

Abstract
Most human idiopathic generalized epilepsies (IGEs) are polygenic, but virtually nothing is known of the molecular basis for any of the complex epilepsies. Recently, two GABAA receptor δ subunit variants (E177A, R220H) were proposed as susceptibility alleles for generalized epilepsy with febrile seizures plus and juvenile myoclonic epilepsy. In human embryonic kidney 293T cells, recombinant hα1β2δ(E177A) and hα1β2δ(R220H) receptor currents were reduced, but the basis for the current reduction was not determined. We examined the mechanistic basis for the current reduction produced by these variants using the hα4β2δ receptor, an isoform more physiologically relevant and linked to epileptogenesis, by characterizing the effects of these variants on receptor cell surface expression and single-channel gating properties. Expression of variant α4β2δ(R220H) receptors resulted in a decrease in surface receptor proteins, and a smaller, but significant, reduction was observed for variant α4β2δ(E177A) receptors. For both variants, no significant alterations of surface expression were observed for mixed population of wild-type and variant receptors. The mean open durations of α4β2δ(E177A) and α4β2δ(R220H) receptor single-channel currents were both significantly decreased compared to wild-type receptors. These data suggest that both δ(E177A) and δ(R220H) variants may result in disinhibition in IGEs by similar cellular and molecular mechanisms, and in heterozygously affected individuals, a reduction in channel open duration of δ subunit-containing GABAA receptors may be the major contributor to the epilepsy phenotypes.
Introduction
Genetic factors play an important role in the pathogenesis of idiopathic generalized epilepsies (IGEs) (Mulley et al., 2003; Gourfinkel-An et al., 2004). Although the majority of mutations associated with monogenic IGEs are in voltage- or ligand-gated ion channel genes (Mulley et al., 2003), they only account for a small portion of genetic epilepsies (Scheffer and Berkovic, 2003), suggesting that the majority of IGEs are polygenic requiring additive action of an unknown number of susceptibility genes (Gargus, 2003; Scheffer and Berkovic, 2003; Mulley et al., 2005).
GABAA receptors are ligand-gated chloride channels that mediate the majority of inhibition in the brain. More than 16 GABAA receptor subunit subtypes have been identified (Olsen and Macdonald, 2002), and it has been proposed that αβγ and αβδ may be the major isoforms present in vivo (McKernan and Whiting, 1996). GABAA receptor stoichiometry has been shown to be 2α, 2β, and 1γ or 1δ (Olsen and Macdonald, 2002). GABAA receptors play a critical role in several animal models of seizures (Sperk et al., 2004), and GABAA receptor subunit mutations have been reported to be associated with human monogenic IGEs (Macdonald et al., 2004). GABAA receptor γ2 subunit mutations were linked to generalized epilepsy with febrile seizures plus (GEFS+) (Baulac et al., 2001; Harkin et al., 2002) and childhood absence epilepsy and febrile seizures (Wallace et al., 2001; Kananura et al., 2002), and a GABAA receptor α1 subunit mutation was associated with juvenile myoclonic epilepsy (JME) (Cossette et al., 2002). Studies of recombinant receptors incorporating the mutant subunits demonstrated reduced GABAA receptor current amplitudes and/or current duration, which would increase neuronal excitability (Baulac et al., 2001; Bianchi et al., 2002; Bowser et al., 2002; Cossette et al., 2002; Harkin et al., 2002; Gallagher et al., 2004).
Recently, a rare variant of the GABAA receptor δ subunit (E177A) was reported to be heterozygously associated with GEFS+, and a polymorphic allele (R220H) was present in both a family with JME and in the general population (Dibbens et al., 2004). The two variants were proposed to be susceptibility alleles of the GABRD gene in complex epilepsy, because whole-cell currents from hα1β2δ receptors containing the δ(E177A) or δ(R220H) variant were significantly smaller than those of wild-type receptors. However, it is not known whether the reduction in whole-cell current was attributable to a decrease in receptor expression and/or a defect in channel gating. Furthermore, α4 and δ subunits preferentially coassemble in the thalamus (Sur et al., 1999), a structure critical for expression of IGEs, and alterations in α4 and δ subunit proteins were reported in experimental temporal lobe epilepsy and proposed to contribute to epileptogenesis (Peng et al., 2004). Therefore, whole-cell current amplitude and time course, surface expression, and gating properties of hα4β2δ receptors containing each of the variant δ subunits were examined to determine the mechanistic basis for these susceptibility variants contributing to the pathogenesis of complex IGEs.
Materials and Methods
Transfection and selection of recombinant GABAA receptors.
Human β2 and δ subunits were subcloned into the pcDNA3.1+ plasmid (Invitrogen, Carlsbad, CA), and the α4 subunit was subcloned into the pRK-5 plasmid (BD Biosciences, San Jose, CA) as well as the pcDNA3.1+ plasmid. Electrophysiological recordings confirmed that whole-cell α4β2δ currents obtained with transfection of α4 subunit in pRK-5 or pcDNA3.1+ plasmids were equivalent (data not shown). To be consistent, all data reported for α4β2δ receptors were derived from the α4 subunit in the pRK-5 plasmid. The δ subunit variants (E177A, R220H) were made using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). A V5 epitope (Gly Lys Pro Ile Pro Asn Pro Leu Leu Gly Leu Asp Ser Thr) was inserted at the N terminus (between the fourth and fifth amino acids) of the wild-type or variant δ subunit to study the receptor expression. Electrophysiological recordings demonstrated that α4β2δ receptors containing the epitope-tagged wild-type or variant δ subunit had currents that were similar to those from nonepitope-tagged δ subunits (data not shown). All wild-type and variant subunit cDNAs were verified by DNA sequencing (Vanderbilt DNA Sequencing Facility, Nashville, TN).
Human embryonic kidney 293T (HEK293T) cells were maintained in DMEM (Invitrogen), supplemented with 10% fetal bovine serum (Invitrogen), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Grand Island, NY) in an incubator with 5% CO2/95% air. Cells were transfected with cDNAs encoding human α4, β2, and wild-type as well as variant δ subunits using a modified calcium phosphate precipitation method (Feng et al., 2004). For wild-type receptor subunit expression on whole cells, 2 μg of each subunit cDNA of wild-type α4, β2, and δ were used (1:1:1 ratio). For variant-containing receptor expression, 2 μg of each subunit cDNA of α4, β2, and variant δ was used. For coexpression of wild-type and variant receptors on whole cells, to simulate the heterozygote individual, 2 μg of α4 and β2 as well as 1 μg of wild-type and 1 μg of variant δ subunit cDNAs were used so that the total amount of “δ” subunit cDNA was still 2 μg. For transfections for electrophysiological recordings, 2 μg of pHOOK (Invitrogen) was cotransfected as a selection marker. The transfected cells were selected using an immunomagnetic bead separation method (Greenfield et al., 1997), and the electrophysiological recordings were performed 24 h later.
Whole-cell and single-channel recordings.
Whole-cell and single-channel currents were recorded at room temperature using the patch-clamp technique. The cells were bathed in an external solution composed of the following (in mm): 142 NaCl, 1 CaCl2, 6 MgCl2, 8 KCl, 10 glucose, and 10 HEPES, pH 7.4 (327–330 mOsm). The recording pipette internal solution consisted of the following (in mm): 153 KCl, 1 MgCl2, 10 HEPES, 2 MgATP, and 5 EGTA, pH 7.3 (301–303 mOsm). The combination of the external and internal solutions produced an ECl value of 0 mV and an EK value of −75 mV. The low resistance recording pipettes (0.8–1.5 MΩ) for whole-cell recordings were pulled from the thin-wall borosilicate glass [inner diameter (i.d.), 1.12 mm; outer diameter (o.d.), 1.5 mm; World Precision Instruments, Sarasota, FL], and the high resistance recording pipettes (6–16 MΩ) for single-channel recordings were pulled from the thick-wall borosilicate glass (i.d., 0.84 mm; o.d., 1.5 mm; World Precision Instruments) on a P-2000 Quartz Micropipette Puller (Sutter Instruments, Novato, CA). The micropipettes were fire polished on an MF-830 Micro Forge (Narishige, Tokyo, Japan) before use. The micropipettes for single-channel recordings were coated with polystyrene Q-dope (GC Electronics, Rockford, IL) to minimize the noise during recordings.
Whole-cell and single-channel currents were low-pass filtered at 2 kHz and were recorded using an Axopatch 200A amplifier (Molecular Devices, Foster City, CA) and Digidata 1322A data acquisition system (Molecular Devices). Cells were voltage clamped at −50 mV for whole-cell recordings and at −75 mV for single-channel recordings. Series resistance was not compensated. Series resistance errors might cause an underestimation of peak current. However, the resistance of the whole-cell recording electrode was low, and the average peak current amplitude of α4β2δ receptors was <300 pA. The voltage drop across the electrode was negligible compared to the holding potential. In addition, it was previously reported that the series resistance errors did not significantly affect the extent of desensitization of currents even in the nanoampere range (Bianchi and Macdonald, 2002).
GABA was purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in water to make stock solutions, which were diluted to the desired concentrations with external solution on the day of the experiment. GABA was applied by gravity using a manually pulled three-barrel square glass connected to a Perfusion Fast-Step system (Warner Instruments, Hamden, CT). The open electrode tip 10–90% rise times were <1 ms (Hinkle et al., 2003). Consecutive applications of GABA were separated by an interval of at least 45 s to minimize accumulation of desensitization. The duration of GABA application was 4 s. GABA-evoked single-channel currents were recorded from excised outside-out patches.
Detection of total and surface receptor proteins.
Biotinylation and Western blot analysis were performed to examine the surface receptor expression as described previously (Gallagher et al., 2004; Kang and Macdonald, 2004). HEK293T cells were transfected with human α4, β2, and epitope-tagged wild-type or variant δ subunit using Lipofectamine Plus reagents (Invitrogen) to enhance transfection efficiency. The cells were washed with cold PBS, pH 7.4 (Invitrogen) and incubated with 1 mg/ml NHS-LC-Biotin (Pierce Biotechnology, Rockford, IL) for 1 h at 4°C. The biotinylation reaction was quenched by washing two times with 100 mm glycine. The cells were washed and lysed in 800 μl of ice cold radioimmunoprecipitation assay buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 0.25% Na-deoxycholate, and 1% Triton X-100) mixed with proteinase inhibitor cocktail tablets (Roche, Indianapolis, IN) at a ratio of one tablet/10 ml for 30 min with constant shaking. The lysate was centrifuged at 14,000 × g for 30 min at 4°C, and the supernatant was incubated with streptavidin (Pierce Biotechnology) for 1 h at room temperature. The streptavidin beads were spun down by centrifugation at 14,000 × g and washed with lysis buffer three times. The captured proteins were then eluted from streptavidin with Laemmli sample buffer at 70°C and centrifuged apart at 14,000 × g at room temperature. The supernatant was separated using 7.5% SDS-PAGE, and the proteins were transferred electrophoretically to pure nitrocellulose membrane (0.2 μm) (Bio-Rad, Hercules, CA). The membrane was blocked in a buffer containing 5% nonfat milk and 0.1% Tween 20 for 1 h at room temperature. The membrane was then incubated with the rabbit anti-V5-HRP polyclonal antibody IgG2a (dilution, 1:1000) (Novus Biologicals, Littleton, CO) in blocking buffer overnight at 4°C. After washing, the membranes were incubated with horseradish peroxidase conjugated secondary antibody (goat anti-rabbit IgG; 1:2000; Upstate Biotechnology, Lake Placid, NY). To detect the expression level of β-actin, the internal loading control, the membranes were reblotted with mouse monoclonal anti-β-actin antibody (1:5000; Sigma-Aldrich) and horseradish peroxidase conjugated with goat anti-mouse secondary antibody (1:2000; Upstate Biotechnology). The immunoreactivity of wild-type or variant δ subunit-containing receptors as well as β-actin was visualized with Hyperfilm (Amersham Biosciences, Buckinghamshire, UK) using a chemiluminescence detection system (Bio-Rad). The total receptor proteins (proteins on cell membrane and in the intracellular compartments) were also examined using Western blot. The surface and total proteins were quantified by normalizing to β-actin using program ImageJ (at http://rsb.info.nih.gov/nih-image).
Data analysis.
Whole-cell currents were analyzed off-line using Clampfit 8.1 (Molecular Devices). The normalized concentration-response data were fitted using the following equation: I = Imax/[1 + 10(LogEC50-Logdrug)*Hill slope]. I is the peak current evoked by a given concentration of GABA. Imax is the maximal peak current of GABA. Whole-cell current desensitization was evaluated using extent of desensitization (%), the percentage of current reduction after 4 s of GABA application. The current deactivation phase was fitted using standard exponential Levenberg–Marquardt method in the form of Σanτn, where a is the relative amplitude, τ is the time constant, and n is the number of exponential components. A weighted τ was calculated to compare the rates of deactivation based on the formula: a1*τ1/(a1 + a2) + a2*τ2/(a1 + a2), where a1 and a2 are the relative amplitudes, and τ1 and τ2 are corresponding time constants of the exponential components. GABA-evoked single-channel currents were acquired at 50 μs intervals and analyzed off-line with Fetchan 6.0 (Molecular Devices). Only open-state properties were analyzed, because some patches exhibited overlapping openings, indicating multiple channel activities, which could result in spurious measurements of other kinetic parameters such as open frequency. Kinetic analysis of open states was performed using Interval 5 (Dr. Barry S. Pallotta, University of North Carolina, Chapel Hill, NC). The system dead time was ∼100 μs.
Data were reported as mean ± SEM. Mann–Whitney test and one-way ANOVA followed by Newman–Keuls multiple comparison test were used to compare the current features or protein relative optical density among wild-type and variant receptors. The difference was considered to be statistically significant if p was <0.05.
Results
EC50 values of α4β2δ GABAA receptors were not altered by the variant δ subunits
GABA concentration-response curves were obtained for wild-type α4β2δ receptors and variant-containing α4β2δ(E177A) or α4β2δ(R220H) receptors (Fig. 1A). Compared with wild-type receptors (EC50 = 0.61 μm), no shift of GABA concentration-response curves was observed for α4β2δ(R220H) receptors (EC50 = 0.60 μm), but a small leftward shift was observed for α4β2δ(E177A) receptors (EC50 = 0.45 μm) (Fig. 1B). The Hill coefficient was 1.0, 1.3, and 0.9 for α4β2δ, α4β2δ(E177A), and α4β2δ(R220H) receptors, respectively. When the EC50 values of individual cells were calculated, the mean EC50 value of the α4β2δ receptor was 0.60 ± 0.08 μm (n = 7), consistent with previous reports (Brown et al., 2002; Wallner et al., 2003). The mean EC50 values of variant-containing α4β2δ(E177A) (0.53 ± 0.09 μm, n = 5) and α4β2δ(R220H) (0.54 ± 0.12 μm; n = 5) receptors were not significantly different from that of wild-type receptors.

GABA concentration responses were similar among α4β2δ GABAA receptors containing wild-type or variant δ subunits. A, Examples of whole-cell current traces evoked by increasing GABA concentrations from wild-type α4β2δ and variant α4β2δ(E177A) or α4β2δ(R220H) receptors. B, The current responses were normalized to the maximal current of each cell. The squares denote the mean amplitudes of currents for wild-type α4β2δ receptors (n = 7), the triangles denote those for variant α4β2δ(E177A) receptors (n = 5), and the circles denote those for variant α4β2δ(R220H) receptors (n = 5). The solid bar above each current trace denotes the duration of GABA application (4 s). wt, Wild-type α4β2δ receptors. The error bars represent SEM. The holding potential was −50 mV for wild-type and variant receptors.
Peak current amplitudes and macroscopic kinetic properties of α4β2δ GABAA receptors were altered by variant δ(E177A) subunits
Saturating (1 mm) GABA-evoked currents were recorded from wild-type α4β2δ, a mixed population of wild-type and variant α4β2δ(E177A), and variant α4β2δ(E177A) receptors (Fig. 2A). The mean amplitude of mixed currents (118.0 ± 56.9 pA; n = 26) was significantly smaller than that of wild-type currents (222.0 ± 42.4 pA; n = 77; p < 0.05) (Fig. 2B). The mean amplitude of variant currents (76.5 ± 20.9 pA; n = 25) was also significantly smaller than that of wild-type currents (p < 0.01). Although the mean current amplitude of variant currents was smaller than that of mixed currents, the difference was not significant (p > 0.5) (Fig. 2B).

The peak GABA current amplitudes were reduced, and the kinetic properties were altered for α4β2δ receptors containing mixed wild-type and variant or pure variant δ(E177A) receptors. A, Representative whole-cell current traces evoked by saturating GABA (1 mm) from wild-type α4β2δ, mixed wild-type and variant, and pure variant α4β2δ(E177A) receptors. B, Compared with wild-type receptors (n = 77), the mean current amplitude was smaller for mixed receptors (n = 26) as well as for pure variant receptors (n = 25). C, The mean extent of desensitization was decreased for mixed and pure variant receptors compared with wild-type receptors. D, Comparison of the mean rate of current deactivation among wild-type, mixed, and pure variant receptors. The solid bar above each current trace denotes the duration of GABA application (4 s). The open bars represent the mean current features for wild-type receptors, the hatched bars represent those for the mixed receptors, and the black bars represent those for the pure variant receptors. wt, Wild-type α4β2δ receptors; mix, presence of both receptors; var, pure variant receptors. The error bars denote SEM. The asterisks indicate significant difference from wild-type α4β2δ receptors (*p < 0.05, ** p < 0.01).
During a 4 s application of GABA, wild-type currents exhibited significant slow desensitization, with a reduction of 38.3 ± 0.9% (n = 48) from peak current amplitude (Fig. 2A,C). The mean extent of desensitization was significantly decreased for the mixed (32.7 ± 3.3%; n = 8) and variant (33.1 ± 2.2%; n = 12) currents when compared with wild-type currents (p < 0.05) (Fig. 2A,C). Compared with wild-type currents, the mean deactivation rate of variant currents was significantly increased, although no significant alteration of deactivation rate was observed for the mixed currents (Fig. 2A,D).
Peak current amplitudes, but not macroscopic kinetic properties, of α4β2δ GABAA receptors were altered by variant δ(R220H) subunits
Whole-cell currents evoked by GABA (1 mm) were also recorded from α4β2δ, mixed wild-type and variant α4β2δ(R220H) receptors, and variant α4β2δ(R220H) receptors (Fig. 3A). Compared with wild-type currents, the mean amplitudes for mixed (112.7 ± 52.0 pA; n = 20) (p < 0.01) and variant (46.9 ± 25.0 pA; n = 24) (p < 0.01) currents were significantly smaller (Fig. 3A,B). Although the mean amplitude of variant currents was smaller than that of mixed currents, the difference was not significant (Fig. 3B). The extent of desensitization and the rate of deactivation were not significantly different among wild-type, mixed, or variant currents (Fig. 3C,D).

The peak GABA current amplitudes were reduced, but the kinetic properties were not altered for α4β2δ receptors containing mixed wild-type and variant or pure variant δ(R220H) receptors. A, Representative whole-cell current traces evoked by saturating GABA (1 mm) from wild-type α4β2δ, mixed wild-type and variant, and pure variant α4β2δ(R220H) receptors. B, Compared with wild-type receptors (n = 77), the mean current amplitude was smaller for the mixed receptors (n = 20) as well as for pure variant receptors (n = 24). C, The mean extent of desensitization was not different among wild-type, mixed, and pure variant receptors. D, The mean rate of current deactivation of mixed, and pure variant receptors was not altered compared with wild-type receptors. The solid bar above each current trace denotes the duration of GABA application (4 s). The open bars represent the mean current features for wild-type receptors, the hatched bars represent those for the mixed receptors, and the black bars represent those for pure variant receptors. wt, Wild-type α4β2δ receptors; mix, presence of both receptors; var, pure variant receptors. The error bars denote SEM. The asterisks indicate significant difference from wild-type α4β2δ receptors (**p < 0.01).
Surface expression of α4β2δ GABAA receptors was decreased by the variant δ subunits
The δ(E177A) or δ(R220H) variant might result in decreased whole-cell current by interfering with receptor assembly and/or trafficking to decrease the surface receptor number. To explore this possibility, HEK293T cells were transfected with: (1) wild-type α4, β2, and V5 epitope-tagged δ subunits (1:1:1 cDNA ratio to simulate the human wild-type “gene dose”); (2) wild-type α4, β2, and a mixture of epitope-tagged wild-type δ and epitope-tagged variant δ subunits (1:1:0.5:0.5 cDNA ratio to simulate the human heterozygote gene dose); or (3) wild-type α4, β2, and epitope-tagged variant δ subunits (1:1:1 cDNA ratio to simulate the human homozygote gene dose). Total receptor proteins were analyzed using Western blot, and the surface receptor proteins were captured by biotinylation followed by Western blotting. An immunoreactive band of ∼50 kDa was detected for the wild-type, mixed wild-type and variant, or variant receptors (Fig. 4A,B). The bands were quantified by normalizing the optical density of receptors to that of β-actin (internal control). No significant differences in total receptor proteins obtained from whole-cell lysates were found among wild-type, mixed, and variant receptors (data not shown).
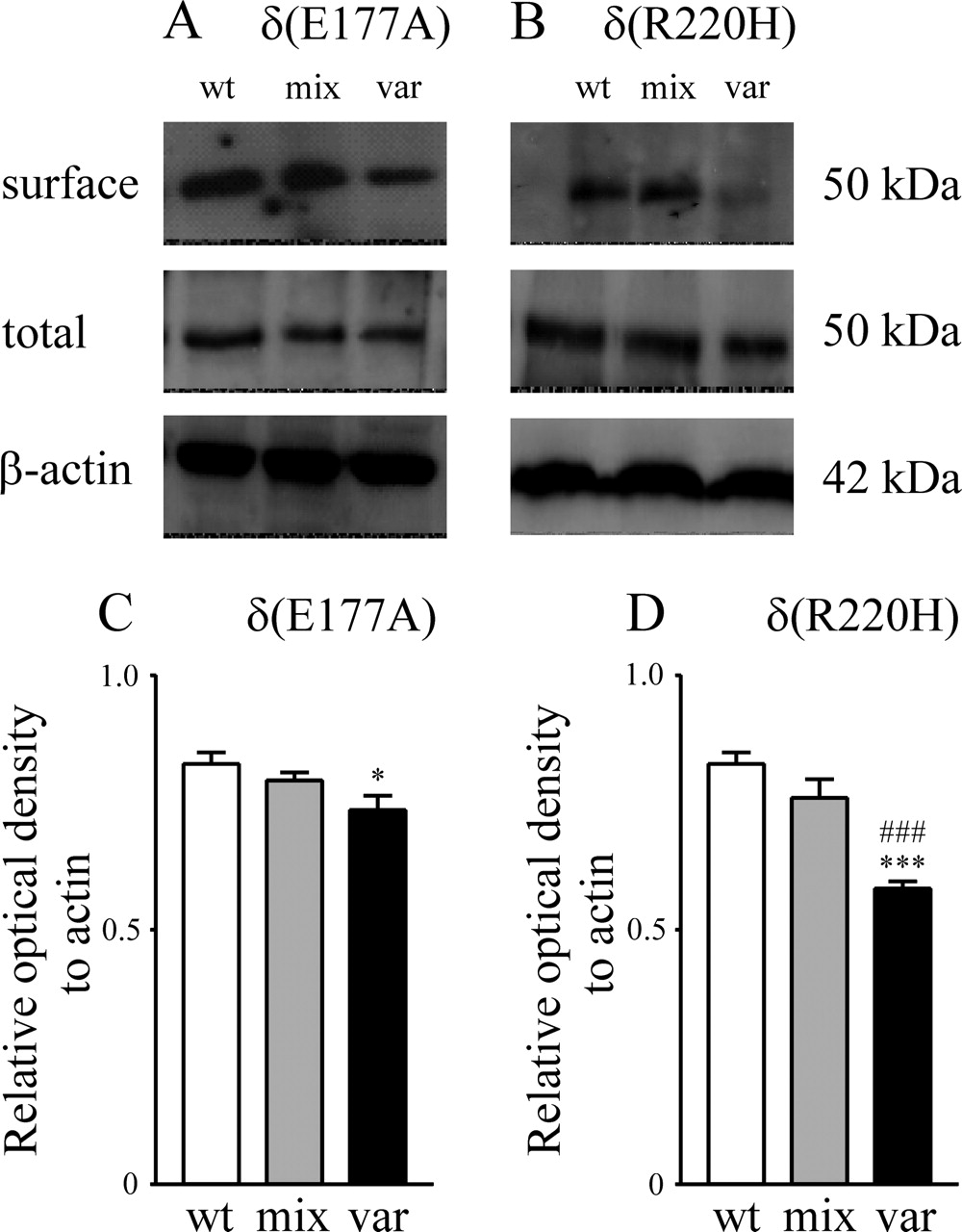
Surface receptor expression was decreased for α4β2δ receptors containing pure variant δ subunits. A, Examples of Western blot bands for wild-type α4β2δ, mixed receptor population, and variant α4β2δ(E177A) receptors are presented. Compared with wild-type receptors, the surface receptor proteins were slightly decreased for pure variant receptors, but those for mixed receptors were not obviously altered. The total receptor proteins (surface plus intracellular compartments) were not different among wild-type and variant receptors. B, Examples of Western blot bands for wild-type α4β2δ, mixed, and pure variant α4β2δ(R220H) receptors. Compared with wild-type receptors, the surface receptor proteins for pure variant receptors were substantially reduced, but those for the mixed receptors were not different from wild-type receptors. The total receptor proteins were not altered for variant receptors compared with wild-type receptors. C, The surface receptor proteins of wild-type and δ(E177A) variant receptors were quantified by normalizing to β-actin and expressed as relative optical density. The relative optical density of pure variant receptors (n = 7) was reduced compared with wild-type receptors (n = 14). The relative optical density of mixed receptors (n = 7) was not different from that of wild-type and pure variant receptors. D, The relative optical density of pure variant α4β2δ(R220H) receptors (n = 7) was substantially reduced compared with wild-type receptors (n = 14). The relative optical density of mixed receptors (n = 7) was not different from that of wild-type receptors but was significantly different from that of pure variant receptors. A V5 epitope was tagged at the N terminus of wild-type or variant δ subunit. The total receptor proteins were analyzed by Western blot using antibody against the V5 epitope. The surface receptor proteins were captured by biotinylation and analyzed by Western blot. A protein band of ∼50 kDa was detected for the wild-type α4β2δ receptor or the α4β2δ receptor containing either of the variant subunits. wt, Wild-type α4β2δ receptors; mix, mixed population of receptors; var, pure variant receptors. The error bars denote SEM. In both A and B, β-actin was applied as internal loading control. The asterisks indicate significant difference from wild-type α4β2δ receptors (*p < 0.05, ***p < 0.001). ###p < 0.001, significant difference from mixed wild-type and variant α4β2δ(R220H) receptors.
Compared with the relative optical density of surface receptor proteins for wild-type receptors (0.83 ± 0.02; n = 14), a significant decrease (∼11%) in surface proteins was observed for variant α4β2δ(E177A) receptors (0.74 ± 0.03; n = 7; p < 0.05) but not for mixed receptors (0.79 ± 0.02; n = 7) (Fig. 4C). No significant difference in surface receptor proteins was observed between variant α4β2δ(E177A) and mixed receptors.
Compared with the relative optical density of surface wild-type receptors, a significant decrease in surface receptor protein (∼30%) was observed for variant α4β2δ(R220H) receptors (0.58 ± 0.01; n = 7; p < 0.001), and a small (8.4%) but nonsignificant change was found for mixed receptors (0.76 ± 0.04; n = 7). The relative optical density for variant α4β2δ(R220H) receptors was also significantly smaller than that for mixed receptors (p < 0.001) (Fig. 4D).
Single-channel gating properties of α4β2δ receptors were modified by the variant δ subunits
The δ(E177A) or δ(R220H) variant might affect the gating properties or conductance of α4β2δ GABAA receptor channels to reduce whole-cell currents. To explore this possibility, single-channel recordings were obtained from wild-type and variant δ subunit-containing α4β2δ receptors. Because only one copy of the δ subunit is assembled in a single δ subunit-containing receptor, excised patches were pulled from cells expressing only the variant δ subunits. Similar to α1β3δ receptors (Fisher and Macdonald, 1997; Wohlfarth et al., 2002; Feng et al., 2004), application of a saturating concentration of GABA (1 mm) evoked single-channel currents from human α4β2δ receptors that exhibited isolated, nonbursting openings (Fig. 5A1). The single-channel current amplitudes of variant α4β2δ(E177A) (1.94 ± 0.07 pA; n = 7) (Fig. 5B1) and α4β2δ(R220H) (1.95 ± 0.05 pA; n = 8) (Fig. 5C1) receptors were not significantly different from those of wild-type α4β2δ receptors (1.98 ± 0.03 pA; n = 14) (Fig. 5A1). Compared with wild-type receptors (26.5 ± 0.5 pS), the mean single-channel conductance was not significantly altered for variant α4β2δ(E177A) (25.9 ± 0.9 pS) or α4β2δ(R220H) (26.0 ± 0.7 pS) receptors. It was previously reported that the single-channel main conductance of αβ receptors was 10–15 pS but that of αβδ receptors was 26.7 pS (Angelotti and Macdonald, 1993; Fisher and Macdonald, 1997). Our single-channel recordings demonstrated that the main conductance of variant receptors was similar to that of wild-type receptors (∼26 pS), suggesting that, like wild-type receptors, variant receptors are ternary αβδ receptors that are unlikely to be contaminated with αβ isoforms. The single-channel currents recorded from variant α4β2δ(E177A) or α4β2δ(R220H) receptors had gating patterns that were similar to those of wild-type α4β2δ receptors, except that the single-channel open durations were shorter for the variant receptors (Fig. 5B1,C1). Mean open durations were significantly shorter for α4β2δ(E177A) receptors (1.3 ± 0.2 ms; n = 7; p < 0.001) and α4β2δ(R220H) receptors (1.6 ± 0.1 ms; n = 8; p < 0.01) compared with mean open duration of α4β2δ receptors (2.6 ± 0.2 ms; n = 14) (Fig. 5D).

Mean open duration of single-channel currents was reduced for α4β2δ receptors containing pure variant δ subunits. A1–C1, Examples of single-channel currents evoked by 1 mm GABA from wild-type α4β2δ, and pure variant α4β2δ(E177A) and α4β2δ(R220H) receptors are presented. The single-channel current open duration was longer for the wild-type α4β2δ receptor than for variant α4β2δ(E177A) or α4β2δ(R220H) receptor. A2–C2, The distributions of open states of the wild-type α4β2δ, and variant α4β2δ(E177A) and α4β2δ(R220H) receptors were plotted. There were 4387 open events used for A2, 2331 open events for B2, and 2931 open events for C2. Each histogram contained data from a single patch. D, The single-channel current mean open duration was decreased for variant α4β2δ(E177A) (n = 7) and α4β2δ(R220H) receptors (n = 8) compared with wild-type α4β2δ receptors (n = 14). E, Comparison of the mean value of τ1, τ2, and τ3 among wild-type α4β2δ and variant α4β2δ(E177A) and α4β2δ(R220H) receptors. F, Comparison of the mean A1, A2, and A3 among wild-type α4β2δ and variant α4β2δ(E177A) and α4β2δ(R220H) receptors. The average open events used for each patch were 2559, 2414, and 2921 for wild-type α4β2δ, variant α4β2δ(E177A), and α4β2δ(R220H) receptors, respectively. wt, Wild-type α4β2δ receptors. The error bars represent SEM. Asterisks indicate significant difference from wild-type α4β2δ receptor (*p < 0.05, **p < 0.01, and ***p < 0.001).
In contrast to the reports of α1β3δ receptors (Fisher and Macdonald, 1997; Wohlfarth et al., 2002; Feng et al., 2004) and a recent report of α4β2δ receptors (Akk et al., 2004) that only two exponential components were observed, the distributions of wild-type α4β2δ, α4β2δ(E177A), and α4β2δ(R220H) single-channel openings were fitted best by three exponential functions (Fig. 5A2,B2,C2). However, the distribution of wild-type α4β2δ currents was shifted toward longer durations while the distributions of the variant α4β2δ(E177A) and α4β2δ(R220H) receptors were shifted toward shorter durations, consistent with the reduced mean open time of the variant receptor single-channel currents.
For single-channel variant α4β2δ(E177A) receptor currents, the mean time constants of the shortest (τ1) (0.20 ± 0.02 vs 0.31 ± 0.02) (p < 0.01) and second exponential component (τ2) (1.85 ± 0.34 vs 2.59 ± 0.21) (p < 0.05) were significantly smaller than those for α4β2δ receptors. For single-channel variant α4β2δ(R220H) receptor currents, mean τ1 (0.24 ± 0.03) (p < 0.05) and τ2 (1.67 ± 0.16) (p < 0.05) were also significantly smaller than for α4β2δ receptors. Mean τ3 for neither variant receptors was significantly altered (Fig. 5E).
For both variant α4β2δ(E177A) and α4β2δ(R220H) receptor single-channel currents, the mean relative areas of the shortest (A1) and second (A2) exponential components were altered, but that of the longest exponential component (A3) was unchanged. Compared with wild-type receptors (29.1 ± 2.8%), mean A1 was significantly increased for both α4β2δ(E177A) (51.1 ± 4.2%; p < 0.001) and α4β2δ(R220H) receptors (44.6 ± 4.2%; p < 0.01), whereas mean A2 was significantly decreased for both α4β2δ(E177A) (38.1 ± 3.2%) (p < 0.01) and α4β2δ(R220H) receptors (37.9 ± 5.3%; p < 0.05) compared with wild-type α4β2δ receptors (55.8 ± 3.6%) (Fig. 5F).
Mean open duration is the sum of the products of time constants and relative areas for each component (ΣAiτi, i = 1, 2, 3). The decrease in mean open duration for variant receptors was predominantly contributed by the reduction in the mean Aτ of second component (A2τ2), which was responsible for 63.4% of the reduction in mean channel open time for α4β2δ(E177A) receptors and 87.9% for α4β2δ(R220H) receptors.
Discussion
Whole-cell current amplitudes were significantly smaller for mixed and pure variant α4β2δ(E177A) and α4β2δ(R220H) receptors, suggesting that these susceptibility variants may result in disinhibition that may contribute to IGEs. For both α4β2δ(E177A) and α4β2δ(R220H) receptors, surface receptor expression was reduced for pure variant but not for mixed receptors, and the mean open duration of variant receptor single-channel currents was decreased (Table 1).
Summary of the changes in variant receptors as compared with wild-type α4β2δ receptors
Reduction of α4β2δ receptor whole-cell currents and its link to IGEs
αβγ and αβδ receptors are the predominant GABAA receptors in vivo (McKernan and Whiting, 1996), with αβγ receptors mediating primarily phasic inhibition, and αβδ receptors primarily mediating tonic inhibition (Saxena and Macdonald, 1994; Haas and Macdonald, 1999; Farrant and Nusser, 2005). δ subunits preferentially coassemble with α4 subunits in the thalamus, a structure critical for pathogenesis of IGEs (Wisden et al., 1992; Sur et al., 1999), and GABRD knock-out animals exhibit spontaneous seizures (Spigelman et al., 2002), reinforcing our finding that impaired α4βδ receptor function is associated with complex epilepsy in humans (Dibbens et al., 2004). Also, α4 and/or δ subunit expression was changed in experimental seizures (Clark et al., 1994; Schwarzer et al., 1997; Brooks-Kayal et al., 1998; Peng et al., 2004), and reduction of δ subunits may contribute to epileptogenesis in experimental epilepsy (Peng et al., 2004). All of these link α4βδ receptor function to epileptogenesis. Demonstration that α4β2δ receptors containing the δ(E177A) or δ(R220H) variant have reduced currents is consistent with the conclusion that they contribute to human IGEs (Dibbens et al., 2004).
Alterations of surface expression of α4β2δ receptors and their link to IGEs
We observed a small decrease of pure variant α4β2δ(E177A) receptors and a substantial decrease of pure variant α4β2δ(R220H) receptors in surface expression but no significant alteration of mixed α4β2δ(E177A) or α4β2δ(R220H) receptors, suggesting that these variants interfered with receptor assembly and/or trafficking of pure variant but not mixed receptors. Another γ2 subunit mutation, γ2(R43Q), reduced surface expression of GABAA receptors (Kang and Macdonald, 2004; Sancar and Czajkowski, 2004; Hales et al., 2005), and like the γ2(R43Q) mutation, both δ(E177A) and δ(R220H) variants are located in the subunit N terminus. The γ2 subunit N terminus interacts with α and β subunits during receptor assembly (Klausberger et al., 2000; Jin et al., 2004), and the γ2(R43Q) subunit mutation resulted in abnormal receptor assembly and trafficking (Kang and Macdonald, 2004; Hales et al., 2005). The present demonstration that surface expression of α4β2δ(R220H) and α4β2δ(E177A) receptors were reduced suggests that the N terminus of the δ subunit may also be involved in receptor assembly and/or trafficking. Neuronal αβδ receptors are localized perisynaptically (Mody and Pearce, 2004; Farrant and Nusser, 2005), and it is unknown whether these variants interfere with perisynaptic targeting of αβδ receptors.
Mechanistic basis for defective function of α4β2δ(E177A) and α4β2δ(R220H) receptors
Although variant receptor single-channel conductance was not altered, mean open durations for both α4β2δ(E177A) and α4β2δ(R220H) receptors were decreased, suggesting that assembly of either variant into α4β2δ receptors resulted in defective channel gating. The δ(E177A) variant is adjacent to one of the invariant cysteines that form the cysteine loop (loop 7), an important feature of cys-loop receptors. It was reported that residues on α1 subunit loop 7 interacted with those on the linker between transmembrane (TM) domains 2 and 3 to affect channel gating (Kash et al., 2003). It is possible that the δ(E177A) variant may interrupt the coupling of loop 7 and TM2–3 linker to compromise channel gating. The δ(R220H) variant is located approximately in the middle between the δ(E177A) variant and the beginning of TM1. It is not known whether the residues in this region can interact with TM2–3 linker to affect channel gating.
The primary effect of the δ(E177A) and δ(R220H) variants was to reduce mean channel open time
Wild-type α4β2δ peak current (222 pA) was greater than pure variant α4β2δ(E177A) (76.5 pA) and α4β2δ(R220H) (46.9 pA) receptor peak currents (Figs. 2, 3). To determine the basis for reduction of variant current, we used the measured reduction in open time and surface receptor expression to estimate their relative contribution to reducing current. Single-channel data used to determine mean open time were obtained from desensitized receptors (prolonged application of 1 mm GABA), and the best estimate of desensitized whole-cell current amplitude can be obtained from the percentage of desensitized data (Fig. 2, 3). Current amplitudes at the end of the 4 s GABA application were 137 pA for wild-type, 51.2 pA for variant α4β2δ(E177A), and 28.9 pA for variant α4β2δ(R220H) receptors.
We determined the reduction of pure variant α4β2δ(E177A) current predicted by their gating and trafficking defects. An 11% reduction of surface expression and a 50% decrease in mean open time would reduce the current to 60.9 pA (56% reduction), which compares quite well with the measured current of 51.2 pA (63% reduction), and 90.1% of the reduction was attributable to reduced gating, and only 9.9% was caused by reduced trafficking. We also predicted the effect of the δ(E177A) variant on mixed current, assuming that half of the translated δ receptors were wild-type and half were variant receptors, the wild-type and variant subunits assembled equally well into receptors, and they were each trafficked to the cell surface as predicted by results obtained with mixed receptors. The desensitized mixed current was predicted to be 101.8 pA (26% reduction), which was somewhat larger than the measured mixed current of 79.4 pA (42% reduction).
We determined the reduction in pure variant α4β2δ(R220H) current predicted by their gating and trafficking defects. A 30% reduction of surface expression and a 38.5% decrease in mean open time would reduce the current to 59 pA (57% reduction), which was larger than the measured variant desensitized current of 28.9 pA (78.9% reduction), and 67.6% of the reduction was caused by reduced gating and 32.4% caused by reduced trafficking. The desensitized mixed current was predicted to be 97.9 pA (29% reduction), which was somewhat larger than the measured 69.5 pA (49% reduction).
Thus, for both mixed and pure variant receptors, ∼80–90% of the reduction in desensitized current could be explained quite well by extrapolating the trafficking and gating deficits for each variant receptor, and the primary basis for the current reduction for both variant receptors was the reduction in mean channel open time.
Implication of GABRD in complex epilepsies
Significant advances have led to some understanding of the genetic basis for the rare monogenic epilepsies. Twin studies have also suggested a strong genetic basis for the complex epilepsies (Berkovic et al., 1998); however, description and validation of the underlying genetic variation for these more common epilepsies has only just begun. Changes in channel properties of CACNA1H are consistent with complex epilepsy (Khosravani et al., 2004, 2005; Vitko et al., 2005). The present study reinforces and extends the previously reported role of GABRD as another susceptibility gene underlying complex epilepsy. The same susceptibility genes may have wider implications as genetic modifiers of penetrance and expressivity of the genes for the so called “monogenic” epilepsies. The boundary between monogenic and complex epilepsy is likely to become blurred with modifiers simply regarded as part of the polygenic spectrum involved in complex epilepsy. Sequencing and subsequent functional validation of putative susceptibility alleles by electrophysiology has so far proven to be the only successful approach to understanding the molecular and mechanistic basis for complex epilepsies (Mulley et al., 2005).
The present finding that α4β2δ receptors containing the δ(E177A) or δ(R220H) variant had reduced current amplitudes in a heterologous expression system suggests that these susceptibility alleles of the GABRD gene may result in reduction of tonic GABAergic inhibition. Tonic inhibition generates a persistent increase in the input conductance of a neuron and thus dampens the size and duration of excitatory synaptic activities, making it less likely to fire action potentials (Farrant and Nusser, 2005). A potential reduction of tonic inhibition by these variants, therefore, would tend to lead to neuronal hyperexcitability and to increased susceptibility to develop epilepsy. It is of note that a heterologous expression system may contain different cellular machinery than neurons and does not form synapses. Therefore, whereas the present findings of variant effects in HEK cells give insights into the effects of these variants on receptor function, it will be important to confirm the effects of both variants on GABAergic tonic inhibition in neurons and in intact animals. Phenotypically, the δ(E177A) variant was heterozygously associated with GEFS+, and the δ(R220H) variant was both heterozygously and homozygously associated with JME. The epilepsy in individuals heterozygous for a variant may be predominantly attributable to the gating defect and in patients homozygous for a variant may be caused by both gating and trafficking defects.
This and previous studies involving variation at CACNA1H and GABRD represent the only mechanistic progress made so far among the likely plethora of susceptibility genes underlying complex epilepsy. Because all of the variants thus far studied represent a small minority of cases, most of the interpersonal variation associated with complex epilepsy remains to be identified and functionally characterized.
Footnotes
This work was supported by National Institutes of Health Grant NS33300 (R.L.M.) and an Epilepsy Foundation postdoctoral fellowship (H.-J.F.).
- Correspondence should be addressed to Dr. Robert L. Macdonald, Department of Neurology, Vanderbilt University Medical Center, 6140 Medical Research Building III, 465 21st Avenue South, Nashville, TN 37232-8552. Email: robert.macdonald{at}vanderbilt.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}