

Abstract
Hyperpolarization-activated pacemaker currents (IH) contribute to the subthreshold properties of excitable cells and thereby influence behaviors such as synaptic integration and the appearance and frequency of intrinsic rhythmic activity. Accordingly, modulation of IH contributes to cellular plasticity. Although IH activation is regulated by a plethora of neurotransmitters, including some that act via phospholipase C (PLC), the only second messengers known to alter IH voltage dependence are cAMP, internal protons (H+Is), and phosphatidylinositol-4,5-phosphate. Here, we show that 4β-phorbol-12-myristate-13-acetate (4βPMA), a stereoselective C-1 diacylglycerol-binding site agonist, enhances voltage-dependent opening of wild-type and cAMP/H+I-uncoupled hyperpolarization-activated, cyclic nucleotide-regulated (HCN) channels, but does not alter gating of the plant hyperpolarization-activated channel, KAT1. Pharmacological analysis indicates that 4βPMA exerts its effects on HCN gating via sequential activation of PKC and diacylglycerol kinase (DGK) coupled with upregulation of MAPK (mitogen-activated protein kinase) and phospholipase A2 (PLA2), but its action is independent of phosphoinositide kinase 3 (PI3K) and PI4K. Demonstration that both phosphatidic acid and arachidonic acid (AA) directly facilitate HCN gating suggests that these metabolites may serve as the messengers downstream of DGK and PLA2, respectively. 4βPMA-mediated suppression of the maximal HCN current likely arises from channel interaction with AA coupled with an enhanced membrane retrieval triggered by the same pathways that modulate channel gating. These results indicate that regulation of excitable cell behavior by neurotransmitter-mediated modulation of IH may be exerted via changes in three signaling lipids in addition to the allosteric actions of cAMP and H+Is.
- phosphatidic acid
- arachidonic acid
- protein kinase C
- diacylglycerol kinase
- phospholipase A2
- mitogen-activated protein kinase
Introduction
Physiological control of the speed and extent of opening of hyperpolarization-activated pacemaker currents (IH) influences the emergence and frequency of rhythmic excitation and the sensitivity of a cell to excitatory or inhibitory input, whereas aberrant perturbation of such regulation may contribute to pathological disruption of normal activity of excitable cells (McCormick and Bal, 1997; McCloskey et al., 1999; Okabe et al., 1999; Robinson and Siegelbaum, 2002). In accord with serving an important role in control of cellular activity, IH activation is sensitive to many neurotransmitters. In light of this, it is perhaps surprising that only three second messengers, cAMP, intracellular protons (H+Is), and phosphatidylinositol-4,5-phosphate (4,5-PIP2), have been clearly established as regulators of the voltage dependence of channel opening (Munsch and Pape, 1999; Santoro and Tibbs, 1999; Kaupp and Seifert, 2001; Zong et al., 2001; Robinson and Siegelbaum, 2002; Pian et al., 2006), with cAMP and 4,5-PIP2 acting to enhance activation and H+Is acting as an inhibitory effector.
Are cAMP, H+Is, and 4,5-PIP2 the sole arbiters of neurotransmitter control of IH? Several lines of evidence suggest that this may not be the case. First, the broad-spectrum protein kinase inhibitors H7 and H8 inhibited IH in cardiac Purkinje fibers and sympathetic neurons (Tokimasa and Akasu, 1990; Chang et al., 1991), whereas phosphatase inhibition with calyculin A resulted in an H7-sensitive depolarization of cardiac Purkinje fiber and ventricular myocyte IH gating (Yu et al., 1993, 1995). Second, inhibition of mitogen-activated protein kinase (MAPK) hyperpolarizes gating of hippocampal IH by ∼25 mV, whereas MAPK activation elicits an 11 mV depolarizing shift from the control level (Poolos et al., 2006). Third, a number of receptors, including neuromedin U (NMU) (Qiu et al., 2003), muscarinic (Colino and Halliwell, 1993; Zhu and Uhlrich, 1998), epidermal growth factor (Wu et al., 2000), and angiotensin-II type 1 (Egli et al., 2002), enhance gating of IH but are either uncoupled from, or negatively coupled to, adenylate cyclase and are positively coupled to phospholipase C (PLC) (activation of which would also be expected to inhibit IH if it only influences the channels by decreasing 4,5-PIP2). Although these observations could result from secondary changes in cAMP, H+I, and 4,5-PIP2 through, for example, PKC activation of adenylate cyclase, alteration of proton pumps, or upregulation of phosphoinositide kinases, these observations may indicate that there is additional complexity in signaling to IH.
To explore the possible role of signaling via pathways sensitive to diacylglycerol without the potential complication of receptor-mediated cleavage of 4,5-PIP2 and changes in calcium signaling as well as PLC-independent receptor signaling (Rajagopal et al., 2005), here we used 4β-phorbol 12-myristate 13-acetate (4βPMA), a high-affinity, stereoselective agonist of the cysteine-rich C-1 DAG/4β-phorbol-binding pocket. We show that 4βPMA selectively facilitates the activation gating of both wild-type hyperpolarization-activated, cyclic nucleotide-regulated (HCN) channels and H+I and/or cAMP-uncoupled REHR and RE mutants thereof (shown schematically in supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and does so independently of phosphoinositide (PI) kinase 3 (PI3K) and PI4K activity. Our data indicate that 4βPMA facilitates HCN gating by recruiting diacylglycerol kinase (DGK) and cytoplasmic/independent PLA2 (c/i-PLA2). An increase in the levels of phosphatidic acid (PA) and arachidonic acid (AA), respective products of DGK and PLA2, may mediate this response via direct interaction with the channels.
Materials and Methods
Molecular biology.
Channels and mutants thereof were subcloned into pGH19 (HCN1) and pGHE (HCN2 and KAT1) vectors. Site-directed mutations were prepared by either two rounds of PCR (followed by subcloning using convenient flanking restriction sites) or by QuikChange II mutagenesis (Stratagene, La Jolla, CA), whereas truncation constructs were as described previously (Wainger et al., 2001). Constructs were amplified in STBL2 (Invitrogen, Carlsbad, CA) or XL1-Blue (Stratagene) cells. Subcloning sites and regions generated by PCR or constructed from linkers were sequenced on both strands. cRNA was transcribed from NheI (HCN1 and KAT1 constructs) or SphI (HCN2 constructs) linearized DNA using T7 RNA polymerase (Message Machine; Ambion, Houston, TX). One to fifty nanograms of RNA were injected into each Xenopus oocyte.
Electrophysiology.
Recordings were made in either two-microelectrode voltage clamp (TEVC) or excised inside-out patch-clamp (IOPC) configurations from Xenopus oocytes 1–5 d after cRNA injection. Cells were maintained in L-15 media without Ficoll (Specialty Media, Phillipsburg, NJ) at 17°C until use. In both configurations, data were digitized using an ITC-18 interface (Instrutech, Port Washington, NY) and recorded with Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany). Ag–AgCl ground wire(s) were connected to the bath solution by 3 m KCl 2% agar salt bridges placed downstream of, but close to, the oocyte. Recordings were obtained at room temperature (22–24°C). In all cases, voltages are reported as the command potentials. In TEVC, the measured deviation was <1%.
TEVC data were acquired using a Warner Instruments (Hamden, CT) OC-725C amplifier, filtered at 1 kHz, and then digitized at 2 kHz. For most experiments, oocytes were bathed in a recording solution of (in mm) 107 NaCl, 5 KCl, 2 MgCl2, and 10 HEPES (free acid), pH 7.4 (NaOH). However, KAT1 was recorded in a solution in which the NaCl and KCl were adjusted to 82 and 30 mm, respectively. Microelectrodes were fabricated from 1B120-F4 borosilicate glass (World Precision Instruments, Sarasota, FL) and had resistances of 0.1–0.5 MΩ (I passing) and 1–4 MΩ (V sensing) when filled with 3 m KCl. An active virtual ground was used to clamp the bath.
IOPC data were acquired using an Axon Instruments (Foster City, CA) 200B patch-clamp amplifier, in resistive mode, filtered at 2.5 kHz using a Warner LPF-8 eight-pole Bessel filter, and then digitized at 5 kHz. The extracellular solution was (in mm) 107 KCl, 5 NaCl, 10 HEPES (free acid), 1 MgCl2, and 1 CaCl2, pH 7.4 (KOH). Two intracellular solutions were used: a Mg solution [(in mm) 107 KCl, 5 NaCl, 1 MgCl2, 1 EGTA-free acid, and 10 HEPES-free acid, pH 7.4 with KOH] or a Mg-free solution (wherein the 1 mm MgCl2 was exchanged for 1 mm EDTA-free acid). Electrodes with resistances between 1 and 2 MΩ were fabricated from Kimax-51 borosilicate glass (Kimble Glass, Vineland, NJ) and coated with Sylgard (Dow Corning, Midland, MI). In patches in which the maximal current exceeded 500 pA, we applied analog series resistance compensation (90% correction; 20 μs lag with the resistance set to the initial resistance of the electrode).
Paradigms and analysis.
Steady-state activation curves were determined from tail currents (measured at 0 mV in TEVC and −40 mV in IOPC) that followed hyperpolarizing voltage steps applied in −10 mV increments. The holding potential was −40 mV in IOPC and −30 mV in TEVC. Tail-current amplitudes were measured by averaging the current during the plateau of the tail (after allowing the voltage clamp to settle and the uncompensated linear capacitance to decay) and subtracting from this the baseline current recorded after the channels had fully deactivated. In cases in which channels were active at the holding potential, some cells were stepped to +20 mV for 0.5 s to fully deactivate the channels immediately before the hyperpolarizing test step. This was sufficient to close all channels but did not alter the determined V1/2. Tail-current amplitudes were plotted versus the hyperpolarization step voltage and fitted with the Boltzmann equation (Eq. 1), where A1 is the current offset, A2 is the maximal amplitude, V is step voltage, V1/2 is the activation midpoint, and s is the slope factor:
 The tail amplitudes and Boltzmann fit were then normalized to the maximal tail amplitude, A2 − A1. Where mean activation curves are shown, the normalized tail-current amplitudes from multiple experiments were averaged, and the mean data were refit with the Boltzmann equation.
The tail amplitudes and Boltzmann fit were then normalized to the maximal tail amplitude, A2 − A1. Where mean activation curves are shown, the normalized tail-current amplitudes from multiple experiments were averaged, and the mean data were refit with the Boltzmann equation.
HCN channel activation drifts hyperpolarized after patch excision. Although extensive perfusion of the patch before data collection will tend to mitigate convolution of drug-mediated changes in gating with drift (see paradigms described in Figs. 5A, 8A), we adopted a protocol that permitted us to follow changes in activation with greater temporal resolution than can be achieved when constructing full activation curves (channel gating is extremely slow). Each 60 s, a patch was stepped to an intermediate voltage (VINT) that would achieve ∼20 or 50% activation (determined from a full activation curve constructed at the beginning of the recording). After determination of the intermediate current (IINT) and the corresponding tail current (IINT TAIL), the patch was stepped to a voltage that saturated channel opening (VMAX), and the maximal current (IMAX) and corresponding tail current (IMAX TAIL) were determined. From these data, we constructed conductance ratios: GINT TAIL/GMAX TAIL and GINT/GMAX (with the latter obtained from the current ratio by correcting for driving force). These nonlinear gating parameters were then converted into linear approximations of the shift in the V1/2, the “ΔV1/2 apparent” (ΔV1/2 APP), by Equation 2 (derived from the Boltzmann equation), where GR is the conductance ratio, V1/2 and s are the initially determined activation midpoint and slope, respectively, and VINT and VMAX are as defined above:
 Analysis of GINT TAIL/GMAX TAIL and GINT/GMAX yielded equivalent estimations of ΔV1/2 APP.
Analysis of GINT TAIL/GMAX TAIL and GINT/GMAX yielded equivalent estimations of ΔV1/2 APP.
Concentration response data were fit with the Hill equation (Eq. 3), where R is the observed response at a particular drug concentration, RMAX is the fit determined maximal response, EC50 is the fit determined concentration required to elicit one-half the maximum response, [A] is the drug concentration, and h is the fit determined Hill coefficient:
 Data analysis was performed in PulseFit (HEKA Elektronik) or using custom analysis routines written in IgorPro (Wavemetrics, Lake Oswego, OR).
Data analysis was performed in PulseFit (HEKA Elektronik) or using custom analysis routines written in IgorPro (Wavemetrics, Lake Oswego, OR).
Statistical analysis.
SigmaStat V3.1 (Systat Software, Point Richmond, CA) was used to perform statistical analysis. When differences between means were determined, the reported error is the sum of the errors of the two populations. Student's t tests were used to determine whether differences between two populations (unpaired) or populations sampled before and after treatment (paired) were significant, whereas comparison of multiple groups was performed using either one-way or two-way (depending on whether a single control or independent control populations were analyzed) ANOVA with Holm–Sidak post hoc analysis.
Chemicals and reagents.
AA, 4βPMA, 4αPMA, 2-[1-(3-(amidinothio)propyl)-1H-indol-3-yl]-3-(1-methylindol-3-yl)maleimide methanesulfonate (Ro31-8220) wortmannin, PA, 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole (SB-202190), bis[amino[(2-aminophenyl)thio]methylene] butanedinitrile (U-0126), 1-[6-([17β-methoxyestra-1,3,5(10)-trien-17-yl]amino)hexyl]-1H-pyrrole-2,5-dione (U73122), 1,6-bis(cyclohexyloximinocarbonylamino) hexane (RHC 80267), arachidonyltrifluoromethyl ketone (AACOCF3), and oleyloxyethyl phosphorylcholine (OPC) were obtained from Biomol (Plymouth Meeting, PA). 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) was obtained from EMD Biosciences (San Diego, CA). Stock solutions of each were prepared in DMSO, ethanol, or H2O as appropriate and diluted into the appropriate buffer immediately before use. Incubations with drugs were performed in glass vials with 3 ml of L-15 plus drug. When activation of channels expressed in a cell was measured before and after incubation, the cell was allowed to recover in L-15 for 10 min before being transferred to a bath of L-15 containing drug. In all cases, vehicle controls were performed in parallel. All drug stock solutions were frozen in aliquots and stored at −80°C. All other reagents used in electrophysiology were obtained from Sigma (St. Louis, MO) and were of the highest available purity.
Results
HCN channel gating is facilitated by the membrane-permeable, nonmetabolizable DAG-mimetic 4βPMA
Figure 1 shows TEVC recordings obtained from two Xenopus oocytes, one expressing HCN1 (Fig. 1A–C) and the other expressing HCN2 (Fig. 1D–F). In Figure 1, A and D, the left records show current families evoked in response to a series of hyperpolarizing voltage steps, whereas the right panels show tail currents observed on stepping back to 0 mV before (top) and after (bottom) incubation of the cell with 200 nm 4βPMA. Inspection of these current records suggests that HCN1 and HCN2 channels opened at more depolarized voltages after incubation with drug but that the total available current was decreased. Thus, HCN1 channels are clearly active at −25 mV in the presence of 4βPMA, whereas under control conditions, there is no channel activation until the cell is stepped to voltages below −35 mV. Similarly, the threshold for activation of HCN2 channels after incubation in the presence of 4βPMA is ∼10 mV depolarized to the threshold of −65 mV seen in the absence of 4βPMA. 4βPMA-mediated depolarization of gating of both channels is accompanied by an approximately threefold to fivefold decrease in current amplitude. To quantify the effects of 4βPMA on HCN function, we constructed current–voltage plots (Fig. 1B,E) and normalized activation curves (Fig. 1C,F). Fits of the activation curves with the Boltzmann relationship reveal that activation gating is depolarized by 27 and 9 mV for the cells shown in A and D, respectively. Comparison of inward current density (Fig. 1B,E) at voltages at which activation is saturated (Fig. 1C,F) confirms that the maximal current is reduced. A comparison of the ratios of the maximally activated inward current to the corresponding instantaneous tail current in cells incubated in the absence or presence of 200 nm 4βPMA (HCN1, 0.36 ± 0.01 vs 0.37 ± 0.02; n = 13 and 11; p > 0.50; HCN2, 0.35 ± 0.03 vs 0.29 ± 0.02; n = 13 and 9; p > 0.12) indicates that the reversal potential of the channels is not altered, suggesting that 4βPMA incubation decreases channel availability, open probability, or the single-channel conductance. Data presented below suggest that a reduction in the number of available channels is the most likely explanation.

The diacylglycerol-mimetic 4βPMA facilitates gating of heterologously expressed HCN1 and HCN2 channels. A, D, TEVC current families (left) and tail currents (right) from cells expressing HCN1 (A) and HCN2 (D), each recorded before (top) and after (bottom) incubation with 200 nm 4βPMA (HCN1 for 30 min; HCN2 for 10 min). The cell expressing HCN1 (A) was stepped to +20 mV for 500 ms immediately before the 3 s activation step to deactivate channels open at the holding potential in the presence of 4βPMA. B, E, Current–voltage relationships of cells shown in A and D. C, F, Steady-state activation curves constructed from the tail currents (It) as a function of the maximal tail current (Itmax) shown in A and D. Fits of the Boltzmann function are superimposed.
Stereoselectivity and high apparent affinity indicates that phorbol ester modification of HCN channel gating requires ligand association with a C-1 diacylglycerol-binding site
To investigate the basis of the modulation of HCN gating by 4βPMA, we incubated cells with varying concentrations of the C-1 agonist, with 4αPMA [which differs from 4βPMA only in the orientation of a single hydroxyl but which, as a consequence, is unable to associate with C-1 sites (Zhang et al., 1995)], and with vehicle control. Figure 2A shows that the enhancement of HCN1 gating in response to 4βPMA developed in a time- and concentration-dependent manner, such that incubation with 20 or 200 nm 4βPMA resulted in equivalent depolarization of gating that reached a half-maximal level in 5–10 min (Fig. 2A), but 2 nm 4βPMA was less effective. A similar time course was observed in all other HCN constructs analyzed (data not shown). Construction of dose–response curves from the data in Figure 2A reveals that the EC50 for gating enhancement was ∼2 nm (Fig. 2B). In contrast to these potent effects of 4βPMA, the 4α stereoisomer was without effect on gating of HCN1 (Fig. 2A). This requirement for the 4β versus 4α orientation of the critical hydroxyl was maintained across all HCN constructs examined as is shown for HCN1-REHR and HCN2-REHR (Fig. 3C).
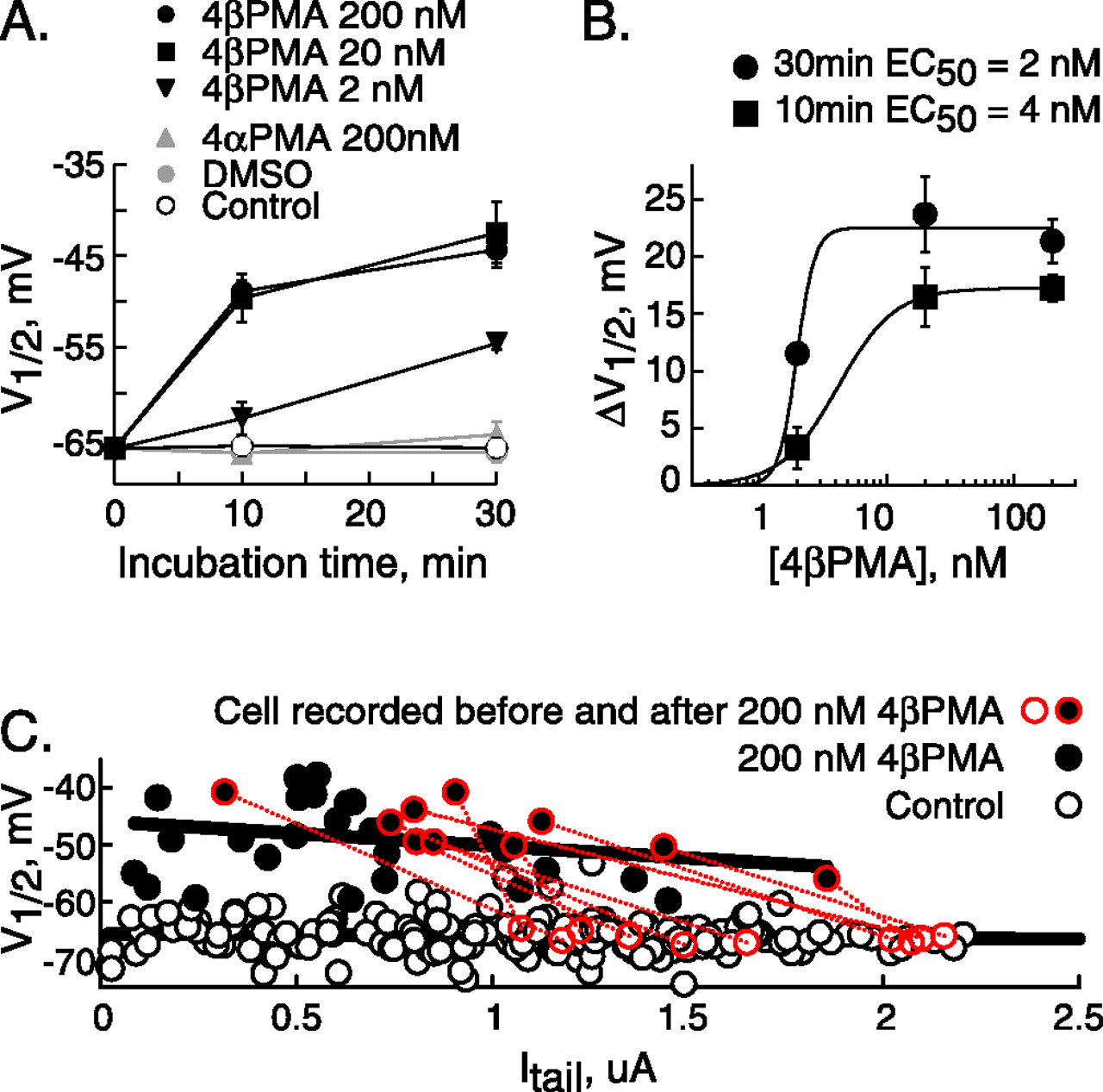
4βPMA acts via a high-affinity, stereoselective site to modify HCN channel gating. A, V1/2 of HCN1 as a function of time in the absence or presence of 4βPMA, 4αPMA, or DMSO vehicle. The V1/2 in 4βPMA was significantly different (p < 0.001) from control and DMSO vehicle groups at all concentrations and times except 2 nm at 10 min. However, control, DMSO, and 4αPMA populations were not different from each other (p = 0.28–0.93) at any time. The number of determinations per point was between 4 and 97. B, ΔV1/2 as a function of 4βPMA concentration. Data are from A and are fit with the Hill equation. C, V1/2 versus maximal tail-current amplitude for HCN1. Data from 97 control recordings and 35 cells recorded after 10 min of incubation in 200 nm 4βPMA. Symbols outlined in, and linked by, red lines are paired recordings obtained before and after incubation with 4βPMA. Solid black lines are linear regressions through the control and 200 nm 4βPMA data. Error bars represent SEM.

4βPMA facilitation of HCN gating is not mediated by cAMP or protons acting at their allosteric sites. A, B, TEVC current families (left) and tail currents (right) recorded from cAMP/H+I-uncoupled HCN1-REHR (A) and HCN2-REHR (B) channels before (top) or after (bottom) 30 min of incubation with 200 nm 4βPMA. C, Steady-state activation curves constructed from cells expressing HCN1-REHR (left) or HCN2-REHR (right) after 30 min of incubation in the absence or presence of 200 nm 4βPMA, 200 nm 4αPMA, or DMSO vehicle. The number of cells per condition is as follows: control, 19 and 226; 4βPMA, 8 and 81; 4αPMA, 6 and 5; and DMSO vehicle, 5 and 38, for HCN1-REHR and HCN2-REHR, respectively. D, ΔV1/2 in response to 200 nm 4βPMA as determined by the difference in the means of the treated and untreated populations for wild-type, RE, and REHR channels. In each case, the difference between the 4βPMA population and the paired control conditions (untreated, DMSO, and 4αPMA) was significant (p < 0.005), whereas the controls were not different from each other (p = 0.20–0.82; data not shown, but see overlapping activation curves in C). Error bars represent SEM.
The maximal current carried by HCN1 and HCN2 was reduced to 19.4 ± 6.1% (p < 0.001; n = 5) and 35.8 ± 6.9% (p < 0.006; n = 8) after 30 min of incubation in the presence of 200 nm 4βPMA but was not altered by the incubation in the presence of 4αPMA (HCN1, 100.5 ± 2.4%; p = 0.769; n = 6; HCN2, 98.1 ± 12.3%; p = 0.969; n = 8) or DMSO vehicle (HCN1, 97.9 ± 3.5%; p = 0.732; n = 6; HCN2, 89.2 ± 8.3%; p = 0.929; n = 8). The EC50 for suppression of the HCN1 current after incubation in varying concentrations of 4βPMA was ∼2 nm (data not shown).
Given that incubation with 4βPMA decreases current density, we were concerned that the shift in gating may arise as a result of a series resistance error rather than a second messenger-mediated alteration in gating. We addressed this issue by considering whether the V1/2 was correlated with current density. Figure 2C plots the saturating tail-current amplitude versus the V1/2 for HCN1 obtained from control and 200 nm 4βPMA-treated cells. Data obtained from a cell recorded before and after incubation with 4βPMA are shown ringed in red, with the corresponding control and drug-treated determinations linked by a dashed red line. These data show that current suppression after incubation with 4βPMA occurs in tandem with the depolarizing shift in gating, but there is no dependency of V1/2 on current amplitude (as shown by the essentially flat regression lines through both control and 4βPMA-treated populations).
Together, these data indicate that HCN gating can be enhanced (and maximal current suppressed) by recruitment of at least one of the small family of proteins that contains the canonical C1 DAG/4β-phorbol-binding site (Ron and Kazanietz, 1999; Brose and Rosenmund, 2002; Kazanietz, 2002; Yang and Kazanietz, 2003).
4βPMA enhancement of HCN channel gating is not mediated by changes in cAMP or H+I acting at their allosteric sites on the HCN subunits
The prototypical DAG/4βPMA-sensitive proteins are the conventional (cPKC) and novel (nPKC) isoforms of PKC. In light of the ability of PKCs to increase cAMP by activation of PKC-sensitive adenylate cyclase (Yoshimura and Cooper, 1993; Jacobowitz and Iyengar, 1994; Zimmermann and Taussig, 1996; Bol et al., 1997; Nasman et al., 2002) and, potentially, alkalinize the intracellular milieu [as is observed when the DAG levels of oocytes are elevated by blockade of DGK (Sadler et al., 1996)], we considered the hypothesis that the action of 4βPMA on HCN gating was mediated through changes in one or both of these known allosteric modulators. To address this directly, we asked whether gating of H+I- and/or cAMP-uncoupled HCN channels retain sensitivity to 4βPMA.
Figure 3 shows recordings from cells expressing the cAMP/H+I-uncoupled HCN1-REHR and HCN2-REHR channels after incubation in the absence or presence of 200 nm 4βPMA. Inspection of the current records indicates that gating of each of these channels is depolarized after incubation with the C-1 agonist. These observations are confirmed by inspection of the mean activation curves for HCN1-REHR and HCN2-REHR (Fig. 3C) and the mean shifts in V1/2 (Fig. 3D) for these channels and their corresponding cAMP-insensitive/H+I-sensitive counterparts (HCN1-RE and HCN2-RE). Thus, 4βPMA facilitates HCN gating by a mechanism that is distinct from cAMP or H+Is acting as allosteric ligands of channel gating. Similarly, the suppression of the maximal current by 4βPMA is not sensitive to the status of the cAMP- or H+I-binding sites (see, e.g., Figs. 3A,B, 7C).
What of the apparent enhancement of the effects of 4βPMA seen in the RE and REHR channels compared with wild-type parents? Inspection of the activation curves in Figures 1 and 3 reveals that under control conditions, the V1/2 of activation of HCN1-REHR (−88.5 ± 0.8 mV; n = 19) and HCN2-REHR (−96.8 ± 0.3 mV; n = 226) are somewhat hyperpolarized relative to their cognate wild-type parents (HCN1, −66.0 ± 0.3 mV; n = 97; HCN2, −77.3 ± 0.4 mV; n = 131), but the V1/2 for the REHR and wild-type channels approach each other after exposure to 4βPMA (HCN1, −44.3 ± 1.9 mV; n = 24 vs HCN1-REHR, −52.9 ± 2.6 mV; n = 8; HCN2, −68.4 ± 1.4 mV; n = 18 vs HCN2-REHR, −79.2 ± 0.8 mV; n = 81). Our interpretation of these findings is that the hyperpolarizing shifts in basal gating reflect a loss of response to background cAMP (after introduction of RE) and internal pH (by introduction of the “pseudo-protonated” permanent charge at the H+I sensor). We suggest that the enhancement of the 4βPMA response is a result of there being a ceiling to the V1/2 above which it cannot be driven but which it can approach by the sum of cAMP-, H+I-, and C-1-dependent signaling. Channels with only the RE mutation also displayed an enhancement in the shift in V1/2 compared with their parental channels, as shown in Figure 3D (HCN1-RE, −75.0 ± 0.3 mV, n = 210 in controls cells; −47.3 ± 3.0 mV, n = 10 after 4βPMA; HCN2-RE, −92.9 ± 0.6 mV, n = 69 in control cells; −78.4 ± 2.1 mV, n = 14 after 4βPMA).
The conserved core region of HCN channels imparts sensitivity to 4βPMA
To gain insight into the possible mechanism of regulation, we next considered what domains of the HCN channels are important for transducing the 4βPMA response. To investigate this, we examined the response of a number of truncation mutants that removed presumptive cytoplasmic domains of HCN1 and HCN2 (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). 4βPMA facilitation of channel activation was not disrupted after deletion of the cyclic nucleotide-binding domain (CNBD) (and distal C-terminal elements), the C-linker, or the variable N-terminal region (Fig. 4). Indeed, gating of the minimal channel HCN1-ΔNvΔC (wherein the most severe deletions of the N and C termini were combined) remained as sensitive to 4βPMA as the parental HCN1 wild-type channel (deletion of the entire C terminus is not tolerated in the background of HCN2, precluding a comparable analysis). Similarly, the maximal current carried by HCN1-ΔNvΔC was reduced to 19.8 ± 3.8%; p < 0.001; n = 9) after 30 min of incubation in the presence of 200 nm 4βPMA, a reduction that is indistinguishable from the phorbol-mediated suppression of wild-type HCN1 (p = 0.954). It is important to note that although these deletions remove up to 65% of the proteins [84% of the cytoplasmic sequence (supplemental Fig. 1, available at www.jneurosci.org as supplemental material)], they do so without causing any major alteration to the channel architecture as reported by unaltered ion permeation (data not shown). These findings both confirm the conclusion that cAMP interaction with the CNBD is not involved in the 4βPMA facilitation and reveal that the domains required to transduce the effect of 4βPMA do not include any of the motifs that have been shown to be involved in intermolecular interactions with cytoplasmic scaffolding or gating modifier proteins (Gravante et al., 2004; Kimura et al., 2004; Santoro et al., 2004; Zong et al., 2005).

Deletion mapping reveals that the conserved transmembrane core of HCN channels is sufficient for 4βPMA facilitation. Left, V1/2 for HCN1 (top) and HCN2 (bottom) and mutants thereof after 30 min of incubation in the absence (○) or presence (•) of 200 nm 4βPMA. Drug-treated values were different from the control (p < 0.005). Right, ΔV1/2 relative to control (top data point and dashed reference line). Numbers on the right are n values. Error bars represent SEM.
4βPMA facilitation of HCN channel activation requires an intact cellular environment
Although high-affinity, stereoselective effects of phorbol esters have not been ascribed to a mechanism other than those acting via C-1 proteins, we were concerned that the gating response of the channels could be a consequence of the ligand binding to a novel site on the channel. To address this, we applied 4βPMA directly to the minimal channel HCN1-ΔNvΔC in cell-free excised inside-out patches. Figure 5A shows the experimental paradigm. Figure 5B shows current families evoked after hyperpolarization of a patch expressing HCN1-ΔNvΔC before and after perfusion with 200 nm 4βPMA. Inspection of the current records (Fig. 5B) and activation curves obtained from these data (Fig. 5C) indicates that direct exposure to 200 nm 4βPMA had no effect on either channel gating or the maximal current. This observation was confirmed by plotting the mean ΔV1/2 APP (Fig. 5F), which was determined as shown in Figure 5D from tail-current records (such as in Fig. 5E) from five independent patches and the maximal current stability (data not shown) determined from the same recordings. These findings demonstrate that the membrane-permeable DAG analog, 4βPMA, does not act directly on HCN channels.

4βPMA facilitation of HCN channel gating requires an intact cellular environment. A, Experimental paradigm. Patches containing HCN1-ΔNvΔC were excised into the Mg-containing intracellular solution. IV indicates collection of current–voltage records shown in B. The bottom time ticks show when ΔV1/2 APP protocol was executed. Asterisks indicate times when current records shown in D and E were obtained. Black bar indicates perfusion with 200 nm 4βPMA. B, Current records (left) and tail currents (right) before (top) and after (bottom) exposure to 200 nm 4βPMA. C, Steady-state activation curves constructed the tail currents (It) as a function of the maximal tail current (It max) obtained from the records shown in B. Fits of the Boltzmann equation are superimposed. D, ΔV1/2 APP voltage protocol (top) and representative current records (bottom) obtained before (gray) and after (black) perfusion with 200 nm 4βPMA obtained from same patch as in B and C at the times indicated by the asterisks in A. E, Tail currents from sweeps shown in D. F, Mean values of ΔV1/2 APP determined in five patches recorded as shown in A–E. Error bars represent SEM.
4βPMA facilitation of HCN gating requires PKC but does not involve phosphorylation of the pore-forming subunit of the channel
We next asked whether 4βPMA facilitation of HCN involves recruitment of cPKCs or nPKCs. To test this, we preincubated oocytes expressing HCN1 with varying concentrations of the bisindolmaleimide pan-PKC inhibitor, Ro31-8220. Figure 6A shows that incubation with Ro31-8220 alone had no effect on HCN1 activation, but the drug blunted channel facilitation during a subsequent 10 min challenge with 200 nm 4βPMA. Although the determined IC50 of the Ro31-8220 suppression of 200 nm 4βPMA enhancement of HCN1 gating (∼2 μm) is above the IC50 determined for Ro31-8220 inhibition of PKC in cell-free assays, it is consistent with concentrations of BIM-I (a closely related bisindolylmaleimide) required to block the effects of PKC on P/Q-calcium channels in Xenopus oocytes (Wu et al., 2002). Similar results were obtained in experiments with the CNBD-disabled channel, HCN1-RE (data not shown).

4βPMA facilitation of HCN gating is abolished by preincubation with the pan-PKC inhibitor Ro31-8220, but elimination of phosphorylatable residues in the conserved channel core does not blunt the response. A, Mean V1/2 (bottom) and ΔV1/2 (top; obtained by subtraction of the means in the bottom panel) of HCN1 preincubated with Ro31-8220 for 6 h before 10 min of incubation in the absence (○) or presence (•) of 200 nm 4βPMA. V1/2 in 0.3, 1, 3, or 10 μm Ro31-8220 was not different from the untreated control (p ∼ 0.67, 0.97, 0.85, and 0.62, respectively). V1/2 after 4βPMA was significantly different from paired incubations in 0 and 0.3 μm Ro31-8220 (p < 0.0005 and ∼0.01, respectively) but not after incubation with 1, 3, or 10 μm PKC inhibitor (p ∼ 0.08, 0.29, and 0.65). Suppression of the 4βPMA response by Ro31-8220 is fit with the Hill equation. The number of cells in each population is shown next to symbols in the bottom panel. B, Serine, threonine (S/T; black circles) and tyrosine (Y; gray circles) residues within the minimal channel HCN1-ΔNvΔC. Residues likely to be exposed to the cytoplasm are shown boxed in green and are numbered according to the cassette followed by position within that cassette. Residues in A2 are in consensus PKC sites. S/T/Y residues in a cassette were simultaneously mutated S/T to N and Y to F. Where multiple mutations were not tolerated (no detectable channel activity), each S/T/Y in a cassette was mutated individually. At two positions (A3:3 and C1:2), data are from constructs bearing alanine rather than asparagine, because the latter mutants did not form functional channels. C, Left, V1/2 after 30 min of incubation in the absence (○) or presence (•) of 200 nm 4βPMA. Right, ΔV1/2 determined as difference of means. Facilitation in the presence of 4βPMA compared with paired controls (p < 0.05) was not different from that observed with the parental channel, HCN1-ΔNvΔC (top data point and blue dashed reference line). wt, Wild-type HCN1-ΔNvΔC. Error bars represent SEM.
The above findings raised the hypothesis that 4βPMA enhancement of HCN gating results from a PKC-mediated phosphorylation of the pore-forming subunit. Figure 6B shows a schematic representation of HCN1-ΔNvΔC and maps onto this all of the serines, threonines, and tyrosines within this minimal 4βPMA-responsive molecule. The residues located in cassette A2 are both consensus PKC phosphorylation sites. Simultaneous elimination of both consensus PKC sites did not blunt the response of HCN1-ΔNvΔC to 4βPMA (Fig. 6C). To test whether phosphorylation by any downstream kinase could account for 4βPMA enhancement of channel gating, we systematically eliminated the remaining 19 serine, threonine, and tyrosine residues that also mapped to presumptive cytoplasmic motifs. Residues were mutated in cassettes (Fig. 6B, green boxes). Where such cassette mutations were found to be nonfunctional, the residues within the cassettes were mutated individually. Although basal gating of several of the constructs was altered relative to the parental molecule (Fig. 6C, left), the enhancement of gating by 4βPMA was not significantly disrupted by any of the replacements (Fig. 6C, right). These data indicate that the action of 4βPMA on HCN gating does not involve direct phosphorylation of the pore-forming subunits. Although it is possible that multiple residues need to be phosphorylated, the absence of any significant reduction in the 4βPMA response in any of the constructs suggests that such a hypothesis is unwarranted.
Activation of DGKs contributes to 4βPMA facilitation of HCN channel gating
The novel and conventional isoforms of the PKC superfamily are not the only signal transduction molecules that can bind 4βPMA via C-1 motifs. C-1 sites are also found in regulators of low-molecular-weight G-proteins (RAS–GRP and chimaerins), in Munc-13 (a protein involved in vesicle cycling) and in several DGK isoforms (Ron and Kazanietz, 1999; Topham and Prescott, 1999; van Blitterswijk and Houssa, 2000; Shindo et al., 2001; Brose and Rosenmund, 2002; Kanoh et al., 2002; Kazanietz, 2002; Topham and Prescott, 2002; Shindo et al., 2003; Yang and Kazanietz, 2003).
Although the C-1 sites in a number of DGK isoforms are degenerate, at least three isoforms bind 4β-phorbols with high affinity (Shindo et al., 2003). Intriguingly, translocation of DGKθ to the plasma membrane requires both occupancy of its C-1 sites by DAG or phorbol ester and an interaction with, and phosphorylation by, PKCε or PKCη (van Baal et al., 2005). In light of these findings and the important location of DGKs in the generation and regulation of membrane signaling lipids (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material), we considered two hypotheses: (1) DGK acts to keep DAG levels below that at which PKC will be activated, thereby preventing tonic facilitation of HCN gating, and (2) DGK recruitment is itself involved in mediating 4βPMA facilitation of HCN gating. These hypotheses predict that block of DGK will either mimic and occlude the action of 4βPMA (1) or block the effect of the tumor promoter (2).
To test these hypotheses, we preincubated oocytes expressing HCN channels with the DGK inhibitor R59949, a high-affinity analog of R59022 [6-E2-E4-[(p-fluorophenyl)phenyl-methylene]-1-piperidinyl(ethyl)-7-methyl-5H-thiazolo(3,2-a)pyrimidine-5-one] (Jiang et al., 2000), an inhibitor that has been shown to stably double DAG levels in Xenopus oocytes within 60 min (Sadler et al., 1996), consistent with this class of inhibitors acting on DGKs in this system.
R59949 had no effect (p > 0.13) on basal activation of HCN2-REHR (V1/2 = −98.5 ± 0.6 mV, n = 31 and −97.3 ± 0.5 mV, n = 38 in the absence and presence of R59949, respectively). The implication of these findings is that elevated DAG cannot alter HCN gating in the absence of functional DGK. However, preincubation with 30 μm R59949 attenuates 4βPMA facilitation of HCN2-REHR gating (Fig. 7A,B), such that the ΔV1/2 with respect to the initial values were 17.9 ± 1.6 mV (n = 7) and 6.5 ± 1.2 mV (n = 16) in the absence and presence of R59949, respectively (p < 0.001). These data indicate that recruitment of DGK is on the path leading to 4βPMA-mediated enhancement of HCN activation.

DGK, MAPK, and PLA2, but not PIK or PLD, contribute to 4βPMA modulation of HCN channel gating. A, ΔV1/2 of HCN2-REHR elicited by 200 nm 4βPMA (30 min) after preincubation in the absence (gray bars) or presence (black bars) of the indicated inhibitor or combination thereof (wortmannin, 10 μm for 60 min; 1-butanol, 30 mm for 60 min; OPC, 3 μm for 60 min; AACOCF3, 50 μm for 60 min; R59949, 30 μm for 60 min; SB-202190, 50 μm for 120 min; U-0126, 50 μm for 120 min). The dashed and dotted lines represent the mean and SEM of the depolarization elicited by 200 nm 4βPMA determined across all recordings in which HCN2-REHR-expressing cells were exposed to 200 nm 4βPMA (81 separate cells). Numbers in each experimental group are indicated next to the appropriate rows. Asterisks indicate that the value determined in the presence of inhibitor is significantly different from its paired control (p < 0.05). B, ΔV1/2 in the presence of inhibitor normalized to the maximal response seen in the absence of inhibitor in paired control recordings. C, Suppression of HCN2-REHR current in response to 200 nm 4βPMA (30 min) after preincubation in the absence (gray bars) or presence (black bars) of the indicated inhibitor or combination thereof. Concentrations and times are as in A. Dashed and dotted lines and asterisks have the same meaning as in A. D, Suppression of current in the presence of inhibitor normalized to the maximal response seen in the absence of inhibitor in paired control recordings (ΔIMAX plus inhibitor/ΔIMAX minus inhibitor). Error bars represent SEM.
During the course of these experiments, we noted that preincubation of oocytes with 30 μm R59949 not only attenuated the 4βPMA shift in activation gating, but also appeared to protect against loss of current. Thus, whereas the maximal current was reduced approximately threefold after 30 min of incubation in 200 nm 4βPMA in cells that received no pretreatment, the current density in cells pretreated with 30 μm R59949 was only reduced ∼30% after subsequent exposure to 4βPMA (p ∼ 0.001) (Fig. 7C,D).
Is activated DGK the final arbiter that controls HCN channel facilitation?
The immediate product of DGK is PA. This low-abundance membrane lipid both is the entry point for DAG into the PI synthesis pathway (leading to the formation of 4,5-PIP2, a polyanionic lipid that can facilitate HCN gating) (our unpublished observations) (Pian et al., 2006) and acts as a signaling lipid in its own right (see Discussion). Accordingly, we considered three alternative hypotheses to account for the DGK sensitivity of 4βPMA enhancement of HCN gating: (1) An elevation of PA leads to an increase in local concentrations of 4,5-PIP2; (2) PA acts to recruit the MAPK/PLA2 pathway, and the action of 4βPMA is exerted downstream of one or both of these enzymes (Poolos et al., 2006); or (3) PA acts as a direct modifier of HCN gating.
To test the hypothesis that 4βPMA recruitment of DGK acts on HCN channels by enhancing synthesis of 4,5-PIP2, we preincubated oocytes expressing HCN2-REHR with 10 μm wortmannin (an inhibitor of PI3 and PI4 kinases with a Ki for the latter of ∼0.5 μm) and subsequently challenged the cells with 200 nm 4βPMA. Preincubation with 10 μm wortmannin had no effect (p > 0.37) on basal gating (V1/2 = −98.5 ± 0.6 mV, n = 31 and −97.8 ± 0.5 mV, n = 34 in the absence and presence of wortmannin, respectively), nor did it blunt the phorbol ester-evoked depolarization of gating such that the ΔV1/2 with respect to the initial values were 17.9 ± 1.6 mV (n = 7) and 18.8 ± 2.7 mV (n = 8), in the absence and presence of wortmannin, respectively (p ∼ 0.74) (Fig. 7A,B). Rather, block of PI3K and PI4K appeared to accelerate the response of the channels to 4βPMA (data not shown), indicating that entry of PA into the phosphoinositide path slows down channel facilitation. Furthermore, inhibition of PI3 and PI4 kinases did not suppress the 4βPMA inhibition of maximal current (p ∼ 0.82) (Fig. 7C,D). These findings suggest that channel activation is controlled by a mechanism that lies downstream of DGK but does not involve phosphoinositide synthesis.
PA is known to play a role in the recruitment of the MAPK pathway by recruiting and enhancing activation of the upstream kinase Raf-1 (Ghosh et al., 1996; Andresen et al., 2002; Anderson, 2006). To investigate a potential role for the MAPK pathway in mediating the 4βPMA response, we preincubated cells expressing HCN2-REHR with a mixture of MEK (an activating kinase of MAPK) and MAPK blockers (50 μm each of U-0126 and SB-202190) and then challenged the cells with 4βPMA (200 nm for 30 min). Preincubation with the MEK/MAPK blockers had no effect on basal gating. Thus, the V1/2 in the presence of U-0126 and SB-202190 (−95.5 ± 1.1 mV; n = 28) was not significantly different (p > 0.4) from paired controls (−96.3 ± 0.8 mV; n = 28). However, inclusion of these inhibitors attenuated the effect of 4βPMA on gating (Fig. 7A,B) such that ΔV1/2 in their presence (10.9 ± 1.7 mV; n = 7) was significantly different (p < 0.01) from that observed in their absence (19.0 ± 2.1 mV; n = 5). Preincubation with U-0126 and SB-202190 also provided a significant (p ∼ 0.002) protection against the 4βPMA suppression of IMAX (Fig. 7C,D).
What is the mechanism by which the MAPK pathway can contribute to 4βPMA-mediated enhancement of HCN gating? Our finding that the action of 4βPMA persists when most cytoplasmically exposed serines, threonines, and tyrosines are removed from the channel by domain deletion and the remaining residues are sequentially eliminated by site-directed mutagenesis (see above) suggests that direct phosphorylation of the channel core does not account for the action of this kinase. Although this conclusion must be qualified because we cannot formally exclude the possibility that MAPK acts by promoting phosphorylation of multiple residues, the failure of any mutation to decrease the 4βPMA response (Fig. 6B,C) would suggest that this hypothesis is unwarranted.
In a variety of cell types, 4βPMA has been shown to elicit a potent upregulation of PLA2 activity that is mediated via activation of MAPK (Lin et al., 1993; Qiu and Leslie, 1994; Xing and Insel, 1996; Xing et al., 1997; You et al., 2005). Additionally, it has been established that conventional and novel isoforms of PKC can activate the MAPK pathway, primarily by phosphorylation of the upstream kinase Raf-1 (Kolch et al., 1993; Ueda et al., 1996; Cai et al., 1997; Schonwasser et al., 1998). To investigate the potential role of PLA2 activity in 4βPMA-mediated facilitation of HCN gating, we incubated oocytes expressing HCN2-REHR with an inhibitor of either the secreted (s-PLA2) or the cytoplasmic/independent (c/i-PLA2) enzyme subfamilies: OPC or AACOCF3, respectively.
The V1/2 of HCN2-REHR in the presence of either 3 μm OPC (−93.1 ± 0.6 mV; n = 39) or 50 μm AACOCF3 (−95.5 ± 0.7 mV; n = 41) were not significantly different from paired untreated (−96.8 ± 0.5 mV; n = 35) or vehicle controls (−95.4 ± 1.2 mV; n = 17), indicating that neither inhibitor altered the basal gating of the channel. However, blockade of c/i- but not s-PLA2s suppressed channel facilitation after exposure to 4βPMA (Fig. 7A,B) such that the ΔV1/2 in the presence of 4βPMA (14.5 ± 2.2 mV; n = 9) was significantly different (p < 0.001) from the effect of 4βPMA determined in the presence of AACOCF3 (ΔV1/2 = 5.2 ± 0.7 mV; n = 13) but not in the presence of OPC (ΔV1/2 = 13.1 ± 2.0 mV; n = 8). It should be noted that whereas 3 μm OPC is only three times the in vitro Ki for inhibition of s-PLA2, higher concentrations could not be tested, because they resulted in cell death. Incubation with AACOCF3 (p ∼ 0.001) but not OPC (p ∼ 0.41) attenuated the 4βPMA-evoked decrease in the maximal current (Fig. 7C,D). These findings suggest that recruitment of c/i-PLA2 isozymes, possibly via the MAPK cascade, is a downstream mechanism by which PA can regulate HCN channel activation and regulation of current density.
Generation of phosphatidic acid by phospholipase D does not contribute to the 4βPMA response
PA is readily generated not only from DAG via DGK, but also from cleavage of phosphatidylcholine by the action of the PKC-activated enzyme phospholipase D (PLD) (Butikofer et al., 1993; Bollag et al., 2005). We therefore asked whether blocking the action of PLD by 1-butanol could occlude the 4βPMA facilitation, which would indicate a role for this mechanism of PA synthesis in the phorbol response. However, incubation with 1-butanol (30 mm for 60 min) had no effect on basal gating (V1/2 in the absence or presence of butanol were −95.9 ± 1.2 mV, n = 19 and −96.6 ± 1.2 mV, n = 23, respectively; p > 0.68), nor did it interfere (p ∼ 0.62) with 4βPMA enhancement of channel gating (ΔV1/2 = 14.2 ± 2.5, n = 7 in the absence of butanol and 12.9 ± 3.5, n = 7 in the presence of the alcohol) (Fig. 7A,B) or suppression of the maximal current (p ∼ 0.6) (Fig. 7C,D).
Phosphatidic acid and arachidonic acid directly facilitate HCN channel gating
The above results indicate that DGK and cPLA2 mediate the 4βPMA-evoked depolarization of HCN gating. To test the hypothesis that the immediate downstream products of these two enzymes, PA and AA, respectively, can act as direct regulators of HCN gating, we applied each of these lipids to excised inside-out patches expressing HCN2. Figure 8A and Figure 8, B and C (top), show the protocols used in these experiments. The patches were initially excised into a Mg-containing intracellular solution to deplete lipase-sensitive lipids such as 4,5-PIP2, and then they were exchanged into a Mg-free solution before lipids were applied. Figure 8, B and C (bottom), shows representative sweeps obtained before and after perfusion with the indicated metabolite. It is apparent from these records that gating in the presence of PA or AA was enhanced at the intermediate voltage but was unaltered at a voltage that would saturate channel opening, indicating that the V1/2 of channel activation had shifted to a more depolarized potential. To quantify the effects of PA and AA on HCN2 gating, we calculated the apparent shift in the V1/2 (Fig. 8D,E, top) and the maximal current relative to the initial value (Fig. 8D,E, bottom). These data show that both metabolites depolarized activation gating, consistent with them acting as messengers downstream of 4βPMA. Modulation of HCN gating occurred with a modest (AA) or no (PA) decrease in maximal current.

Phosphatidic acid and arachidonic acid, the products of DGK and PLA2, respectively, both directly facilitate HCN gating. A, Schematic representation of the experimental paradigm. Data were obtained from inside-out patches excised into a Mg-containing intracellular solution from cells expressing HCN2. IV and bottom time ticks indicate determination of full activation curve and execution of the ΔV1/2 APP protocol, respectively. Black bar indicates perfusion with 5 μm PA or AA. B, C, Voltage protocol (top) and representative sweeps (bottom) acquired before (gray traces) and after (black traces) inclusion of 5 μm PA (B) or AA (C). D, E, Mean values of ΔV1/2 APP (top) and maximal current amplitude relative to the initially determined value (bottom) determined from seven (D) and six (E) separate recordings as shown in B and C. Open circles indicate absence of PA or AA; filled circles indicate presence of PA or AA. Error bars represent SEM.
To ask whether DGK- and PLA2-mediated increases in PA and AA combine to enhance HCN gating, we pretreated cells expressing HCN2-REHR with a combination of R59949 and AACOCF3. Preincubation with 30 μm R59949 and 50 μm AACOCF3 had no effect (p > 0.95) on basal gating (V1/2 = −96.0 ± 1.3 mV, n = 12 and −96.1 ± 0.7 mV, n = 65 in the absence and presence of the inhibitor mixture, respectively), but it greatly diminished the phorbol ester-evoked depolarization of gating (ΔV1/2 after 30 min of incubation with 200 nm 4βPMA was 13.3 ± 1.0 mV, n = 15 in the absence and 3.1 ± 0.8 mV, n = 22 in the presence of the inhibitor mixture) (Fig. 7A,B) and suppression of the maximal current (p < 0.001) (Fig. 7C,D). The effect of these inhibitors together appeared to be somewhat more efficacious than the effects of either inhibitor alone (compare the normalized effects in Fig. 7B,D), but this enhancement did not reach the level of statistical significance.
The PLC/DAG lipase pathway of arachidonic acid generation does not play a role in the 4βPMA response in Xenopus oocytes
We have shown that c/i-PLA2-generated AA appears to contribute to the 4βPMA-evoked alteration of HCN channel gating. A second route by which AA can be generated is through DAG lipase cleavage of PLC-generated DAG. To determine whether this alternative path may also contribute to HCN regulation by independently elevating AA, we tested the sensitivity of HCN channel enhancement by 4βPMA to the PLC inhibitor U73122 (10 μm) or the DAG lipase inhibitor RHC 80267 (40 μm). Neither inhibitor had significant effects on basal gating or the 4βPMA-mediated depolarization of gating of HCN1-REHR (data not shown), indicating that this pathway does not contribute to regulation of HCN channels in this system.
Specificity of the 4βPMA facilitation of HCN channel gating
Although both AA and PA are well described regulators of cellular function, including actions on other ion channels (see Discussion), the suggestion that they are acting to enhance gating of voltage-dependent channels raises several important questions: (1) Do these anionic lipids act on channel gating through a generalized change in surface charge or do they act on HCN channels via specific interactions? (2) What is the basis of current suppression, given that these metabolites appear to have little (AA) or no (PA) effect on the maximal current carried by HCN channels in excised patches? The data presented in Figure 9 offer a window into these issues.

Activation of the plant hyperpolarization-activated channel KAT1 is insensitive to 4βPMA. A, TEVC current families (left) and tail currents (right) recorded from a cell expressing KAT1 before (top) and after (bottom) incubation with 200 nm 4βPMA for 30 min. B, Top, I–V curves constructed from records shown in A. Bottom, Mean steady-state activation curves for KAT1 before (V1/2 = −107.8 ± 4.8 mV; n = 12) and after (V1/2 = −115.2 ± 5.7 mV; n = 6) 30 min of incubation in 200 nm 4βPMA. Data are fit with the Boltzmann equation. The hyperpolarization was not statistically significant (p = 0.34).
Figure 9A shows TEVC recordings obtained from a Xenopus oocyte expressing the plant hyperpolarization-activated channel, KAT1, before (top) and after (bottom) 30 min of incubation in 200 nm 4βPMA. It is apparent from inspection of tail-current records that although 200 nm 4βPMA reduced the available current (as seen with HCN channels), KAT1 gating is not depolarized. Indeed, opening of KAT1 appeared to require more extreme hyperpolarization after incubation with 4βPMA. We quantified these effects by constructing current–voltage and steady-state activation curves (Fig. 9B). This analysis confirmed that 4βPMA suppresses the KAT1 current to 34.1 ± 9% (p < 0.02), but this was not associated with a significant alteration of the voltage dependence of channel opening (p ∼ 0.48). These findings indicate that 4βPMA facilitation of HCN gating is mediated by a channel-specific mechanism rather than a generalized change in surface charge, whereas the suppression of the maximal current appears to be significantly mediated by a channel-independent mechanism.
Discussion
IH channels are regulated by a plethora of neurotransmitter receptors, some of which are unlikely to exert control via changes in cAMP, H+I, or 4,5-PIP2, the only messengers known to alter the voltage dependence of channel opening. In this study, we explored whether DAG-sensitive signaling cascades could alter gating of expressed HCN channels. A summary of the signaling pathways considered here is presented in supplemental information Figure 2 (available at www.jneurosci.org as supplemental material).
We show that 4βPMA evokes a potent (EC50, ∼2 nm) and stereoselective (4αPMA was ineffective) enhancement of HCN activation that requires an intact cellular environment (4βPMA was ineffective when applied in cell-free patches) but which is independent of allosteric actions of cAMP or H+I (cAMP/H+I-uncoupled channels retain 4βPMA sensitivity). Inhibition of this response by Ro31-8220 and R59949 implicates a role for both PKC and DGK.
How does block of either PKC or DGK diminish the HCN response if binding of 4βPMA to their respective integral C-1 sites can activate each enzyme? An attractive possibility was suggested from the observation that DGKθ acts as a coincidence detector such that its translocation and activation requires both an interaction with, and phosphorylation by, PKCε or PKCη and occupancy of its own three C-1 sites (van Baal et al., 2005) [however, see also Luo et al. (2003) for examples of inhibitory interactions between PKC and DGK]. Although DGK trafficking has not been studied in Xenopus oocytes, it is interesting to note that both PKCε and PKCη are present (Xiao et al., 2001; Gundersen et al., 2002; Maeno-Hikichi et al., 2003) and the half time for 4βPMA-mediated enhancement of HCN gating (∼5–10 min) is similar to the half time (∼6 min) for DGKθ to migrate to the membrane in A431 cells (van Baal et al., 2005). We thus consider it likely that signaling downstream of DGKs contributes to 4βPMA regulation.
In light of these results, we asked whether PA, the immediate product of DGK, could control HCN gating. We considered two hypotheses: (1) PA can control HCN channels directly, and (2) PA recruits downstream cascades. Our results suggest that both processes may be occurring. Thus, analysis of the effects of PA in cell-free patches shows that the lipid can directly enhance HCN channel activation, whereas inhibitor studies suggest that signaling cascades sensitive to PA are also important.
What indirect effects of PA may be relevant? PA is the entry point for phosphoinositide synthesis, suggesting that 4βPMA could act by elevating 4,5-PIP2, an acidic lipid that is known to enhance HCN function (Pian et al., 2006; Zolles et al., 2006). PA-rich membranes activate the MEK/MAPK cascade by recruiting Raf-1 kinase to the membrane, where it can interact with Ras (Seger and Krebs, 1995; Ghosh et al., 1996; Schonwasser et al., 1998; Andresen et al., 2002; Anderson, 2006), suggesting that an increase in PA could enhance MAPK signaling. Through the use of inhibitors of PI kinases and the MAPK cascade, we show that MAPK is involved in 4βPMA facilitation of HCN gating, but upregulation phosphoinositide synthesis is not.
Does MAPK effect a change in HCN channel function directly? Although our deletion and sequential site-directed mutagenesis studies do not formally exclude the possibility that phosphorylation of multiple residues across mutation boundaries may contribute to the 4βPMA-mediated alterations in HCN channel function, the finding that inhibition of DGK and PLA2 essentially eliminates the phorbol response supports the hypothesis that phosphorylation of HCN channels by PKC and MAPK is not a major mechanism of regulation within the Xenopus oocyte expression system. It should be noted that these results do not preclude the possibility that, in native cells, IH can be controlled by these kinases acting directly.
How may MAPK effect an indirect change in HCN gating? In a variety of cells, 4βPMA has been shown to evoke a p38-MAPK/PLA2-mediated release of AA (Lin et al., 1993; Qiu and Leslie, 1994; Xing and Insel, 1996; Xing et al., 1997; You et al., 2005), an activation that involves PKC but may also be dependent on an increase in PA. Through the use of inhibitors of PLA2 s, we show that activation of c/i-PLA2, but not s-PLA2, is involved in 4βPMA facilitation of HCN gating, consistent with PLA2 acting as a downstream effector of MAPK. We further show that AA, a metabolite liberated by PLA2 and known to control activation of other channels (Nilius and Voets, 2004; Oliver et al., 2004; Besana et al., 2005), is a direct regulator of HCN activation.
We conclude that two PKC-activated pathways, DGK upregulation of PA and Ras/Raf/MEK/MAPK/PLA2 liberation of AA, act in concert to modify both channel gating and current density. The extent to which each of these pathways contributes to regulation of IH in native cells is an interesting question that merits study.
Do these pathways alter HCN gating via a generalized change in surface charge or do they act on HCN channels by modifying specific channel–lipid interactions? A generalized surface charge mechanism requires that all voltage-sensitive channels respond with an equivalent shift along the voltage axis. In contrast with this prediction, we show that KAT1 gating is either insensitive or hyperpolarized in response to 4βPMA. Although we cannot formally rule out that a depolarizing surface charge effect on KAT1 is offset by a competing effect of some other form of regulation, it is interesting to note that KCNQ1 and KCNQ2 also display isoform-selective responses to 4βPMA (Nakajo and Kubo, 2005). It is also interesting to note that some actions of PA are mediated via specific coupling of the lipids to polybasic clusters in target proteins (Limatola et al., 1994; Frank et al., 1999; Grange et al., 2000; Sergeant et al., 2001; Nakajo and Kubo, 2005), as has been described for 4,5-PIP2 (Zhang et al., 1999; Shyng et al., 2000), whereas regulation of several voltage-gated channels by AA and its metabolites appears to involve lipid interaction with membrane-buried elements of the pore (Hamilton et al., 2003; Oliver et al., 2004).
What is the likely basis for the suppression of the maximal current? Clearly, such a reduction could arise from changes in n, Po, or i (number of channels, channel open probability, and single-channel current, respectively). Although we have not addressed this extensively, we consider the most parsimonious interpretation of the data to be that this process is dominated by a reduction in n as a consequence of a retrieval of the plasma membrane. We hypothesize that the current suppression represents a retrieval of PA- and AA-rich regions of the membrane [both lipids are proendocytotic (Llorente et al., 2000; Andresen et al., 2002; Rizzo and Romero, 2002; Carattino et al., 2003; Anderson, 2006)], an effect that may be enhanced by MAPK [as this enzyme is recruited to, and helps assemble, endocytotic machinery (Rizzo and Romero, 2002; Anderson, 2006)]. This conclusion is supported by the observations that 4βPMA suppresses the maximum current carried by KAT1 (although the gating of this functionally related hyperpolarization-activated channel is insensitive to the ligand) and that 4βPMA does resculpt the oocyte plasma membrane, reducing the number and length of oocyte microvilli (Vasilets et al., 1990). It should, however, be noted that the results in Figure 8E suggest that AA or related metabolites may contribute to the reduction in the HCN current by altering channel function directly.
Overall, our findings suggest that PA, AA, and metabolites thereof may serve as physiological regulators of native IH, and imply that pathways leading to activation of DGK, PLD, MAPK, and PLA2 may be able to control IH in native cells. Intriguingly, it has recently been reported that MAPK is a potent regulator of gating of IH in hippocampal pyramidal neurons, such that inhibition of MAPK (by either SB-202190 or SB-203580) hyperpolarized channel gating by ∼25 mV, whereas upregulation of MAPK activity depolarized IH gating by an additional 11 mV from rest (Poolos et al., 2006), whereas NMUR2 [NMU receptor 2; stimulation of which facilitates IH gating (Qiu et al., 2003)] positively couples to the MAPK/PLA2 cascade (Brighton et al., 2004). In this regard, it is interesting to note that our finding that SB-202190 does not alter basal gating of HCN channels in Xenopus oocytes (thereby demonstrating that the drug does not have a nonspecific effect on the channels) supports the conclusion of Poolos et al. (2006) that activated MAPK contributes to the gating of hippocampal IH.
Given the significant contribution of IH to the subthreshold behavior of many cells in both the CNS and periphery (including the PNS, heart, and smooth muscle), our evidence that the voltage dependence of HCN channels is sensitive to AA and PA opens up avenues of investigation in a number of realms, including synaptic transmission (Sang and Chen, 2006), neuropathic pain (Woolf and Salter, 2000; Chaplan et al., 2003), and cardiac ischemia (Mancuso et al., 2003), in which both IH and changes in these metabolites have been independently implicated.
Footnotes
-
This work was supported by Whitehall Foundation Grants S98-23 and 2003-05-02-REN and Johnson & Johnson Pharmaceutical Research and Development (G.R.T.), and by National Institutes of Health Grant F31 MH070202-02 (K.J.F.). We thank Jay Yang, Sandra Chaplan, Geoffrey Pitt, and Steven Siegelbaum for helpful discussions and Margaret Wood and faculty in the Department of Anesthesiology for their continuing support. We thank members of Steven Siegelbaum's laboratory for generously providing us with Xenopus oocytes. HCN2-RE and KAT1 were generous gifts from Drs. Siegelbaum and Hoshi, respectively.
- Correspondence should be addressed to Gareth R. Tibbs, Department of Anesthesiology, Eye Institute Research Annex, EI3-305, 160 Fort Washington Avenue, New York, NY 10032. grt1{at}columbia.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}