panel, A vertical central section of the theoretical (lower-limit) diffraction-limited excitation probability profile (see Materials and Methods). Right panel, A vertical central section of the 2P PSF measured using 0.17 nm fluorescence beads and a three-dimensional reconstruction of 42 consecutive optical sections 0.25 μm apart. Linear brightness scale, 0–1; scale bar applies to A–C. B, C, Characteristic electron micrographs of giant MF bouton profiles, modified from Chicurel and Harris (1992) (B, yellow shade) and from Acsady et al. (1998) (C, dark staining indicates rabbit anti-substance P immunoreaction identifying a single presynaptic axon). Comparing these to the PSF (dashed ellipse) suggests that optical recordings normally deal with fluorescence volume-averaged across the MFB lumen. D, F, Fluorescence recordings and computed kinetic reaction components at three Ca2+ indicator settings, as indicated. The top trace inset illustrates five simulated AP-evoked pulses of Ca2+ influx; relative scale. Orange traces, Experimental recordings: the average fluorescence time course (global mean for n = 18, 5, and 5 cells in D, E, and F, respectively). The black lines represent simulated data: the best-fit predictions for the recorded fluorescence [CaI] + [Rct], bound Ca2+ buffer [CaB], and free Ca2+ [Ca2+]. Best-fit values for total Ca2+ entry Δ[Ca2+]tot and removal rate P are shown.
The model included several independently estimated quantities: (1) [Ca2+]rest ≈ 110 nm; (2) a direct measure of the main endogenous buffer (calbindin-28K) concentration [B]tot = 160 μm (Muller et al., 2005), which is in line with an estimate derived from Ca2+ imaging (Jackson and Redman, 2003); (3) the Ca2+ entry half-duration of ∼0.5 ms (Bischofberger et al., 2002); and (4) indicator concentration [I] clamped by the whole-cell pipette (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The remaining two free parameters were the total Ca2+ influx Δ[Ca2+]tot and the Ca2+ removal rate P. Adjusting these, however, had virtually independent effects on, respectively, the amplitude and the decay constant of the evoked Ca2+-sensitive fluorescence transient: changing Δ[Ca2+]tot twofold produced a comparable change in the predicted ΔF/F amplitude but a <5% change in the decay constant; conversely, changing the Ca2+ removal constant P twofold produced a comparable change in the predicted decay constant but <5% alteration in the ΔF/F amplitude (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In these conditions, fitting the computed fluorescence kinetics to the experimental ΔF/F is relatively straightforward, as discussed previously (Rusakov et al., 2005). To cross-validate the unknowns of Ca2+ dynamics under different buffer configurations, we conducted similar experiments at two different concentrations of Fluo-4 (200 and 50 μm) and also with a lower-affinity indicator Fluo-5F. In all three cases, optimization with the two parameters gave a very good fit, yielding consistent estimates of Δ[Ca2+]tot and P (Fig. 4D–F). This consistency further argues that the previous estimates of the endogenous Ca2+ buffer concentration in the soma and proximal boutons (Jackson and Redman, 2003; Muller et al., 2005) are relevant for giant MFBs, as indeed expected from the long-term diffuse equilibration of soluble calbindin-28K. Together, the data thus predict Δ[Ca2+]tot = 51 ± 2 μm and an average Ca2+ removal rate, p = 0.37 ± 0.03 ms−1.
According to morphological observations, the volume of giant MFBs is in the region of 20–50 μm3 (Chicurel and Harris, 1992; Acsady et al., 1998), or 2–5 × 10−14 L. In electron micrographs, synaptic vesicles and mitochondria appear to occupy ∼50% of the giant MFB profile (Fig. 4B,C); this is likely to scale down twofold the volume available to Ca2+ ions. The ∼50 μm step concentration increase (3 × 1019 ions/L) would then correspond to 3–8 × 105 Ca2+ ions per bouton. This is in excellent agreement with electrophysiological observations showing that 3.7 × 105 Ca2+ ions flow into a patched giant MFB following an AP (Bischofberger et al., 2002). The estimated Ca2+ dynamics is also in correspondence with the unchanged Ca2+ entry during short 20 Hz AP trains (Geiger and Jonas, 2000). Together, these data provide quantitative insights into the presynaptic Ca2+ kinetics in the absence of exogenous buffering imposed by Ca2+ indicators (see Fig. 6) (see Discussion).
Internal Ca2+ stores and presynaptic Ca2+ signaling
The role of presynaptic Ca2+ stores at MF synapses remains controversial (Carter et al., 2002; Liang et al., 2002; Lauri et al., 2003; Breustedt and Schmitz, 2004). To address this, we imaged giant MFBs loaded with either Fluo-4 or Fluo-5F (200 μm) (Fig. 5A,B) and, once the Ca2+ responses were stable (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), applied either ryanodine (20–60 μm) or thapsigargin (1 μm). These blockers interfere with Ca2+-induced ryanodine receptor-dependent Ca2+ release or with the endoplasmic reticulum Ca2+-ATPase (Ca2+ pump), respectively. Because Ca2+ stores might not respond to a single AP (Lauri et al., 2003), we routinely evoked five or more APs at 20 Hz and measured the integrated ΔF/F signal over a 500 ms window from the first response onset.

Ca2+ store depletion increases resting Ca2+ but decreases Ca2+ transients. A–C, Singe-cell example. A reconstructed granule cell axon (A); dashed square, the giant MFB of interest enlarged in B (Alexa Fluor 594 channel). Line scans in C (Fluo-4 channel) show recordings from the bouton in B, an average of seven traces. Cntrl, Control; Rya, ∼10 min after application of 50 μm ryanodine. D, Effect of Ca2+ store blockers ryanodine (n = 8) or thapsigargin (n = 5) on Ca2+ signaling in experiments with 200 μm Fluo-4 (applies to D–F). The average fluorescence kinetics in response to five APs (global mean; n = 13) in control (blue) and test (red) conditions. E, Relative changes in baseline signal F, ΔF/F (integrated over five pulses), and fluorescence decay time τ (single exponent) after application of ryanodine (n = 8; black circles) or thapsigargin (n = 5; green circles) in individual cells. The overall changes are as follows (n = 13; see Results for separate statistics on ryanodine and thapsigargin): F, 12 ± 2% (∗∗∗p < 0.001); ΔF/F, −13 ± 5% (∗p < 0.03); τ, 21 ± 8% (∗p < 0.03); the dashed lines connect data points from the same MFB. F, The time course of changes in the resting F (normalized values) after ryanodine application (gray segment) in the experiments shown in D and E. The circles represent global mean; error bars indicate SEM. G, H, Experiments using 200 μm Fluo-5F; notations are the same as in D and E for ryanodine application; average changes are as follows: F, 6 ± 6% (n = 7; NS); ΔF/F, −17 ± 4% (n = 7; ∗∗∗p < 0.004); τ, 34 ± 12% (n = 5; ∗p < 0.04). I, The time course of changes in the resting F (normalized values) after drug application (gray segment) in experiments shown in G and H. The circles represent global mean; error bars indicate SEM.
Although ryanodine and thapsigargin interfere with different mechanisms of internal Ca2+ storage, the longer-term consequence is thought to be the blockade/depletion of endoplasmic reticulum Ca2+ stores (Verkhratsky, 2005). Indeed, both ryanodine and thapsigargin produced a small yet significant increase in the resting fluorescence F (11 ± 3%, n = 8, p < 0.02; and 12 ± 3%, n = 5, p < 0.02, respectively) and a decrease in ΔF/F (18 ± 6%, n = 8, p < 0.02; and 8 ± 7%, n = 5, NS, respectively), recorded in experiments with the high-affinity Fluo-4 (Fig. 5D–F). In contrast, ryanodine application in experiments with the lower-affinity Fluo-5F produced only insignificant increases in F, yet it decreased ΔF/F robustly (17 ± 4%, p < 0.004; integrated over six APs) (Fig. 5G–I). In both cases, the significant changes exceeded at least threefold the experimental fluctuations of F and ΔF/F documented in baseline conditions on a similar timescale (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material).
These results are consistent with an elevation in the resting Ca2+ concentration, [Ca2+]rest: the higher affinity Fluo-4 is more sensitive than Fluo-5F to changes in [Ca2+]rest, yet it saturates to a greater degree and therefore is less sensitive to changes in ΔF/F. According to Equation 3, in baseline conditions of Fluo-4 fluorescence, a ∼12% increase in F corresponds to an increase in [Ca2+]rest of 40–50 nm. In the case of lower-affinity Fluo-5F, however, the same increase in [Ca2+]rest should produce a much smaller elevation of F, which is fully consistent with our observations. In line with the buffering role of Ca2+ stores, their blockade also slowed down the decay of the ΔF/F signal following five or more APs: by 21 ± 8% in Fluo-4 experiments (the apparent decay time constant changed from 730 ± 111 to 856 ± 110 ms; n = 10; p < 0.03) and by 34 + 12% in Fluo-5F experiments (time constant changed from 1106 ± 121 to 1533 ± 295 ms; n = 65; p < 0.004); the fluorescent decay in these multi-AP recordings was substantially slower than that following a single AP (Fig. 2), partly because of the indicator saturation.
Discussion
In this study, we have evaluated the main determinants of Ca2+ signals generated in individual giant MFBs by electrical activity in a single granule cell. Somatic voltage, which affects Ca2+ responses in proximal axonal compartments, does not influence rapid Ca2+ transients in giant MFBs. We find that Ca2+ stores take part in maintaining the resting concentration of presynaptic Ca2+, a mechanism which has not previously been reported in cortical synapses. Rapid Ca2+ release from internal stores also contributes to Ca2+ transients in giant MFBs (this could be masked in multiple MF recordings, at least in part, if other MF compartments were insensitive to Ca2+ store blockade). The data provide a basis for deciphering the kinetics of free Ca2+ at giant MFBs, a key to presynaptic mechanisms of use-dependent plasticity at the MF–CA3 pyramidal cell synapse, as discussed below.
Electrotonic control of presynaptic Ca2+ entry
The regulatory role of presynaptic ionotropic receptors in neurotransmitter release has attracted much interest (Engelman and MacDermott, 2004). However, to what extent presynaptic electrotonic influences spread along the terminal remains poorly understood. Although the granule cell somatic voltage affects Ca2+ kinetics at MF axonal boutons in the hilus (Ruiz et al., 2003), we detected no such influence at giant MFBs in CA3. This was consistent with the estimated electrotonic length constant of Ca2+ entry control in MFs, λ = 150–200 μm, also agreeing with the lack of somatic voltage control over MF excitability in stratum lucidum (Schmitz et al., 2000). However, very recent observations of presynaptic EPSCs in giant MFBs have proposed an MF electrotonic length constant of ∼450 μm (Alle and Geiger, 2006). The most parsimonious explanation for the difference is that AP-evoked Ca2+ entry is not sensitive to small changes in the local membrane potential. Indeed, somatic voltage has no effect on Ca2+ currents in giant MFBs (Alle and Geiger, 2006). In any case, the value of λ suggests that the Ca2+-dependent release machinery acting at synapses between granule cells and interneurons or mossy cells in the hilus could be influenced by the resting somatic membrane voltage. Conversely, presynaptic ionotropic action could propagate between neighboring giant MFBs occurring 100–200 μm apart (Acsady et al., 1998).
At the calyx of Held, moderate presynaptic depolarization increases the resting Ca2+ concentration, thus augmenting the release probability (Turecek and Trussell, 2001; Awatramani et al., 2005). The depolarization-induced elevation of resting Ca2+ was also reported in proximal MFBs (Ruiz et al., 2003). If similar phenomena occur locally in giant MFBs, an increase in the postsynaptic responses could follow (Alle and Geiger, 2006). This might reconcile the facilitatory effect of low kainate concentration on MF transmission (Schmitz et al., 2001) with the depolarizing action of kainate receptors. Furthermore, presynaptic depolarization could initiate AP broadening (Geiger and Jonas, 2000), thus boosting Ca2+ entry and the release probability further.
Kinetics of free Ca2+ and short-term synaptic facilitation
We have used several Ca2+ indicator configurations to estimate and cross-validate Ca2+ kinetics at giant MFBs. Although our experiments rely on fluorophores that directly interfere with Ca2+ buffering, the resulting kinetic model allows evaluation of the free Ca2+ kinetics in the absence of exogenous buffers (Fig. 6A). The data predict substantial use-dependent facilitation of both rapid Ca2+ transients and residual Ca2+ rises, with the latter extending beyond 500 ms postpulse (Fig. 6A, gray line). However, the volume-integrated kinetics could underrepresent concentration microdomains near Ca2+ entry sites, thus underestimating local buffer saturation and hence overestimating use-dependent facilitation of local free Ca2+ (Meinrenken et al., 2003). Nonetheless, even modest facilitation would increase the release probability substantially if neurotransmitter release at MFBs depended on Ca2+ in a highly supralinear manner, as reported in the calyx of Held (Schneggenburger and Neher, 2000). This is, however, not the case in giant MFBs: increasing presynaptic Ca2+ transient through either AP broadening, elevation in external Ca2+, or changed buffering conditions enhances MF–CA3 pyramidal cell transmission with the less than second power relationship (Regehr et al., 1994; Geiger and Jonas, 2000; Blatow et al., 2003; Mori-Kawakami et al., 2003). The question therefore arises whether the kinetics of free presynaptic Ca2+ evaluated here could explain short-term synaptic facilitation at these synapses.
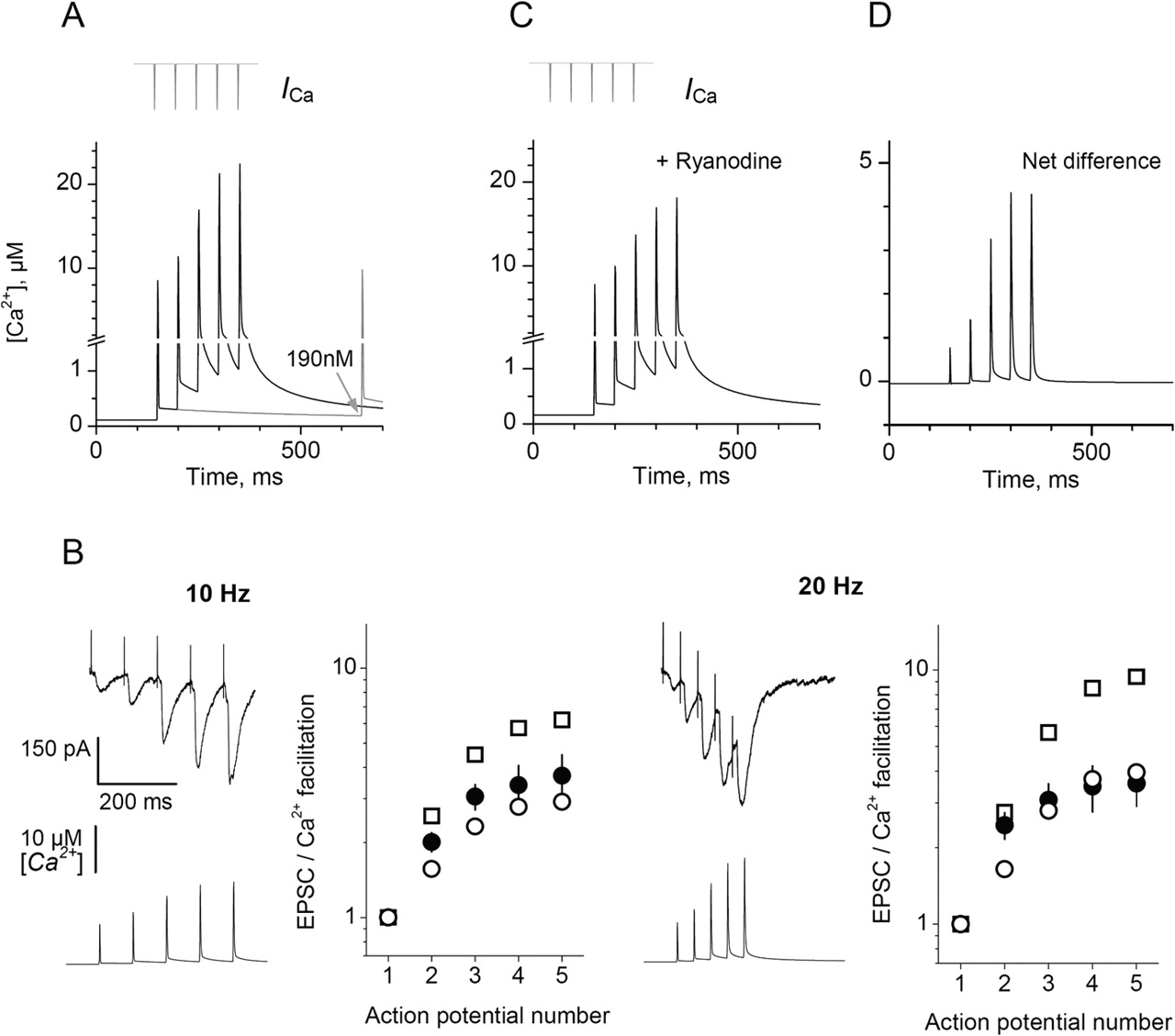
Kinetics of free Ca2+ and synaptic facilitation at MF–CA3 pyramidal cell synapses. A, According to the predicted kinetics, Ca2+ entry driven by five APs at 20 Hz (inset) corresponds to a progressive increase in peak and residual free [Ca2+]; two spikes 500 ms apart (gray line) show modest facilitation of the peak [Ca2+] value (15%) and a more substantial increase of the integrated [Ca2+] spike (34%; averaged over 4 ms during the AP) and residual [Ca2+] elevation (73%; gray arrow). B, Insets show a single-cell example of recorded responses (top trace) and the corresponding calculated time course of free presynaptic [Ca2+] (bottom trace) at two stimulation frequencies, as indicated. Plots compare the use-dependent facilitation of the computed rapid spikes of free Ca2+ (average [Ca2+] over 4 ms during an AP; open circles), the computed residual prepulse [Ca2+] (open squares), and the average experimental EPSC amplitude (filled circles) in CA3 pyramidal cells in response to five APs (n = 5 cells). All values are normalized with respect to the first response. C, Same as in A but the blockade of Ca2+-induced Ca2+ release from stores is mimicked as a reduction in evoked Ca2+ entry by 20% and an increase in resting Ca2+ by 40% (see Results). D, The difference between A and C illustrating net contribution of Ca2+ store releases to free Ca2+ transients.
To address this, we calculated use-dependent increases of residual [Ca2+] and rapid [Ca2+] transients (average [Ca2+] elevation over 4 ms during an AP) in giant MFBs and compared them with the average facilitation of EPSCs evoked in CA3 pyramidal cells by short trains of APs at 10 and 20 Hz (elicited by extracellular stimuli in dentate gyrus) (Fig. 6B). The comparison suggests that, in conditions of our experiments, synaptic facilitation follows more closely rapid [Ca2+] transients than residual [Ca2+] rises (with an exception of the second pulse). Although these data suggest a causal link between facilitation of presynaptic Ca2+ signals and postsynaptic responses, it would be important to establish how the presynaptic Ca2+ kinetics reflects synaptic adaptation to different firing patterns (Zucker and Regehr, 2002).
A role for Ca2+ stores
We found that blockade of internal Ca2+ stores in giant MFBs elevates resting Ca2+ by ∼40%. Although this phenomenon has not previously been reported in cortical synapses, it is not surprising: the Ca2+ concentration inside endoplasmic reticulum stores is critical for cytosolic Ca2+ homeostasis (Verkhratsky, 2005). Indeed, Ca2+ leakage from the stores is negatively coupled with Ca2+ uptake, producing a kinetic equilibrium (Solovyova et al., 2002), which is likely to reflect the resting Ca2+ level. By taking Ca2+ stores out of the equilibrium, ryanodine blockade should reduce the Ca2+ removal capacity of giant MFBs, thus elevating resting Ca2+concentration. The present data allow us to evaluate this quantitatively.
Ca2+ removal depends primarily on the two families of Ca2+ pumps, the plasma membrane Ca2+-ATPase and the sarco/endoplasmic reticulum Ca2+-ATPase; mitochondria and a low-affinity Na+/Ca2+ exchanger also contribute to Ca2+ removal, albeit on a slower timescale (for review, see Parekh, 2003; Mata and Sepulveda, 2005; Verkhratsky, 2005). Because pumps operate through binding and translocation, Ca2+ uptake kinetics could be generally represented by the following two-stage reaction:  where P and Cat2+ denote pumps and intralumen Ca2+, respectively, CaP stands for the Ca-bound pump complex, and kon, koff, and ku stand for the average rate constants of Ca2+ binding, unbinding, and translocation (coupled with the reappearance of available pumps P), respectively. These reactions (Eq. 9) evoke the following simple kinetic equations:
where P and Cat2+ denote pumps and intralumen Ca2+, respectively, CaP stands for the Ca-bound pump complex, and kon, koff, and ku stand for the average rate constants of Ca2+ binding, unbinding, and translocation (coupled with the reappearance of available pumps P), respectively. These reactions (Eq. 9) evoke the following simple kinetic equations: 
 with the mass conservation law
with the mass conservation law  where brackets denote concentration, LCa is the rate of overall Ca2+ leakage into the cytosol, and [P]tot is the total amount of available pumps. In the steady-state case (time derivatives zeroed), Equations 10a, 10b, and 10c reflect equilibration between uptake and leakage as follows:
where brackets denote concentration, LCa is the rate of overall Ca2+ leakage into the cytosol, and [P]tot is the total amount of available pumps. In the steady-state case (time derivatives zeroed), Equations 10a, 10b, and 10c reflect equilibration between uptake and leakage as follows:  The equilibrium corresponds to the resting Ca2+ level, [Ca2+]rest, evoking the following relationship:
The equilibrium corresponds to the resting Ca2+ level, [Ca2+]rest, evoking the following relationship:  Direct monitoring of Ca2+ store uptake in dorsal root ganglion neurons and in pancreatic acinar cells has indicated that the maximum uptake rate (after caffeine-induced Ca2+ release) is approximately four times greater than the resting uptake rate (Mogami et al., 1998; Solovyova et al., 2002). Because the maximum uptake is likely to reflect Ca2+ pump saturation, which implies [CaP] → [P]tot, Equation 11a predicts the relationship ku[P]tot ≈ 4LCa. Substituted in Equation 11b, this gives the following:
Direct monitoring of Ca2+ store uptake in dorsal root ganglion neurons and in pancreatic acinar cells has indicated that the maximum uptake rate (after caffeine-induced Ca2+ release) is approximately four times greater than the resting uptake rate (Mogami et al., 1998; Solovyova et al., 2002). Because the maximum uptake is likely to reflect Ca2+ pump saturation, which implies [CaP] → [P]tot, Equation 11a predicts the relationship ku[P]tot ≈ 4LCa. Substituted in Equation 11b, this gives the following:  When Ca2+ stores are blocked, this should reduce the concentration of available pumps to [P]tot∗, thus elevating [Ca2+]rest by ∼40% (see Results). Equations 12a and 11b recast in terms of the elevated Ca2+ level [Ca2+]rest = 1.4[Ca2+]rest and [P]tot∗, thus yield ku[P]tot∗ ≈ 3.1LCa, or [P]tot∗ ≈ 0.78[P]tot. Store blockade, therefore, removes ∼22% of the buffering/uptake capacity that maintains the low resting Ca2+ concentration. This value is consistent with a ryanodine-induced prolongation of the ΔF/F decay, which depends mainly on Ca2+ removal.
When Ca2+ stores are blocked, this should reduce the concentration of available pumps to [P]tot∗, thus elevating [Ca2+]rest by ∼40% (see Results). Equations 12a and 11b recast in terms of the elevated Ca2+ level [Ca2+]rest = 1.4[Ca2+]rest and [P]tot∗, thus yield ku[P]tot∗ ≈ 3.1LCa, or [P]tot∗ ≈ 0.78[P]tot. Store blockade, therefore, removes ∼22% of the buffering/uptake capacity that maintains the low resting Ca2+ concentration. This value is consistent with a ryanodine-induced prolongation of the ΔF/F decay, which depends mainly on Ca2+ removal.
We also find that blockade of Ca2+ stores reduces by up to 20% the Ca2+ transients evoked in giant MFBs by a short train of APs. This is consistent with the progressive decrease of EPSCs recorded in CA3 pyramidal cells after Ca2+ store blockade (Lauri et al., 2003). Incorporating a 20% reduction of Ca2+ entry and a 40% increase in resting Ca2+ concentration into the indicator-free model (Fig. 6A) predicts the free Ca2+ kinetics depicted in Figure 6C. Interestingly, the difference between the two cases (Fig. 6A,C) suggests little impact of stores on the first two Ca2+ transients (Fig. 6D). This is because the reduction in Ca2+ entry is compensated for by the reduced buffering capacity (attributable to the elevated resting Ca2+). The phenomenon might explain, at least partly, why blockade of Ca2+ stores has little influence on single or paired synaptic responses (Carter et al., 2002; Liang et al., 2002; Lauri et al., 2003). Reduction in Ca2+ buffering capacity through elevation of resting Ca2+ could also contribute to the release probability increase at the calyx of Held after presynaptic depolarization (Turecek and Trussell, 2001; Awatramani et al., 2005).
Although this mechanism acts at the giant MFBs representing synapses on CA3 pyramidal cells, the main synaptic target of MFs are interneurons (Acsady et al., 1998; Lawrence and McBain, 2003; Mori et al., 2004). The MF–interneuron synapses in CA3 are formed either by filopodial extensions of giant MFBs or by en passant boutons (Acsady et al., 1998). These presynaptic specializations show clear AP-evoked Ca2+ signals that are insensitive to ryanodine (our unpublished observations), whereas the corresponding synapses exhibit use-dependent plasticity distinct from that at MF–CA3 pyramidal cell synapses (Toth et al., 2000; Pelkey et al., 2005). Whether this functional diversity of MF synapses in CA3 could explain why Ca2+ store blockade has little effect on Ca2+-dependent fluorescence recorded in bulk-loaded MFs (Carter et al., 2002; Breustedt and Schmitz, 2004) remains to be established.
Footnotes
-
This work was supported by the Wellcome Trust, the Medical Research Council UK, the European Union (HPRN-CT-2000-00082; PROMEMORIA LSHM-CT-2005-512012), and the Human Frontier Science Program. We thank Dimitri Kullmann, Kirill Volynski, Alex Verkhratsky, and colleagues at University College London for valuable comments on this manuscript.
- Correspondence should be addressed to Dmitri A. Rusakov, Institute of Neurology, University College London, Queen Square, London WC1N 3BG, UK. Email: d.rusakov{at}ion.ucl.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}