

Abstract
Encephalitis and dementia associated with acquired immunodeficiency syndrome (AIDS) are characterized by leukocyte infiltration into the CNS, microglia activation, aberrant chemokine expression, blood–brain barrier (BBB) disruption, and eventual loss of neurons. Little is known about whether human immunodeficiency virus 1 (HIV-1) infection of leukocytes affects their ability to transmigrate in response to chemokines and to alter BBB integrity. We now demonstrate that HIV infection of human leukocytes results in their increased transmigration across our tissue culture model of the human BBB in response to the chemokine CCL2, as well as in disruption of the BBB, as evidenced by enhanced permeability, reduction of tight junction proteins, and expression of matrix metalloproteinases (MMP)-2 and MMP-9. HIV-infected cells added to our model did not transmigrate in the absence of CCL2, nor did this condition alter BBB integrity. The chemokines CXCL10/interferon-gamma-inducible protein of 10 kDa, CCL3/macrophage inflammatory protein-1α, or CCL5/RANTES (regulated on activation normal T-cell expressed and secreted) did not enhance HIV-infected leukocyte transmigration or BBB permeability. The increased capacity of HIV-infected leukocytes to transmigrate in response to CCL2 correlated with their increased expression of CCR2, the chemokine receptor for CCL2. These data suggest that CCL2, but not other chemokines, plays a key role in infiltration of HIV-infected leukocytes into the CNS and the subsequent pathology characteristic of NeuroAIDS.
Introduction
Human immunodeficiency virus 1 (HIV-1) enters the CNS early in the course of infection and has been detected in the brain (Davis et al., 1992) and CSF (Chiodi et al., 1988) of individuals with neurological complications. Approximately 20% of individuals with acquired immunodeficiency syndrome (AIDS) develop HIV encephalitis (HIVE), and 15% develop HIV-1-associated dementia (HAD) in the United States (Albright et al., 2003). Despite the initial drop in the incidence of encephalitis and/or dementia as a result of highly active antiretroviral therapy (HAART), these percentages are again increasing as HIV-infected individuals are living longer. Despite extensive evidence of pathological changes in the CNS of infected individuals, the mechanism(s) of viral entry into the brain is still not completely understood. Studies suggested that this mechanism is dependent, in part, on the transmigration of infected leukocytes across the blood–brain barrier (BBB) into the CNS (Price and Brew, 1988; Persidsky et al., 1997; Weiss et al., 1999). The BBB is composed mainly of specialized endothelial cells (ECs) in contact with astrocytes and is characterized by the presence of tight junction proteins (TJPs) between EC–EC and EC-astrocytes and selective permeability to physiological ions (Goldstein, 1988; Risau et al., 1990; Rubin and Staddon, 1999). Passage of migrating immune cells across this specialized vasculature during immune surveillance is believed to be a rapid process that does not damage the integrity of the BBB. In pathological conditions, such as HIV-1 infection of the CNS, accumulation of monocytes/macrophages within the CNS parenchyma has been demonstrated, as well as significant structural and functional abnormalities of the BBB. TJP disruption has also been detected in HIV encephalitic brain tissue (Petito and Cash, 1992; Dallasta et al., 1999). However, it has not yet been demonstrated that these alterations are a consequence of transmigration of HIV-infected leukocytes into the CNS, and the mechanisms involved in the disruption of BBB integrity are unknown.
Chemokines mediate the recruitment and activation of leukocytes by binding to specific cell surface receptors. They play an important role in the pathogenesis of CNS diseases. CCL2 is a chemoattractant for monocytes and binds to the CCR2 receptor (Rollins, 1996). Elevated levels of CCL2, CCL5, and CXCL10 have been detected in the brain and CSF of patients with HIVE and HAD (Conant et al., 1998; Kelder et al., 1998; Kolb et al., 1999; McManus et al., 2000a), suggesting that glial cells and EC, major sources of chemokines, may play a key role in the recruitment of uninfected and HIV-infected leukocytes into the CNS.
In this study, we used different HIV isolates to determine whether infection of human peripheral blood mononuclear cells (PBMC) alters their transmigration properties across our model of the BBB. We also analyzed whether BBB permeability, TJP, and matrix metalloproteinases (MMPs) are altered in our model as a result of transmigration of HIV-infected PBMC. Based on our data, we propose that HIV-infected leukocyte infiltration of the CNS and BBB disruption are critically dependent on both viral infection and the presence of CCL2 specifically within the brain parenchyma and may contribute to the pathology of NeuroAIDS.
Materials and Methods
Isolation of PBMC.
Anticoagulated blood was obtained from healthy volunteers, and PBMCs were isolated by underlayering with Ficoll-Paque. The purified PBMC populations used in these studies were ∼90% lymphocytes and 10% monocytes, as determined by fluorescence-activated cell sorting (FACS).
Viral isolates used.
Cell-free viral inocula were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Germantown, MD). HIV infection occurs by binding of circulating virus through its gp120 to CD4 and either the CCR5 (R5 virus) or CXCR4 (X4 virus) coreceptor on the target cell, resulting in fusion between lipid layers of the viral envelope and the host plasma membrane. In this study, we used HIVADA, an R5 isolate that infects human monocytes/macrophages, and was isolated from PBMC of a patient with HAD. HIVJR-CSF, an R5 isolate that infects primary human peripheral blood lymphocytes and some populations of mononuclear phagocytes, was obtained from the CSF of a patient with Kaposi’s sarcoma and severe AIDS encephalopathy. HIV92UG021, an X4 virus that infects human peripheral blood lymphocytes, was obtained from an asymptomatic patient from Uganda. In addition, for some experiments, HIV92UG029 (X4), HIV92BR021 (R5), and HIV92RW009 (R5X4) were used also.
HIV infection of PBMC.
PBMCs were isolated and activated with phytohemagglutinin (PHA) (5 μg/ml) plus interleukin 2 (IL-2; 5%) in Roswell Park Memorial Institute 1640 for 48 h in polypropylene tubes at a density of 2 × 106 cells/ml. Cells do not adhere to these tubes, and therefore monocytes do not differentiate into macrophages. Some cells were incubated with high titers of HIV-1 (ADA, JR-CSF, 92BR021, 92UG021, 92UG029, 92RW009, or 92UG021) for this 1–2 h period, washed, resuspended in fresh medium, and maintained in polypropylene tubes for an additional 7 d to facilitate viral replication. Other cells, which serve as controls, were activated with PHA plus IL-2 for 48 h, washed, and maintained in polypropylene tubes in fresh media for an additional 7 d without any virus. To determine the levels of HIV infection, HIV-1 p24 was determined by ELISA (PerkinElmer, Boston, MA) 7 d after infection.
Astrocyte and EC cultures.
Human fetal cortical tissue was obtained as part of an ongoing research protocol approved by the Albert Einstein College of Medicine. Astrocytes and ECs were isolated as described previously (Eugenin and Berman, 2003).
BBB model.
Our BBB model consists of ECs and astrocytes cocultured on opposite sides of gelatin-coated tissue culture inserts with 3 μm pores (BD-Falcon, Franklin Lakes, NJ) that permit astrocyte processes to penetrate the insert and establish contact with the EC as described previously (Hurwitz et al., 1993; Weiss et al., 1998; Eugenin and Berman, 2003). In this model, ECs differentiate and express BBB markers as a result of contact with astrocytes, such as the glucose transporter (GLUT-1) and γ-glutamyltranspeptidase, and have enhanced expression of TJPs. Cocultures exhibit barrier resistance to 3H-inulin and albumin. In previous studies, we compared the transmigration of monocytes and lymphocytes across our model using human umbilical vein-derived EC or human brain-derived microvascular EC (Cell-Systems, Kirkland, WA). We did not detect any differences in transmigration properties of PBMC (Weiss et al., 1998, 1999; E. A. Eugenin and J. W. Berman, unpublished data) between these two types of cocultures.
Assay of PBMC transmigration across the model of the human BBB.
HIV-infected or uninfected PBMCs (3 × 105 cells) were added to the top of each tissue culture insert. CCL2, CXCL10, CCL3, CCL5 (100 or 500 ng/ml), or medium alone was added to the bottom chamber. After 24 h, the number of PBMCs that transmigrated into the bottom chamber was analyzed by FACScan using premixed human CD45 and CD14 monoclonal antibodies conjugated to FITC and phycoerythrin (PE), respectively, according to the instructions of the manufacturer (1:50; Caltag Laboratories, Burlingame, CA) (Weiss et al., 1998, 1999; Eugenin and Berman, 2003).
Permeability analysis of BBB model.
After 24 h of uninfected or HIV-infected PBMC transmigration across the BBB model, the integrity of the barrier was determined by a permeability assay. In other experiments, HIV-tat protein (100 ng/ml; 1–72 aa) (a generous gift from Dr. Avindra Nath, Johns Hopkins Medical Center, Baltimore, MD) and/or HIV-1JRFL (X4) and HIV-1HxB (R5) gp120 (100 nm) (generous gifts from Dr. Robert Doms, University of Pennsylvania, Philadelphia, PA) were added directly to cocultures for 24 h. Inserts were washed with phenol red-free DMEM and placed in 24-well tissue culture plates containing 400 μl of phenol red-free DMEM/10% FBS in each well. Albumin (0.45%) conjugated to Evans blue dye (200 μl) was added to the top of the insert, and after 30 min of incubation at 37°C, media was collected from the lower chamber and read spectrophotometrically at 620 nm to quantify the passage of albumin through the BBB model. Because the conjugation of Evans blue dye to albumin was variable, 200 μl of each preparation was read at 620 nm to determine the maximal OD reading that could be obtained with this preparation. This value was set as that obtained after complete disruption of permeability, signifying the passage of all Evans blue dye-conjugated albumin through cocultures. Before transmigration, representative cocultures were assayed for permeability and were completely impermeable to Evans blue dye coupled to BSA. After PBMC transmigration, permeability assays were performed on cocultures, and the OD reading of the media in the bottom chamber was converted to the percentage of permeability (permeability, %) with respect to the maximal OD reading that signifies complete disruption of the barrier.
Immunofluorescence.
After 24 h of leukocyte transmigration, BBB membranes were analyzed by immunostaining for occludin, claudin-1, zona occludens-1 (ZO-1) (Zymed, San Francisco, CA), GFAP, Von Willebrand factor (VWF), MMP-2, or MMP-9 (Sigma, St. Louis, MO) using our previously described protocol (Eugenin et al., 2003). Briefly, inserts were incubated in blocking solution for 30 min and then in diluted primary antibody (anti-occludin, anti-claudin-1, anti-ZO-1, anti-GFAP, anti-VWF, anti-MMP-2, or anti-MMP-9 at 1:800, 1:800, 1:800, 1:1000, 1:400, 1:400, and 1:400 dilution, respectively) overnight at 4°C. BBB samples were mounted and examined by confocal microscopy. In addition, p24 (1:20; National Institutes of Health Repository, Bethesda, MD) and CD14 (1:300) staining were performed on leukocytes to analyze the percentage of HIV-infected monocytes and lymphocytes before and after transmigration.
Western blot analysis.
HIV-infected and uninfected leukocytes, or BBB cells, isolated by scraping off the cells from 16 inserts after 24 h of leukocyte transmigration, were sonicated in solubilization buffer containing protease and phosphatase inhibitors. Protein concentration was measured using the Bio-Rad (Hercules, CA) protein assay. Western blot analyses were performed as described previously (Eugenin and Berman, 2003).
Flow cytometry.
CCR2 expression on the surface of leukocytes was determined by immunofluorescence staining, followed by flow cytometry as described previously (Eugenin and Berman, 2003). Briefly, cells were fixed with 4% paraformaldehyde for 30 min, washed, blocked, and treated with anti-CCR2 antibody conjugated to PE (clone 48607, 1:40; R & D Systems, Minneapolis, MN) or irrelevant isotype matched control antibodies conjugate to PE for 60 min on ice. Cells were washed and analyzed by FACScan.
Statistical analysis.
Student’s one-tailed, paired t test was used to compare leukocyte transmigration under different conditions. Mean differences were tested by nonparametric Kruscal–Wallis analysis. If a significant F value was obtained, means were compared with Bonferroni–Dunn multiple comparison test. A value of p < 0.05 was considered significant.
Results
HIV-infected PBMCs transmigrated to a greater extent across the BBB model in response to CCL2 compared with uninfected cells
We examined chemokine-induced transmigration of activated or HIV-1-infected PBMCs (HIV-1 strains; ADA, JR-CSF, 92UG021, 92UG029, 92BR021, or 92RW009) across our BBB model. For most experiments, three representative HIV-1 strains were used, two R5 (ADA and JR-CSF) and one X4 (92UG021). In addition, for some experiments, we tested three other HIV isolates, 92BR021 (R5), 92UG029 (X4), and 92RW009 (R5X4).
We first analyzed the transmigration of uninfected PBMCs across our model of the BBB. PBMCs were activated with PHA (5 μg/ml) and IL-2 (5%) for 48 h, washed, and then cultured in fresh media for 7 d in polypropylene tubes so cells would not adhere or differentiate but would still proliferate (see Materials and Methods). These are the same conditions used for HIV-infected cells and therefore serve as control cells (C). Three × 105 cells were added to the top chamber of the BBB model, with or without CCL2 (100 ng/ml) as chemoattractant in the lower chamber, and monocyte and lymphocytes that had transmigrated after 24 h were collected from the bottom chamber, immunostained with CD14 and CD45 antibodies, and analyzed by FACScan.
The concentration of chemokine used in our studies was determined by comparing the transmigration of HIVADA-infected leukocytes in response to different concentrations of CCL2 (50, 100, and 500 ng/ml). We detected a significant reduction in the transmigration of HIV-infected lymphocytes (p < 0.05; n = 3) and monocytes (p < 0.003; n = 3) when 50 ng/ml was used as chemoattractant compared with when 100 ng/ml was used. In contrast, when the concentration of chemokine was increased to 500 ng/ml, the numbers of transmigrating lymphocytes and monocytes was not statistically different from those found when 100 ng/ml CCL2 was tested. Therefore, we chose to use 100 ng/ml for our experiments.
Next, we analyzed the transmigration of HIV-infected PBMCs in response to CCL2 when HIV-infected PBMCs were added to cocultures in the presence of CCL2. For these experiments, PBMCs were cultured in polypropylene tubes with PHA and IL-2 for 48 h and then infected with different isolates of HIV for 1–2 h. After that time, the cells were washed and resuspended in polypropylene tubes in fresh media for 7 d to allow viral replication. There were significantly higher numbers of transmigrated monocytes (Fig. 1A, open bars) and lymphocytes (Fig. 1B, open bars) when HIV-infected cells were used in our model compared with uninfected cells (Fig. 1A, B, labeled C on the x-axis). The magnitude of increased monocyte transmigration depended on the viral strain (Fig. 1A). HIV-1 infection with the R5 viruses, ADA and 92BR021, induced significantly higher monocyte transmigration in response to CCL2 compared with HIVJR-CSF (R5), HIV92UG029 (X4), HIV92UG021 (X4), or HIV92RW009 (R5X4). HIVJR-CSF was unusual in that, although an R5-tropic virus, it induced high transmigration of lymphocytes but lower transmigration of monocytes when compared with HIVADA or HIV92BR021. This may be attributable to the fact that HIVJR-CSF infection of PBMC caused extensive aggregation of monocytes after transmigration, reducing their ability to be detected by FACS analysis. Transmigration across the BBB was totally chemokine dependent, because very low levels of transmigration were detected in the absence of CCL2 (Fig. 1, closed bars). Infection with all HIV strains was associated with higher lymphocyte transmigration in response to CCL2 (Fig. 1B, open bars) and was not statistically different among cells infected with R5-, X4-, or R5X4 HIV isolates. Our results indicate the addition of activated PBMCs or HIV-infected PBMCs to the top chamber of our model does not result in significantly increased transmigration of these cells across the BBB model in the absence of a chemotactic stimulus. The establishment of a CCL2 chemotactic gradient, however, resulted in greatly enhanced transmigration of HIV-infected PBMCs when compared with uninfected/activated cells.

HIV-infected PBMC transmigration across an in vitro BBB model in response to CCL2 is greatly increased compared with that of uninfected cells. Uninfected (C) or HIV-infected human PBMCs were added to the top chamber of the BBB model, consisting of cocultured human ECs and astrocytes in the absence (filled bars) or presence (open bars) of CCL2 (100 ng/ml) in the bottom chamber. PBMCs were infected with the R5 viral strains ADA, 92BR021, and JR-CSF, the X4 viral strains 92UG029 and 92UG021, and the R5X4 strain 92RW009. After 24 h, transmigrated cells in the bottom chamber were immunostained with PE-conjugated anti-CD14 (a monocyte marker) and FITC-conjugated anti-CD45 (a pan leukocyte marker) antibodies and analyzed by flow cytometry to quantify the number of monocytes (CD14+) (A) and lymphocytes (CD14−) (B) in the transmigrated population of cells. The data are expressed as mean ± SD of 21 experiments. *p < 0.005 of CCL2 compared with no CCL2 in each group, #p < 0.005 of HIV-infected cells with CCL2 compared with uninfected (C) cells with CCL2; &p < 0.005 of cells infected with JR-CSF, 92UG029, 92UG021, and 92RW009 with CCL2 compared with ADA-infected cells with CCL2.
The enhanced transmigration of HIV-1-infected PBMC in response to CCL2 resulted in a significant increase in BBB permeability
The permeability of our BBB model was analyzed after leukocyte transmigration by quantifying the passage of albumin conjugated to Evans blue dye through cocultures. Before transmigration of cells, these cocultures are highly impermeable to the passage of albumin. The addition of uninfected PBMCs to the top chamber of our model in the absence of CCL2 did not affect the baseline permeability of cocultures (Fig. 2, closed bars). Transmigration of uninfected cells in response to CCL2 (100 ng/ml) (Fig. 2C, x-axis, open bar) resulted in a small, nonsignificant increase in BBB permeability, when compared with cocultures without CCL2 (Fig. 2, closed bar).

Transmigration of HIV-infected PBMCs across the BBB in response to CCL2 increases BBB permeability. The permeability of the BBB model was analyzed after uninfected (C) or HIV-infected PBMC transmigration for 24 h in the absence (filled bars) or presence (open bars) of CCL2 (100 ng/ml) by quantifying the passage of albumin conjugated to Evans blue dye through cocultures. EDTA (4 mm) treatment (E) of cocultures was used as a positive control for disruption of the BBB. The data are expressed as a percentage of permeability as described in Materials and Methods. The data represent the mean ± SD. *p < 0.005 of CCL2 compared with no CCL2 in each group (n = 21); #p < 0.005 of HIV-infected cells with CCL2 compared with uninfected (C) cells with CCL2 (n = 7); &p < 0.005 of cells infected with JR-CSF, 92UG029, 92UG021, and 92RW009 with CCL2 compared with ADA-infected cells with CCL2 (n = 19).
PBMC infected with the HIV strains ADA, 92BR021, JR-CSF, 92UG029, 92UG021, or 92RW009 were added to the top chamber of BBB cocultures without CCL2 in the bottom chamber. After 24 h, there was neither significant transmigration, as described above (Fig. 1), nor disruption of BBB impermeability under these conditions (Fig. 2, closed bars). The addition of CCL2 to the lower chamber induced high levels of HIV-infected cell transmigration, as described above (Fig. 1), and resulted in very significant increases in BBB permeability that were detected with all of the HIV strains tested (Fig. 2, open bars), compared with cocultures after uninfected PBMC transmigration (Fig. 2, labeled C on the x-axis, open bar). BBB cocultures after CCL2-mediated transmigration of HIVADA- or HIV92BR021-infected PBMC showed significantly higher permeability compared with that obtained when leukocytes were infected with HIVJR-CSF, HIV92UG029, HIV92UG021, or HIV92RW009, suggesting that, as was the case with transmigration, barrier disruption was also more dependent on PBMC infection with R5 rather than X4 or R5X4 strains. These differences in transmigration and permeability were not associated with different levels of viral infection, because p24 levels in the PBMC supernatants were similar for all of the viral isolates used in this study (879 ± 193 ng/ml). In control experiments, CCL2 alone (100 or 500 ng/ml) in the bottom chamber of cocultures without PBMC in the top chambers did not affect BBB permeability (data not shown). Also, the addition of CCL2 to the top and bottom chambers of cocultures to eliminate the establishment of a chemotactic gradient toward the bottom chamber did not result in significant leukocyte transmigration or alterations in BBB permeability (data not shown). These results suggest that the adhesion of HIV-infected cells to the BBB model, in the absence of significant leukocyte transmigration resulting from the absence of a CCL2 chemotactic gradient, was not sufficient to induce BBB disruption. BBB permeability was increased only when the combination of HIV-infected cells and a CCL2 chemotactic gradient were present.
CCL2 enhanced the transmigration mainly of HIV-infected monocytes
We determined whether CCL2 preferentially induced the transmigration of HIV-infected cells from the mixed population of uninfected and HIV-infected PBMC used in our transmigration assays. We analyzed input HIV-infected PBMCs, before their transmigration, by double immunofluorescence for CD14 and p24 to quantify the percentage of monocytes (CD14+) and lymphocytes (CD14−) that were HIV infected (p24+). We then analyzed the cells that transmigrated across the BBB model in response to CCL2 to determine the percentage of HIV-infected monocytes and lymphocytes in this population after transmigration.
For the input PBMC cultures infected with the R5 virus, HIVADA, ∼61.5 ± 3.6% of the monocytes and 5.2 ± 1.3% of the lymphocytes were infected. In contrast, for input PBMCs infected with the X4 virus, HIV92UG021, 98.5 ± 0.9% of the lymphocytes and only 1.97 ± 0.5% of the monocytes were infected. Transmigration assays using HIVADA-infected PBMC and CCL2 resulted in a population of transmigrated leukocytes for which the percentage of HIV-infected monocytes was increased to 92.1 ± 2.3%, suggesting a CCL2-mediated enrichment process for transmigration of HIV-infected monocytes. However, we did not detect enrichment in HIVADA-infected lymphocytes. CCL2-mediated transmigration of HIV92UG021-infected cells could not result in enrichment of HIV-infected lymphocytes, because the percentage of input HIV-infected lymphocytes was already 98.5 ± 0.9%. In addition, we did not detect enrichment of HIV92UG021-infected monocytes from the starting population of 1.9 ± 0.5%. These results suggest that CCL2 enhances transmigration of HIV-infected monocytes when they are infected with an R5 virus but not with an X4 virus. Additional studies in our laboratory are addressing this finding.
Enhanced HIV-infected PBMC transmigration and BBB disruption are specifically CCL2 dependent
To analyze whether increased transmigration of HIV-infected PBMC and the associated increase in BBB permeability was specific to CCL2, we determined whether addition of the chemokines CXCL10, CCL3, or CCL5 (100 ng/ml) to the bottom chamber of the BBB model replicated the results obtained with CCL2. After the addition of HIV-infected PBMC, the levels of monocyte (Fig. 3A) and lymphocyte (Fig. 3B) transmigration elicited by these chemokines were significantly less than those induced by CCL2 and were similar to those of uninfected PBMCs (data not shown). Additional assays were performed with 500 ng/ml CXCL10, CCL3, or CCL5 to address the possibility that these results were attributable to a suboptimal concentration of these chemokines. We did not detect increased chemoattraction at this dose (data not shown). BBB permeability was also not altered after 24 h of transmigration of HIV-infected PBMCs in response to these chemokines (Fig. 3C), suggesting that increased transmigration levels and associated changes in BBB permeability are specifically mediated by CCL2.

Unlike CCL2, CXCL10, CCL3, and CCL5 did not induce high levels of HIV-infected PBMC transmigration or increase BBB permeability. After the addition of HIV-infected PBMC to cocultures, monocyte (A) and lymphocyte (B) transmigration (24 h) in the presence of CCL2, CXCL10, CCL3, or CCL5 (100 ng/ml) was quantified by flow cytometry. The permeability of the BBB model (C) was analyzed after HIV-infected PBMC transmigration for 24 h in the presence of CCL2, CXCL10, CCL3, or CCL5 (100 ng/ml). The data represent the mean ± SD. *p < 0.005 of CXCL10, CCL3, or CCL5 compared with CCL2 (n = 5).
The fact that CXCL10, CCL3, or CCL5 did not enhance transmigration of HIV-infected leukocytes in the same numbers as CCL2 was not because the HIV-infected cells lack the appropriate receptors, CXCR3 and CCR5. Our data and those of others indicate that CXCR3 and CCR5 are maintained or upregulated on HIV-infected leukocytes (Wetzel et al., 2002; Lane et al., 2003) (E.A.E. and J.W.B., unpublished data). In addition, HIV-infected cells did transmigrate in our model in response to these chemokines, just not in high numbers, suggesting that these chemokine receptors are functional.
CCL2-mediated transmigration of HIV-infected PBMC reduced TJP expression in BBB cells
We analyzed the distribution and protein levels of the TJPs, ZO-1, claudin-1, and occludin in BBB cells after 24 h of transmigration of uninfected or HIV-infected PBMCs, with and without CCL2, as determined by immunofluorescence and Western blot analysis. Cocultures were immunostained with antibodies to occludin, claudin-1, and ZO-1, and confocal analysis of the Z-series, which corresponds to the EC layer of cocultures, was performed. After transmigration of uninfected PBMCs in the absence (data not shown) or presence of CCL2, ECs and astrocytes were positive for occludin (Fig. 4A), claudin-1 (Fig. 4D), and ZO-1 (Fig. 4G). Occludin and claudin-1 were distributed mainly in cell-to-cell apposition membranes between BBB cells, whereas ZO-1 showed a diffuse pattern of immunoreactivity as well as being detected at intercellular junctions. This diffuse pattern of immunoreactivity is attributable to the BBB model, which has end feet processes of astrocytes in the apical side that also have ZO-1 reactivity. Therefore, the ECs in our coculture model do not have the typical honey comb distribution normally seen in epithelial cells or endothelial cells cultured alone. An example of this astrocyte end feet reactivity can be observed in Figure 4H (inset). For this illustration, the cocultures were stained with antibodies to VWF to identify ECs and with GFAP to identify astrocyte processes. The top surface of the coculture was visualized by confocal microscopy to show the presence of astrocyte end feet between ECs (Fig. 4H, inset).

Immunofluorescence and Western blot analysis of BBB cocultures after HIV-infected PBMC transmigration in response to CCL2 indicates that TJP expression in BBB cells is reduced. After transmigration (24 h) of uninfected (Control) or HIV-infected PBMC in response to CCL2, cocultures were immunostained with occludin, claudin-1, and ZO-1 antibodies and examined by confocal microscopy. After transmigration of uninfected cells, surface scanning of our coculture model showed a characteristic distribution of occludin (A) and claudin-1 (D) in cell-to-cell apposition membranes, whereas ZO-1 immunoreactivity (G) exhibited a diffuse pattern of expression. This is attributable to the reactivity of astrocyte foot processes, positive for TJP, that protrude into the EC layers. After HIVADA-infected (similar results were obtained for HIVJR-CSF) or HIV92UG021-infected PBMC transmigration, the expression of occludin (B, C), claudin-1 (E, F), and ZO-1 (H, I) was greatly reduced. The inset in H represents the staining for VWF (red) and GFAP (green) to demonstrate astrocyte end foot processes on the endothelial monolayer. Scale bar, 50 μm. These results are representative of 12 separate experiments. Protein lysates of BBB cells were prepared from cocultures by scraping off the cells from 16 inserts after transmigration for 24 h of uninfected (Con) and HIV-infected PBMCs in the absence (−) or presence of CCL2 (M). Lysates were analyzed by Western blotting for occludin, claudin-1, and ZO-1. Occludin, claudin-1, and ZO-1 expression in cocultures was reduced only after HIVADA-, HIVJR-CSF-, and HIV92UG021-infected PBMC transmigration in the presence of CCL2 (J). EC protein lysate (S) was used as a positive control to identify the electrophoretic mobility of TJP reactive bands. Blots were stripped and incubated with a histone-1 antibody to document equivalent protein loading. These results are representative of five separate experiments.
CCL2-mediated HIVADA-infected (Fig. 4B, E, H), HIVJR-CSF-infected (data not shown), and HIV92UG021-infected (Fig. 4C, F, I) PBMC transmigration resulted in a dramatic reduction in TJP immunoreactivity. There was a significant reduction in the number of cells expressing these proteins, as well as in the fluorescence intensity of each cell. Western blot analysis of TJPs in BBB cells after transmigration of HIV-infected PBMCs confirmed our immunofluorescence data (Fig. 4J). These results show a direct correlation between transmigration of HIV-infected PBMCs in response to CCL2, BBB permeability, and decreased TJP expression. The transmigration of HIVADA-, HIVJR-CSF-, or HIV92UG021-infected PBMCs in the absence of CCL2 did not alter TJP expression or distribution (data not shown).
CCL2-mediated HIV-infected PBMC transmigration induced MMP-2 and MMP-9 in BBB cells
MMPs are expressed in inflammatory conditions by many cell types, including leukocytes, ECs and astrocytes (Welgus et al., 1990; Werb, 1997), and degrade components of the BBB, such as the EC basement membrane, thereby increasing permeability (Werb, 1997). Previous reports showed that elevated levels of MMP-2, -7, and -9 in the CSF were associated with HIV dementia (Conant et al., 1999; Suryadevara et al., 2003). We examined whether CCL2-mediated transmigration of HIV-infected PBMC affected MMP-2 and MMP-9 expression on BBB cells by confocal analysis of cocultures immunostained with antibodies to MMPs. Phase pictures of BBB membrane cross sections are shown in Figure 5, A, C, E, and G. MMP-2 or -9 staining was not detected in cocultures after transmigration of uninfected cells in the absence of CCL2 (Fig. 5B). After CCL2-mediated cell migration, low levels of MMP-2 staining (Fig. 5D) and no detectable staining for MMP-9 was observed (data not shown). The presence of CCL2 in the lower chamber for 24 h without PBMC in the upper chamber did not induce expression of MMP-2 or MMP-9 (data not shown). After CCL2-mediated transmigration of HIVADA-infected PBMCs, cocultures showed bright staining and broad distribution for both MMPs compared with cocultures after CCL2-mediated transmigration of uninfected leukocytes. MMP-2 expression was increased in both cell layers and in the pores of the BBB model, where astrocyte foot processes are located, as well as transmigrating leukocytes (Fig. 5F). MMP-9 immunoreactivity was limited to the EC and astrocyte layers and was not detected in the pores (Fig. 5H). No differences were detected in the intensity or distribution of MMP-2 or -9 staining when PBMCs were infected with different strains of virus (data not shown).

Immunofluorescence analysis of BBB cocultures after HIV-infected PBMC transmigration in response to CCL2 shows MMP-2 and MMP-9 expression in BBB cells. After 24 h of transmigration of uninfected and HIVADA-infected PBMCs in response to CCL2, cocultures were immunostained with MMP-2 and MMP-9 antibodies and examined by confocal microscopy. A, C, E, and G are phase contrast views of cocultures with the asterisk indicating the EC monolayers of the BBB model. After uninfected PBMC transmigration in the absence of CCL2, MMP-2 staining was not detected (B). After CCL2-mediated uninfected cell migration, low levels of MMP-2 staining by ECs and astrocytes were observed (D). MMP-2 (F) and MMP-9 (H) staining was greatly increased after transmigration of HIVADA-infected PBMCs in response to CCL2 and was localized to both ECs and astrocytes. The arrows indicate pores in the insert membrane. Scale bar, 90 μm. These results are representative of seven separate experiments.
Treatment of the BBB with HIV-tat and/or gp120 or infectious supernatants from HIV-infected leukocytes did not dramatically increase permeability
Indirect effects of HIV infection, including the elaboration of neurotoxic viral proteins such as tat and gp120, may contribute to alterations in BBB cells (Corasaniti et al., 2001a, b; Galey et al., 2003). To address whether these HIV proteins alone or infectious supernatants from HIV-infected leukocytes may contribute to BBB disruption in our model, we added tat (100 ng/ml) and/or gp120 (100 nm; R5 or X4) or supernatants from HIV-infected leukocytes (obtained 7 d after infection) directly to the top chamber of our model for 24 h, in the absence or presence of CCL2 in the bottom of the chamber, and then assayed for permeability. These treatments modestly increased BBB permeability, whether CCL2 was present or not, compared with untreated cocultures (Table 1). These data suggest that the direct effects of HIV-soluble proteins on the BBB, in the presence or absence of CCL2, may contribute to but are not sufficient for the extensive disruption of the BBB seen with transmigration of HIV-infected cells.
BBB permeability after HIV-tat and/or gp120 treatment or treatment with supernatants from HIV-infected PBMCsa
Leukocytes infected with HIV expressed higher levels of CCR2
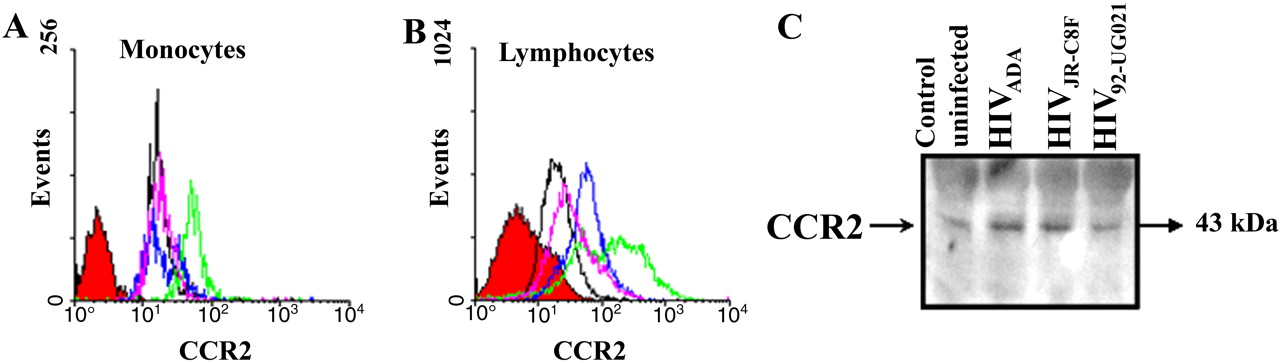
To determine why HIV-infected PBMCs exhibited such exuberant transmigration across the BBB model in response to CCL2, we examined by FACS analysis the surface expression of CCR2, the receptor for CCL2, on uninfected and HIV-infected cells. CCR2 on the surface of monocytes was greatly enhanced with HIVADA infection (Fig. 6A, green line), minimally increased with HIVJR-CSF infection (Fig. 6A, blue line), and unchanged with HIV92UG021 infection (Fig. 6A, pink line) compared with uninfected cells (Fig. 6A, black line). These results support our finding that monocyte transmigration after HIVADA infection is higher than that observed after infection with the other HIV isolates, perhaps because these monocytes are more sensitive to CCL2 because of higher CCR2 expression. CCR2 on the surface of lymphocytes was increased regardless of which HIV isolate was used (Fig. 6B). These results are concordant with the high-lymphocyte transmigration induced by CCL2, regardless of which HIV strain was used to infect PBMC. For each condition, leukocytes were incubated with an isotype-matched mouse myeloma antibody as a negative control (Fig. 6, red-filled curve). Flow cytometry results were confirmed by Western blot analysis of whole-cell lysate, including membrane and intracellular CCR2 protein, of uninfected (control) and HIVADA-, HIVJR-CSF-, or HIV92UG021-infected PBMCs (Fig. 6C), further indicating that HIV infection increases CCR2 expression.

Infection of PBMC with different HIV strains increases CCR2 expression. FACScan analysis of nonpermeabilized monocytes (A) and lymphocytes (B) immunostained with a CCR2 antibody indicate that infection of PBMC with ADA (green line), JR-CSF (blue line), and 92UG021 (pink line) viral strains increases the surface expression of CCR2 on lymphocytes compared with uninfected cells (black line), whereas ADA and JR-CSF viral strains increased CCR2 on monocytes. Cells were also incubated with an isotype-matched mouse myeloma antibody as a negative control for nonspecific staining (red-filled curve). These results of surface FACS analyses were corroborated by quantification of total CCR2 protein by Western blot analysis for CCR2 in uninfected (Control) and HIVADA-, HIVJR-CSF-, and HIV92UG021-infected PBMCs. These results are representative of five separate experiments.
Discussion
In the present study, we demonstrate that HIV-infected leukocytes transmigrate in greater numbers across a tissue culture model of the human BBB in response to CCL2 than do uninfected cells, resulting in increased BBB permeability. This process was characterized by four major findings. First, BBB permeability was increased only when the combination of HIV-infected cells and a CCL2 chemotactic gradient was present. The addition of CCL2 alone or the adhesion of HIV-infected cells alone to the BBB model was not sufficient to induce BBB disruption. Second, the mechanism of BBB disruption and enhanced transmigration of HIV-infected cells was specifically CCL2 dependent, because other chemokines did not replicate the CCL2 effect. Third, the high HIV-infected leukocyte transmigration induced by CCL2 was associated with increased BBB permeability that correlated with a reduction in TJP (ZO-1, claudin-1, and occludin) and an increase in MMP-2 and MMP-9 in BBB cells. Fourth, HIV-infected leukocytes expressed increased surface CCR2.
HIV entry into the CNS occurs early after primary infection and is believed to be due mainly to the transmigration of HIV-infected leukocytes into the brain, providing a source of virus that can then infect CNS resident cells, such as microglia and perivascular macrophages (Wiley et al., 1986; Jordan et al., 1991). This transmigration process is characterized by leukocyte-endothelial interactions using a variety of molecules, including integrins, adhesion molecules, proteases, TJPs, and cytokine/chemokines and their receptors. Our hypothesis is that the specific presence of CCL2 within the CNS plays a key role in the recruitment of HIV-infected leukocytes, as well as in the impairment of the BBB, so as to enhance leukocyte transmigration into the brain. This hypothesis may be relevant to several stages leading to NeuroAIDS.
First, our current results provide a possible mechanism for early HIV entry into the brain, because high HIV replication within leukocytes, as occurs during the initial acute stage of infection, and the presence of even low levels of CCL2 within the CNS parenchyma may be critical to enhanced HIV-infected leukocyte transmigration into the CNS and associated transient BBB disruption. In addition, the increased sensitivity of HIV-infected leukocytes to CCL2, as a result of their increased CCR2 expression, suggests that these virally infected cells can respond to the presence of CCL2 in the CNS that may not be detected by uninfected cells. Previous reports demonstrated that under normal conditions, there are baseline chemokine gradients within the CNS during microglia turnover and migration into the CNS and during development/synaptic plasticity and neuronal/glial migration (Eglitis and Mezey, 1997; Rezaie and Male, 1997, 1999; Rezaie et al., 1997; Aarum et al., 2003). We propose that HIV-infected cells respond to the constitutively expressed CCL2 within the CNS to enter the brain by transmigrating across the CNS vasculature and causing transient BBB disruption, thereby remaining protected within the CNS, even after HAART treatment (Chun et al., 1997; Chang et al., 2003; Sevigny et al., 2004).
Viral amplification within the CNS involves the recruitment of CNS resident cells, such as uninfected microglia and astrocytes, to sites of HIV-infected leukocyte infiltration and active infection. Our published data demonstrated that HIV-tat protein was a mediator of chemotaxis of uninfected microglia by a mechanism that is CCL2 dependent. This finding suggested that CCL2 may play an important role in facilitating the recruitment of uninfected cells to the regions within the CNS that have HIV-infected monocytes, resulting in viral spread within the CNS (Eugenin et al., 2005). The resulting activation and/or infection of CNS resident cells, including perivascular macrophages and microglia, triggers a chronic inflammatory response that may contribute to ongoing CNS impairment. As the pathogenesis of NeuroAIDS progresses, there is ultimately a dramatic increase in inflammation and viral production that is also associated with BBB disruption and infiltration of leukocytes into the brain, resulting in severe cognitive impairment. These proposed stages are associated with the clinical evolution of HIV infection resulting in CNS damage and dementia (Adams and Ferraro, 1997).
We and others have demonstrated that the expression of chemokines and their receptors is increased in HIV-1 and simian immunodeficiency virus encephalitic brain tissue compared with normal brain, as well as in cultured human fetal microglia, astrocytes, and neurons (Sasseville et al., 1996; Gabuzda et al., 1998; Kelder et al., 1998; Sanders et al., 1998; Albright et al., 1999; Conant, 2000; McManus et al., 2000a, b; Zink et al., 2001; Mankowski et al., 2004). Experiments in transgenic mice and humans indicate that CCL2 is an essential chemokine for monocyte transmigration into the brain (Fuentes et al., 1995; Gonzalez et al., 2002), and elevated levels of CCL2 have been detected in the CSF of individuals with AIDS dementia (Conant et al., 1998; Kelder et al., 1998). Thus, CCL2 appears to play an important role in neurological aspects of HIV disease pathogenesis. Our results that HIV-infected leukocytes transmigrated in higher numbers than uninfected cells in specific response to CCL2, but not to other chemokines, suggest that these infected cells have an enhanced sensitivity to local and specific concentrations of CCL2 in the CNS compared with uninfected leukocytes. This increased transmigration directly correlated with increased BBB disruption, with the most significant changes in permeability elicited by monocytes infected with R5 viruses, suggesting that these cells are most involved in alterations in BBB permeability during the process of transmigration. For all of these experiments, we chose to assay transmigration at 24 h, because this was the time point at which we detected maximal monocyte transmigration. Our in vitro findings correlate with the pathogenesis of viral disease in the CNS as HIV strains isolated from CNS and CSF are notably R5-tropic (Cheng-Mayer et al., 1989; Liu et al., 1990). We also found that when monocyte transmigration was assayed after infection with X4 virus, it was lower than with R5 but higher than for uninfected cells. This suggests that the few monocytes infected with the X4 virus (2%) or HIV-infected lymphocytes facilitate, in some way, increased monocyte transmigration. Nonetheless, the degree of monocyte transmigration is far more extensive when R5 viruses are used, suggesting that viral tropism is important in facilitating monocyte transmigration and subsequent BBB disruption. The number of HIV-infected lymphocytes that transmigrated in response to CCL2 was relativity constant regardless of whether R5 or X4 isolates were used. This suggests that either infected monocytes are producing a factor that facilitates T-cell migration or that infected monocytes disrupt the BBB as they transmigrate and that this disruption facilitates T-cell transmigration. Additional experiments will address these possibilities.
The BBB disruption resulting from CCL2-mediated transmigration of HIV-infected leukocytes was associated with high levels of MMP-2 and MMP-9 in the BBB model. Whereas MMP-2, but not MMP-9, was detected during the transmigration process of uninfected cells in response to CCL2, both MMPs were detected in higher intensity and broader distribution after the transmigration of HIV-infected leukocytes in response to this chemokine, suggesting that the combination of CCL2 plus HIV-infected cell transmigration enhanced the expression of these MMPs. In previous studies, BBB permeability was shown to be altered by increased expression of MMPs. Microinjection of MMPs into the brain led to extensive leakage of the BBB that was reduced by MMP inhibitors. After the microinjection of MMP-2 into rat brain, MMP-9 expression was induced and BBB permeability was increased (Rosenberg et al., 1994; Romanic et al., 1998). In our study, the enhanced expression of MMP-2 and -9 may indicate proteolysis of proteins that contribute to the integrity of the BBB.
The increased transmigration of HIV-infected leukocytes in response to CCL2 was correlated with enhanced expression of CCR2 on the surface of these cells. Analysis of CCR2 by FACS, or total expression by Western blot, in HIV-infected leukocytes demonstrated that HIV infection upregulated CCR2 expression. These findings suggest that HIV, in some way, induces or maintains the expression of CCR2 on the surface of leukocytes. Note that CCR2 expression on lymphocytes exposed to a M-tropic virus was also increased. This may be because of the effects of virus on the lymphocytes or to the presence of infected monocytes within the PBMC population that may be producing a factor that increases CCR2 expression on lymphocytes. Future experiments will address these possibilities. We propose that increased CCR2 expression on both monocytes and lymphocytes may facilitate their ability to invade the brain parenchyma and to respond to lower concentrations of CCL2 within specific regions of the CNS. Our findings suggest CCL2/CCR2 as potential therapeutic targets to reduce or prevent the early infiltration of HIV-infected leukocytes into the CNS and their subsequent devastating consequences.
Footnotes
-
This work was supported by the National Institutes of Mental Health Grants MH52974 and MH070297 (E.A.E., T.M.C., J.W.B.), National Institute of Neurological Disorders and Stroke Grant NS39201 (H.G., K.O.), National Institute of Allergy and Infectious Diseases Grant Al48466 (H.G., K.O.), National Institutes of Health (NIH) Grant NS11920 (E.A.E., T.M.C., J.W.B.), and NIH Centers for AIDS Research Grant AI-051519 (to all authors). We thank the Fetal Tissue Repository at the Albert Einstein College of Medicine.
- Correspondence should be addressed to Dr. Joan W. Berman, Department of Pathology, F727, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. Email: berman{at}aecom.yu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}