

Abstract
The vesicle-associated membrane protein-associated proteins (VAPs) VAPA and VAPB interact with lipid-binding proteins carrying a short motif containing two phenylalanines in an acidic tract (FFAT motif) and targets them to the cytosolic surface of the endoplasmic reticulum (ER). A genetic mutation (P56S) in the conserved major sperm protein homology domain of VAPB has been linked to motor-neuron degeneration in affected amyotrophic lateral sclerosis (ALS) patients. We report that in the CNS, VAPB is abundant in motor neurons and that the P56S substitution causes aggregation of mutant VAPB in immobile tubular ER clusters, perturbs FFAT-motif binding, and traps endogenous VAP in mutant aggregates. Expression of mutant VAPB or reduction of VAP by short hairpin RNA in primary neurons causes Golgi dispersion and cell death. VAPA and VAPB are reduced in human ALS patients and superoxide dismutase 1 (SOD1)-ALS-transgenic mice, suggesting that VAP family proteins may be involved in the pathogenesis of sporadic and SOD1-linked ALS. Our data support a model in which reduced levels of VAP family proteins result in decreased ER anchoring of lipid-binding proteins and cause motor neuron degeneration.
- protein aggregation disease
- amyotrophic lateral sclerosis
- ALS
- endoplasmic reticulum
- lipid-binding protein
- VAP family proteins
- motor neuron disease
Introduction
Many late-onset neurodegenerative diseases are characterized by aggregate-prone proteins (Taylor et al., 2002). The failure of a protein to adopt its proper structure usually confers to its dominant toxic properties and threatens normal cell function and viability (Taylor et al., 2002; Ross and Poirier, 2005). In both sporadic and familial amyotrophic lateral sclerosis (ALS) patients, protein inclusions in motor neurons are a constant and pathognomic feature. ALS-linked mutations in superoxide dismutase 1 (SOD1), the most common and best-characterized form of familial ALS, lead to misfolding and aggregate formation resulting in dominant toxic functions of mutant SOD1 (Boillee et al., 2006). Previously, a missense mutation (P56S) in the gene-encoding vesicle-associated membrane protein (VAMP)-binding protein B (VAPB) has been identified in a familial form of ALS (ALS8) (Nishimura et al., 2004). Expression of P56S mutant VAPB (VAPB-P56S) in cultured cells results in the formation of cytosolic aggregates (Nishimura et al., 2004); however, the cellular mechanism leading to VAPB-linked motor neuron degeneration is not known.
The VAP family of proteins was originally identified in Aplysia californica as a binding partner of VAMP/synaptobrevin (Skehel et al., 1995); it consists of two genes in mammals, VAPA (VAP-33) and VAPB (ALS8, VAPC) (Nishimura et al., 1999). VAPs are ubiquitously expressed, type II integral membrane proteins that localize to the endoplasmic reticulum (ER) and pre-Golgi intermediates (Skehel et al., 2000), and have been proposed to regulate transport between the ER and the Golgi (Soussan et al., 1999; Amarilio et al., 2005). Moreover, VAPs have been shown to target lipid-binding proteins carrying a short motif containing two phenylalanines in an acidic tract (FFAT motif) to the ER (Kaiser et al., 2005; Loewen and Levine, 2005). In yeast, the VAP homolog Scs2 binds the FFAT motif and in absence of Scs2, the FFAT-containing proteins mislocalize to the cytoplasm (Loewen et al., 2003). The FFAT motif consists of the consensus amino acid sequence EFFDAxE; it was identified because of its conservation in several lipid-binding protein families implicated in the transfer of lipids between the ER and other organelles, such as the Golgi, endosomes, and plasma membrane (Olkkonen, 2004; Holthuis and Levine, 2005; Levine and Loewen, 2006). Because the lipid-binding properties and supposed physiological roles of FFAT motif-containing proteins are very diverse, VAPs are likely involved in multiple metabolic pathways (Lev, 2004; Olkkonen, 2004; Kawano et al., 2006; Levine and Loewen, 2006; Perry and Ridgway, 2006).
To gain insight into the events leading from VAP alterations to motor neuron disease, we investigated the distribution of VAPB in the CNS and focused on the biochemical and cellular effects of the ALS-linked P56S mutation in VAPB. Our data suggest that VAPB-P56S causes motor neuron degeneration via a dominant-negative mechanism whereby mutant aggregates trap endogenous wild-type (wt) VAP, reduce cytosolic VAP levels, and impair lipid-binding protein function. Because VAP expression is also reduced in the spinal cord of human ALS patients and SOD1-ALS transgenic mice, we propose that VAP may be an important factor involved in the pathogenesis in sporadic and SOD1-linked ALS. The data support a model in which reduced levels of VAP family proteins cause neuron degeneration.
Materials and Methods
GST/His-VAP constructs and antibody generation.
Nucleotide sequences encoding VAPB amino acids 1–225 and VAPA and VAPB amino acids 132–225 were cloned into pGEX-4T (GE Healthcare Bio-Sciences, Piscataway, NJ) to create glutathione-S-transferase (GST) fusion proteins, and into pET-32A (Novagen, Madison, WI) to create His-tagged fusion proteins. GST-VAP fusion proteins were induced in BL21 Escherichia coli cells by isopropyl β-d-1-thiogalactopyranoside and purified using glutathione-Sepharose 4B beads (GE Healthcare Bio-Sciences) according to the manufacturer's instructions. Purified proteins were concentrated using Centricon (Bio-Rad, Hercules, CA) and injected into New Zealand White Rabbits in a suspension of TiterMax Gold adjuvant (Sigma, St. Louis, MO). Sera #1006-00 and #1006-01 are against VAPB amino acids 1–225, sera #1006-02 and #1006-03 are against VAPB amino acids 132–225, and sera #1006-04 and #1006-05 are against VAPA amino acids 132–225. His-VAP fusion proteins were induced in Rosetta bacteria, and purified using Nickel beads (Qiagen, Hilden, Germany) according to the manufacturer's protocol. His-tagged fusion proteins were coupled to cyanogen bromide-activated Sepharose 4B-columns (GE Healthcare Bio-Sciences) and used to purify VAP antibodies.
Expression constructs.
The following mammalian expression plasmids have been described previously: tsVSVG-YFP (Toomre et al., 1999), ORP3-GFP (Lehto et al., 2005), Htt(Q74)-GFP and GFP-heat shock protein 70 (Hsp-70) (gifts from Dr. H. Kampinga, University of Groningen, Groningen, The Netherlands), Hsp-104 (gift from Dr. S. Lindquist, Whitehead Institute for Biomedical Research, Cambridge, MA), and the protein–biotin ligase BirA (Lansbergen et al., 2006). Full-length human VAPA and VAPB constructs were generated by PCR using IMAGE clones 2822547 (BC002992) and 3543354 (BC001712) as templates and cloned into hemagglutinin (HA)- and myc-tagged pGW1-expression vectors. P56S, K87D, and M89D mutations were generated by site-directed mutagenesis. Green fluorescent protein (GFP)-VAPB-transmembrane domain (TMD) (amino acids 213–245), N-terminal GFP-VAPB (GFP-VAPB-N) (amino acids 1–213), and GFP-VAPB-major sperm protein (MSP) (amino acids 1–158) were cloned by PCR in a modified pβactin-16 pl vector. GFP-VAPB was obtained by cloning VAPB-N into GFP-VAPB-TM. GFP-FFAT was obtained by cloning the amino acid sequence of the FFAT domain of the human Nir2-protein and flanking amino acids (NSSEE EFFDAHE GFSDS) into pEGFP-C1 (Clontech, Mountain View, CA). For GFP-Scrambled (GFP-SCR), the amino acids were mixed (FESSE EDNFAHE GFSDS). Bio-HA-VAPB constructs were generated by incorporating a biotinylation-tag (MSGLNDIFEAQKIEWHE) into the HA-tagged VAP-constructs. VAPA4 (gcatgcagagtgctgtttc), VAPA2 (ggaaactgatggaagagtg) and VAPB1 (ggtgatggaagagtgcagg) short hairpin RNAs (shRNAs) were designed against rat VAPA and VAPB sequences (NM_031631 and NM_019806 respectively), annealed and inserted into pSuper-vector (Hoogenraad et al., 2005).
Antibodies and reagents.
The following primary antibodies were used for immunocytochemistry. Mouse monoclonal antibodies against protein disulfide isomerase (PDI; 1:300; Affinity BioReagents, Golden, CO), GM130 (1:1000; BD Biosciences, Franklin Lakes, NJ), HA (1:500; Roche, Indianapolis, IN), lysosome-associated membrane protein 1 (LAMP1; 1:50; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), Ubiquitin FK1 (1:300; Biomol, Plymouth Meeting, PA), vimentin (1:200; Sigma) and myc (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), β-galactosidase (β-gal; 1:2000; Promega, Madison, WI) were used. Polyclonal rabbit antibodies used were against VAPA (#1006-04; 1:500), VAPB (#1006-00; 1:500), calreticulin (1:1000; Affinity BioReagents), HA (1:500; Santa Cruz Biotechnology), γ-adaptin (1:500; BD Biosciences), BiP/GRP78 (1:500; Stressgen Biotechnologies, San Diego, CA), Eif2α (α-subunit of eukaryotic initiation factor 2; 1:500; Sigma), β-galactosidase (1:2000; MP Biomedicals, Irvine, CA) and Atg8/light chain 3 (LC3) Atg12 (1:1000; gifts from Dr. R. Kopito, Stanford University, Stanford, CA) (Iwata et al., 2005). The following primary antibodies were used for Western blot analysis: rabbit anti-VAPA (#1006–04; 1:500), rabbit anti-VAPB (#1006-02; 1:1000), mouse anti-α-tubulin (1:5000; Sigma), rabbit anti-HA (1:500; Santa Cruz), mouse anti-β-actin (1:5000; Millipore, Temecula, CA), rabbit anti-SOD101 (1:1000; Stressgen Biotechnologies), rabbit anti-GFP (1:1000; Abcam, Cambridge, MA), rabbit anti-myc (1:500; Cell Signaling Technology, Beverly, MA), rabbit-anti-calreticulin (1:1000; Affinity BioReagents). Nocodazole and brefeldin A were obtained from Sigma.
HeLa cell transfection and immunocytochemistry.
HeLa cells were cultured in DMEM/HAm'sF10 (50/50%) medium containing 10% FCS and 1% penicillin/streptomycin. One day before transfection, cells were plated at 1:25 in Lab-tek chamber slides (Nunc, Roskilde, Denmark). Cells were transfected with SuperFect transfection reagent (Qiagen) according to the manufacturers protocol and grown for 16 h. Two hours before fixation, cells were serum-stimulated with fresh medium. Cells were fixed in 4% paraformaldehyde for 10 min at room temperature followed by 5 min in 0.1% Triton X-100 in PBS. Slides were blocked in 0.5% BSA/0.02% glycine in PBS and labeled with primary antibody for 2 h at room temperature, as described by Hoogenraad et al. (2000).
COS-1 cell transfection and Western blot analysis.
COS-1 cells were cultured in DMEM/HAMF10 (50/50%) medium containing 10% FCS and 1% penicillin/streptomycin. COS-1 cells were transfected by the DEAE-dextran method (Hoogenraad et al., 2000). Cells were harvested 24 h after transfection, by scraping the cells in PBS and lysating cell pellets in lysis buffer (25 mm Tris-HCl, pH 8.0, 100 mm NaCl, 0.5% Triton X-100, and protease inhibitors; Roche) followed by sonification. Supernatant and pellet fractions were separated by centrifugation at 13,000 rpm for 15 min; pellets were dissolved in an equal amount of lysis buffer. Samples were mixed with 4× sample buffer (8% SDS, 25% glycerol, 0.05 m Tris, pH 6.8, 200 mm DTT, and 40 mg/L bromophenol blue) and boiled. Equal amounts of proteins were loaded onto 10% SDS-PAGE gels and subjected to Western blotting on polyvinylidene difluoride membrane. Blots were blocked with 2% bovine serum albumin/0.05% Tween 20 in PBS and incubated with primary antibodies at 4°C overnight. Blots were washed with 0.05% Tween 20 in PBS three times for 10 min at room temperature and incubated with either anti-rabbit or anti-mouse IgG antibody conjugated to horseradish peroxidase (Dako, High Wycombe, UK). Blots were developed with enhanced chemiluminescent Western blotting substrate (Pierce, Rockford, IL).
Primary neuron cultures and transfection.
Primary rat hippocampal neurons were plated at a density of 75,000/well on 15 mm glass coverslips and transfected at 13 d in vitro (DIV13) with HA-tagged wild-type VAPB (HA-VAPB-wt), P56S, VAP-shRNA, β-galactosidase, or GFP using Lipofectamine-2000 as described previously (Hoogenraad et al., 2005). After 2 d of transfection, neurons were fixed and stained with anti-HA, anti-GM130, and anti-PDI antibodies, and the number of transfected neurons per coverslip was counted. The appearance of the Golgi apparatus was investigated and differences were analyzed using the χ2 test. Representative cells were imaged using a confocal microscope.
Immunoprecipitation.
COS-1 cells were cultured, transfected, and harvested as described above. To increase the solubility of VAPB-P56S, transfected cells were incubated for 2 h before lysis. Cells were lysed in lysis buffer (25 mm Tris-HCl, pH 8–8.5, 50 mm NaCl, 0.5% Triton X-100, and protease inhibitors; Roche) and centrifuged at 13,000 rpm for 15 min. Supernatants were mixed with an equal amount of lysis buffer, protein-A-agarose beads (GE Healthcare Bio-Sciences), and 3–5 μg of mouse anti-HA (Covance, Princeton, NJ), mouse anti-GFP, rabbit anti-HA (Santa Cruz), or control IgG (Sigma). Samples were incubated overnight rotating at 4°C, centrifuged at 2000 rpm and pellets were washed twice with lysis buffer. Proteins were eluted by boiling in 4× sample buffer. Analysis by Western blotting was performed as described above.
Biotin-streptavidin pull-down.
For biotin-streptavidin pull-down assays, HeLa-cells were transfected with biotin-tagged VAPB-wt and P56S using Lipofectamine-2000 (Invitrogen, Eugene, OR) transfection reagent according to the manufacturer's instructions. Cells were lysed 16 h later in 20 mm Tris-HCl, pH 8.0, 150 mm KCl, 1% Triton X-100, and protease inhibitors (Roche). Cell lysates were centrifuged at 13,000 rpm for 15 min and the supernatants were incubated with Dynabeads M-280 streptavidin (Dynal; Invitrogen) for 45 min. Beads were separated by using a magnet (Dynal; Invitrogen) and washed five times in lysis buffer. For protein elution, the beads were boiled in NuPAGE LDS 4 sample buffer (Invitrogen), separated, and supernatants were run on a 10% NuPAGE Bis-Tris gel (Invitrogen). The gel was stained with the Colloidal Blue staining kit (Invitrogen) and analyzed by Western blotting. Mass spectrometry was performed as described previously (Lansbergen et al., 2006).
GST-pull down.
Full-length VAPB and VAPB-N (amino acids 1–213) with or without the P56S-mutation were obtained by PCR and cloned into pGEX-4T (GE Healthcare Bio-Sciences) to create GST fusion proteins. Proteins were induced and purified as described previously and analyzed by SDS-PAGE to control for induction and quantities. HeLa-cells were transfected with GFP-FFAT and GFP-FFAT-SCR by using Lipofectamine-2000 transfection reagent (Invitrogen) and lysed in 50 mm Tris-HCl, 100 mm NaCl, and 1% Triton X-100 containing protease inhibitors (Roche). Lysates were incubated with GST beads (GE Healthcare Bio-Sciences) for 2 h at 4°C, washed four times with lysis buffer, and analyzed by SDS-PAGE and Western blotting as described previously. Blots were incubated with rabbit anti-GFP antibodies (1:1000; Abcam).
Photobleaching experiments.
For quantitative fluorescence recovery after photobleaching (FRAP) experiments, HeLa-cells were grown in six-wells plates on 25 mm coverslips (Menzel-Gläser, Braunschweig, Germany), transfected with GFP-tagged VAPB as described above, and imaged on a 37°C temperature-controlled stage of a Zeiss (Oberkochen, Germany) LSM510 laser-scanning microscope with a 60 mW Argon laser (488 nm) and a 40× 1.2 numerical aperture (NA) or 63× Planapochromat 1.4 NA oil-immersion lens (Zeiss) as described previously [modified from the method described by Essers et al. (2006)]. The microscopes were equipped with an objective heater. A region of interest was bleached with high laser power, followed by imaging of a larger part of the cell for 300 s at low laser power to prevent bleaching. The fluorescence intensity of the bleached region was measured every second to obtain fluorescence recovery curves. Fluorescence intensity after photobleaching was calculated relative to the intensity before bleaching. By measuring a nonbleached region in the same cell, we normalized for the decrease in fluorescence that appears trough bleaching in any case. Average fluorescent intensities were calculated for VAPB-wt, VAPB-P56S, and control regions.
Immunohistochemistry and confocal immunofluorescence.
The brain and spinal cord were sectioned at 40 μm with a freezing microtome. Sections were processed free floating, using a standard avidin-biotin-immunoperoxidase complex method (ABC; Vector Laboratories, Burlingame, CA) with diaminobenzidine (0.05%) as the chromogen, or double-labeling immunofluorescence (Jaarsma et al., 2001). The VAPB-antibodies were diluted at 1:4000 in Tris-buffered saline, pH 7.6, containing 1% normal horse serum and 0.2% Triton X-100. Biotinylated secondary goat-anti rabbit (Vector Laboratories) diluted at 1:200 was used as a secondary antibody. For double-labeling immunofluorescence, sections were incubated with VAPB antibodies (diluted 1:1000), combined with mouse anti-ubiquitin (FK2; Biomol), mouse anti-synaptophysin (Sigma), mouse anti-GM130 (BD Biosciences), or human anti-P0 (1:200; Immunovision). Immunoperoxidase-stained sections were analyzed and photographed using a Leica (Nussloch, Germany) DM-RB microscope and a Leica DC300 digital camera. Sections stained for immunofluorescence were analyzed with a Zeiss LSM510 confocal laser-scanning microscope.
Electron microscopy.
For EM, 24 h before fixation, HeLa cells were transfected with HA-VAPB-P56S with Lipofectamine-2000 transfection reagent (Invitrogen) according to the manufacturers instructions. Cells were fixed in 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, for 30 min, scraped, collected as a pellet, and left overnight in fresh fixative. Cells were further fixed with osmium tetroxide and embedded in Durcupan (Fluka, Saint Quentin Fallavier, France) by standard procedures. Ultrathin sections (70 μm) were cut, mounted on Formvar-coated nickel grids, and processed for immunogold labeling using the procedure described previously (Jaarsma et al., 2001). Primary and secondary antibodies were mouse anti-HA-11 (1:500; clone 16812; Babco, Richmond, CA) and 10 nm gold-conjugated goat anti-mouse (1:30; Aurion, Wageningen, The Netherlands). Gold-labeled sections were analyzed in a Philips (Aachen, Germany) CM10 electron microscope at 80 kV.
G93A-SOD1 mice.
G93A-SOD1 mice descendent from the Gurney G1del line that carry approximately eight transgene copy numbers per haploid genome were housed and handled in accordance with the Principles of Laboratory Animal Care (National Institutes of Health publication No. 86–23) and the guidelines approved by the Erasmus University animal care committee (protocol number 115-97-01 and 115-99-03). Nontransgenic mice were used as controls. The mice develop weakness in one or more limbs from age 24–30 weeks, and reach end-stage disease 2–10 weeks after the onset of limb weakness (Jaarsma et al., 2001). For immunocytochemistry, mice of different ages and disease stages were anesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde.
Human tissue.
Paraffin sections from L3 lumbar spinal cord from four ALS patients (average age 56 years; range, 42–68 years) and three controls (average age 57 years; range, 45–65 years) were obtained from files of The Netherlands ALS tissue bank. Tissue was obtained and used in a manner compliant with the Declaration of Helsinki. Informed consent was obtained for the use of brain tissue. All autopsies were performed within 8 h after death of patients. The ALS cases were sporadic with either bulbar (n = 2) or spinal onset (n = 2). Average duration of disease after diagnosis was 33 months (range 20–72 months). Control cases died from myocardial infarct (n = 1) or cancer (n = 2). One control case showed mild Alzheimer's pathology.
Results
VAPA and VAPB localize to the ER and are widely distributed
Human VAPA and VAPB show 60% amino acid similarity and are composed of three domains, including a highly conserved cytosolic N-terminal MSP domain, a central coiled-coil region, and a C-terminal TMD (Nishimura et al., 1999) (Fig. 1A). We raised antibodies against the variable central domain of VAPA (#1006-04) and VAPB (#1006-02), and the whole cytosolic domain of VAPB (#1006-00) (Fig. 1A), and tested them using lysates of COS-1 cells expressing HA-tagged VAPA or VAPB. The VAPA antibody strongly reacted with wild-type VAPA (VAPA-wt) and P56S mutant VAPA (VAPA-P56S), but did not recognize VAPB or control proteins, N-terminal portion of Bicaudal D2 (BICD2-N) and SOD1 (Fig. 1D). Alternatively, both VAPB antibodies showed a very strong reaction with VAPB-wt and VAPB-P56S, but did not detect VAPA or other control proteins (Fig. 1D). In mock-transfected COS-1 cells, both VAPB antibodies detected a major band of ∼33 kDa (Fig. 1D), which likely represents the endogenous VAPB. We conclude that the new VAP antibodies specifically recognize either VAPA or VAPB.

Distribution and localization of VAPB in vivo. A, Antisera against VAPA and VAPB were made against GST-VAP fusion proteins containing amino acids 1–225 of VAPB (antibody #1006-00) and amino acids 132–225 of VAPA (#1006-04) and VAPB (#1006-02). cc, Coiled-coil region; TM, transmembrane domain. B, C, Cells were fixed and processed for immunofluorescence using antibodies to VAP (green) and PDI (red). D–F, Lysates of COS-1 cells expressing VAP and control proteins (D), cultured cells (E), and mouse CNS (F) were analyzed by immunoblotting using VAP antibodies. G–I, Low- (G1, H1, I1) and high-magnification (G2, H2, H3, I2) of immunoperoxidase stainings using anti-VAPB antibody #1006-00 in murine dorsal hippocampus (G), cerebellum (H), and spinal cord (I). VAPB is expressed at high levels in motor neurons (I2) and large neurons in the cerebellar nuclei (CN; H1, H3) compared with neurons in cerebellar cortex (H1) including cerebellar Purkinje cells (PCs; H2) and hippocampus (G), including hippocampal pyramidal cells (G2). DG, Dentate gyrus; pyr, pyramidal layer; rad, stratum radiatum of CA1 hippocampal subfield; gl, granule-cell layer; ml, molecular layer; wm, white matter. J, K, Double-labeling confocal immunofluorescence of VAPB immunoreactivity (green) and ribosomal protein P0 (red) in mouse spinal motor neurons (J) and dorsal horn neurons (K). VAPB (J1, K1) colocalizes with P0 (J2, K3) in motor neurons, but shows low expression levels in most dorsal horn neurons. A merge is shown on the right. L, Immunoperoxidase staining using anti-VAPB antibody #1006-00 in human lumbar motor neurons (L1), ventral root motor axons (L2), and CA1 hippocampal pyramidal neurons (L3). As in mouse, in human VAPB-immunoreactivity in motor neurons is considerably higher than in other neurons, showing a Nissl body-like distribution (L1). Scale bars: G1, I1, 200 μm; H1, 100 μm; G2, H2, H3, I2, 10 μm; J3, K3, 20 μm; L1, L2, L3, 30 μm.
VAPs have been reported to localize to the ER in cultured cells (Soussan et al., 1999; Skehel et al., 2000; Amarilio et al., 2005). Accordingly, immunofluorescence microscopy revealed that in HeLa cells, VAPA and VAPB antibodies produce a typical reticulate staining pattern, which partially coincides with PDI, an ER resident protein (Fig. 1B,C). Western blot analysis showed that VAPB was present in all cell lines examined, and VAPA was present in all but COS-1, COS-7, and Madin-Darby canine kidney cells (Fig. 1D,E). Accordingly, analysis of mouse tissues including kidney, heart, skeletal muscle, liver, spleen, lung, and different brain areas, showed that VAPA and VAPB are expressed in all tissues and throughout the CNS (Fig. 1F) (data not shown). These results are consistent with previous Northern blot analyses showing a broad tissue distribution of VAPA and VAPB (Nishimura et al., 1999; Skehel et al., 2000). These data indicate that both VAPA and VAPB localize to the ER and are widely expressed in neuronal and non-neuronal tissues and cell lines.
VAPB is highly abundant in motor neurons in mouse and human spinal cord
Next, we investigated the distribution of VAPB in the mouse and human CNS. We used the two new VAPB antibodies, which produced very similar staining patterns. In all brain areas, VAPB immunoreactivity was predominantly localized to neuronal cell bodies and their proximal dendrites, whereas little or no labeling was found in glial cells. The highest levels of immunoreactivity emerged in motor neurons in spinal cord and caudal brainstem (Fig. 1I). Neurons in the cerebellar nuclei (Fig. 1H) and large neurons throughout the reticular formation (data not shown) also stained strongly for VAPB, whereas lower staining intensities occurred in cortical and hippocampal neurons (Fig. 1G) or cerebellar Purkinje cells (Fig. 1H). Within the white matter, staining was predominantly associated with axonal profiles and light VAPB staining also was observed in motor axons in the ventral roots. The distribution of VAPB in sections from human hippocampus, cerebellar cortex, and spinal cord was consistent with that in mouse CNS in that high levels of labeling occurred in motor neurons (Fig. 1L1), whereas low to moderate staining occurred in hippocampal (Fig. 1L2) and cerebellar neurons.
The subcellular distribution of VAPB in human and murine motor neurons strongly resembled the distribution of Nissl substance (Fig. 1L) (i.e., orderly arrays of broad cysternae of ER and polyribosomes) (Peters, 1991). Accordingly, double labeling showed that VAPB immunoreactivity grossly colocalized with ribosomal protein P0 in murine and human motor neurons (Fig. 1J,K), but poorly colocalized with the cis-Golgi marker GM130 (data not shown). Because VAP proteins originally were identified as VAMP-associated proteins, we also have examined whether VAPB codistributed with presynaptic proteins in the neuromuscular endplates and central synapses; however, we did not observe any synaptic VAPB labeling in these structures (data not shown). In sum, the data indicate that VAPB is highly expressed in motor neurons in mice and humans with a dominant localization in the rough ER.
The P56S mutation leads to aberrant aggregation of VAPB
To characterize the properties of VAPB carrying the ALS-linked P56S mutation, we established a cellular VAP aggregation model in cultured cells. In HeLa cells, HA-VAPB-wt predominately localized to the ER, similar to endogenous VAPB (Fig. 2A) and P56S mutant HA-VAPB protein-formed cytosolic aggregates, which did not colocalize with ER markers, such as calreticulin and PDI (Fig. 2B) (Nishimura et al., 2004). Aggregate formation was seen in all transfected cells independently of expression levels. Eighteen hours after transfection, 26 ± 4 VAPB-P56S aggregates with an average diameter of 1.2 μm2 (range, 0.8–2.5 μm) could be found per HeLa cell. Other cell types were also susceptible to HA-VAPB-P56S aggregate formation: although the average cluster size and number could vary significantly, the P56S-mutated VAPB protein formed cytosolic aggregates in all cell types examined (data not shown). These data indicate that the P56S mutation leads to abnormal clustering of VAPB independently of cellular context.

The P56S mutation leads to aberrant aggregation of VAPA and VAPB. A–D, HeLa cells expressing HA-tagged VAPB-wt (A), VAPB-P56S (B, D), and VAPA-P56S (C) were fixed and processed for immunofluorescence using antibodies against HA (green) and the ER marker calreticulin (red). HeLa cells in D were incubated for 2 h at 20°C before fixation. Enlargement of boxed regions indicated in the merged pictures is shown at right. E, Lysates (L) of COS-1 cells transfected with HA-VAPB-wt and HA-VAPB-P56S were solubilized with Triton X-100, fractionated in supernatant (S), and pellet (P) under nonreducing (−DTT) or reducing (+DTT) conditions and analyzed by immunoblotting using anti-HA antibodies. Cells were either maintained at 37°C or incubated for 2 h at 20°C before lysis. F, Quantification of the number of VAPB-P56S clusters per cell, incubated at 15 or 20°C for 3 h or treated with 5 μg/μl BFA or 10 μm Nocodazole (Noco) for 30 min, as indicated. Error bars indicate SEM. **p < 0.005%. Scale bar, 10 μm.
Because VAPB-P56S clusters do not coincide with the ER, we tested whether they colocalize with other organelle or vesicular trafficking markers. Expression of HA-VAPB-P56S in HeLa cells had no detectable effect on the cis- or trans-Golgi morphology, endosomes, or lysosomes as determined by localization of their respective markers, GM130, γ-adaptin, EEA1 (early endosomal autoantigen 1), and LAMP1 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Block of vesicular trafficking by 15°C incubation, brefeldin A (BFA), or nocodazole treatment had no significant influence on the number or size of VAPB-P56S aggregates (Fig. 2F, supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Next, we investigated transport through the secretory pathway by using the temperature-sensitive mutant of vesicular stomatitis virus-glycoprotein (ts045-VSVG) fused to yellow fluorescent protein (VSVG-YFP) in cells containing VAPB-P56S aggregates. Using temperature shifts, we found that VAPB-P56S aggregates did not substantially interfere with the VSVG-YFP transport and never colocalized with VSVG-YFP at any of its trafficking steps (data not shown). Together, these data indicate that VAPB-P56S aggregates constitute no part of the secretory or other vesicular trafficking routes in HeLa cells.
The proline at position 56 in VAPB is highly conserved among the VAP family proteins from various species (Nishimura et al., 1999). To investigate whether the P56S mutation in VAPA (VAPA-P56S) forms cytosolic aggregates as well, we expressed HA-VAPA-P56S in HeLa cells. Transfected cells showed, on average, 9 ± 2 VAPA-P56S clusters, which did not coincide with ER markers and were similar in appearance to VAPB-P56S aggregates (Fig. 2C). However, in contrast to HA-VAPB-P56S-expressing cells, a significant proportion of HA-VAPA-P56S localized to the ER in addition to aggregates (Fig. 2C). In all cell lines examined, the average cluster size was significantly larger and the number of clusters was smaller in VAPA-P56S- compared with VAPB-P56S-expressing cells (data not shown).
To gain better understanding of the biochemical properties of the wild-type and mutant VAPB proteins, we tested their solubility in nonionic detergents. Wild-type VAPB was present in both the Triton X-100-soluble and -insoluble fractions, whereas VAPB-P56S was only present in the detergent-insoluble fraction (Fig. 2E). Similar results were obtained when the samples were run under nonreducing conditions (−DTT) (Fig. 2E). Incubation of cells at 20°C has been shown to reverse aggregation of proteins like mutant cystic fibrosis transmembrane conductance regulator (Denning et al., 1992) and prion protein (Singh et al., 1997). We tested whether a similar effect could be observed for the mutant VAPB and found that the average number of VAPB-P56S aggregates decreased by ∼50% in HeLa cells incubated at 20°C compared with control cells at 37°C (Fig. 2F, supplemental Fig. 1, available at www.jneurosci.org as supplemental material); moreover, at 20°C, some of the VAPB-P56S localized to the ER (Fig. 2D). In line with these data, Western blots showed that VAPB-P56S partly shifted toward the soluble fraction if the transfected COS-1 cells were maintained for 2 h at 20°C (Fig. 2E). This effect was not observed at other temperatures, such as 15 and 30°C (Fig. 2E) (data not shown). We conclude that at 20°C VAPB mutant proteins partially solubilize and distribute within the ER similarly to the wild-type VAPB.
P56S mutant VAPB forms protein aggregates containing ER tubules
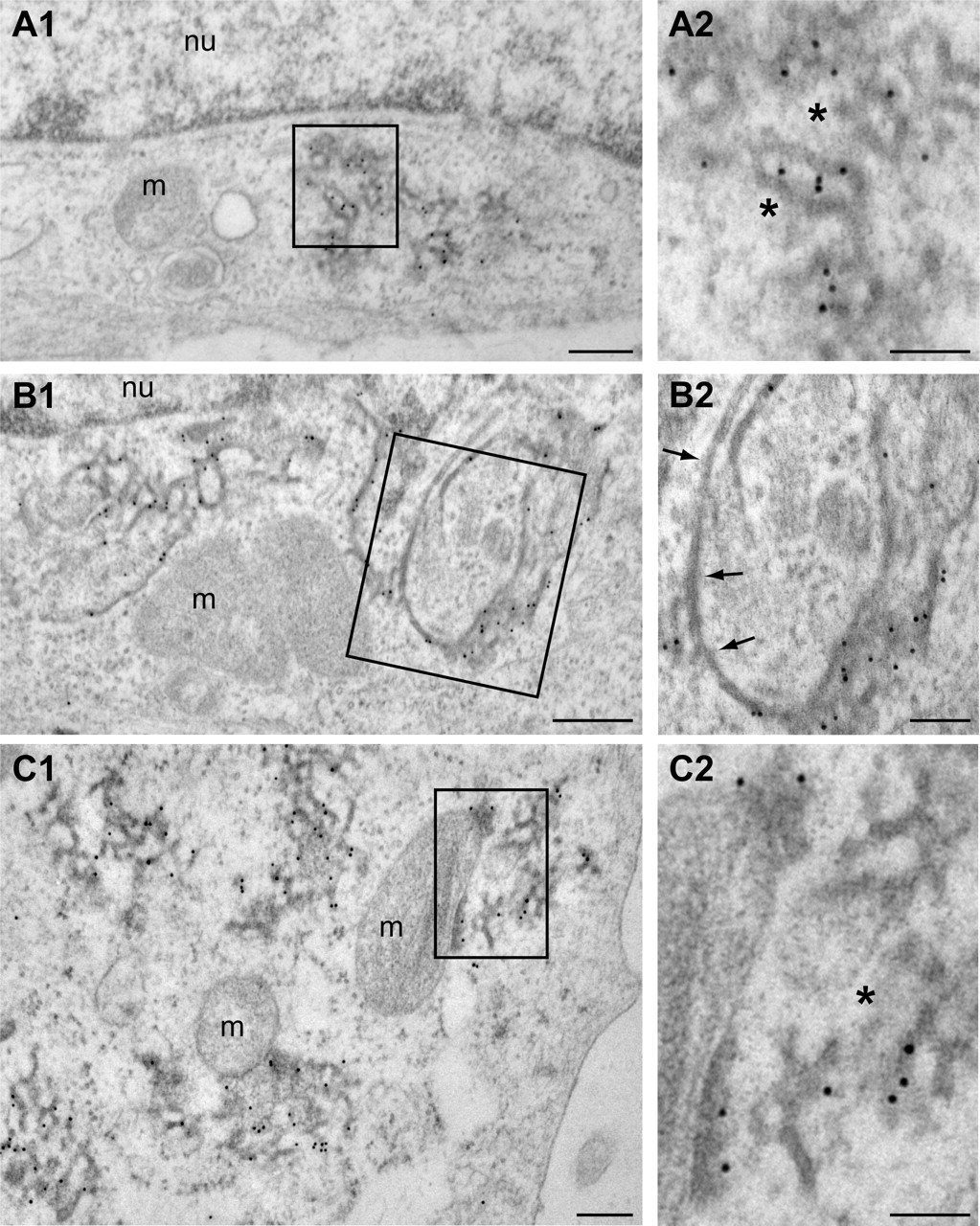
To further characterize the structure of the HA-VAPB-P56S aggregates, we used anti-HA postembedding immunogold electron microscopy. No anti-HA labeling was observed in nontransfected HeLa cells. In HA-VAPB-P56S-transfected HeLa cells, immunogold labeling was specifically associated with clusters of electron-dense tubular profiles with diameters of 20–40 nm that formed irregular branched networks (Fig. 3A). Their morphology strongly resembled the anastomizing networks of smooth ER observed in cells specialized in steroid synthesis (Fawcett, 1981). However, the borders of the tubules showed a more diffuse and electron-dense appearance, strongly suggestive of the presence of high protein levels (Fig. 3A). Notably, the tubular profiles usually were loosely packed and were intermingled with cytoplasm (Fig. 3A, asterisks).

Electron microscopy analysis of VAPB-P56S aggregates in HeLa cells. A1–C2, Transmission electron photomicrographs of anti-HA immunogold labeled ultrathin sections of HeLa cells transfected with HA-VAPB-P56S. Immunogold labeling is specifically associated with clusters of electron dense tubular profiles (A). Occasionally, labeled profiles are continuous with unlabeled profiles reminiscent of ER tubules (B) or with mitochondrial outer membranes (C). Arrows indicate unlabeled ER-tubules; asterisks indicate normal appearing cytoplasm in VAPB-P56S aggregates. m, Mitochondria; nu, nucleus. Scale bars: A1, B1, C1, 200 nm; A2, B2, C2, 100 nm.
In agreement with immunofluorescence analysis of the VAPB-P56S aggregates, the size of the tubule clusters was variable. Interestingly, in cells expressing large numbers of clusters, the HA-VAPB-P56S-positive tubules were often continuous with the normal-appearing ER profiles (Fig. 3B) or the outer membranes of mitochondria (Fig. 3C). In the latter case, parts of the mitochondrial outer membranes showed both a diffuse electron-dense appearance and immunogold labeling, indicating that they contained VAPB aggregates. Immunogold labeling did not occur in the rough ER and mitochondrial outer membranes with normal morphology, nor could it be found in the Golgi apparatus, lysosomes, aggresomes, and other intracellular structures. We conclude that VAPB-P56S aggregates include tubular membrane structures, which most likely originate from the ER. Their appearance was clearly distinct from the organized smooth ER (OSER) (Snapp et al., 2003) induced by the coexpression of VAPs and FFAT-motif proteins (Amarilio et al., 2005; Lehto et al., 2005).
P56S mutant VAPB forms immobile aggregates
To characterize the dynamics of wild-type and mutant VAPB proteins, we generated their GFP-tagged versions. Because fusion of GFP to either the N or C terminus of VAPB leads to an abnormal cellular localization and aberrant mobility in SDS-PAGE electrophoresis (data not shown), we inserted the GFP tag between the TM domain and the cytosolic tail of the VAPB protein (Fig. 4A). These internally GFP-tagged wild-type and P56S mutant VAPBs behaved similarly to untagged-VAPB constructs (Fig. 4C) (data not shown). We monitored the behavior of GFP-VAPB-wt and GFP-VAPB-P56S in HeLa cells 24 h after transfection. Although GFP-VAPB-wt was very dynamic within the cell, as described for other ER transmembrane proteins (Lippincott-Schwartz et al., 2001), the overall location of VAP-P56S aggregates did not change significantly during a 300 s period (supplemental movies 1, 2, available at www.jneurosci.org as supplemental material).

VAPB-P56S clusters are immobile porous aggregates. A, Diagram of GFP-tagged VAPB constructs. B, Fluorescent recovery plots compare the rates of recovery of GFP-VAPB-wt (black triangle), GFP-VAPB-P56S (black box), as well as a nonbleached control region (open circles). Fluorescence intensity was normalized to intensity before bleaching. C–E, Representative confocal images of HeLa cells expressing GFP-VAPB-wt and GFP-VAPB-P56S. To visualize the mobility of the GFP-chimeras, small regions of interest of identical size (outlined square in C) were photobleached and monitored for recovery for 300 s (D). Half the area of a VAPB-P56S aggregate was bleached in E. F, Projection of confocal images of a HeLa cell expressing HA-VAPB-P56S and GFP, fixed and stained with anti-HA (red) antibody. Enlargement of the boxed region containing a single confocal stack (0.6 μm) is shown at the bottom. Scale bars, 10 μm.
Next, we used FRAP to examine the motility of VAP molecules. A 4 × 4 μm region of the cell was bleached by high laser power and the fluorescence intensity was measured over a period of 300 s. Notably, fluorescence recovered very rapidly into the bleached area of GFP-VAPB-wt-expressing cells (Fig. 4B,D, supplemental Movie 1, available at www.jneurosci.org as supplemental material), consistent with the high motility of ER-resident proteins (Lippincott-Schwartz et al., 2001). In GFP-VAPB-P56S-transfected cells, only 35% of the fluorescent signal associated with the aggregates recovered within 300 s after photobleaching, indicating that the mutant proteins are mostly immobilized (Fig. 4B,D, supplemental movie 2, available at www.jneurosci.org as supplemental material). Comparable results were obtained after photobleaching half of an area of a typical VAPB-P56S aggregate (Fig. 4E, supplemental movie 3, available at www.jneurosci.org as supplemental material). The lack of fluorescent recovery for VAPB-P56S aggregates is very similar to that observed for other mutant proteins that form immobile aggregates (Chai et al., 2002; Kim et al., 2002), but differs from proteins inducing organized ER architectures (Snapp et al., 2003).
Because EM analysis revealed that VAPB-P56S aggregates are intermingled with normal-appearing cytosol, we examined whether noninteracting soluble proteins, such as GFP, can diffuse through mutant VAPB aggregates. GFP displayed a uniform pattern of localization both within and outside of VAPB-P56S clusters (Fig. 4F) in accordance with a porous architecture, but unlike solid mutant huntingtin aggregates (Matsumoto et al., 2005).
VAPB-P56S aggregates do not overlap with chaperone, proteasome, autophagy, and ER stress markers
Protein aggregate formation is often influenced by molecular chaperones (Sherman and Goldberg, 2001) and protein degradation routes such as the ubiquitin-proteasome system, and aggresome and autophagosome pathways (Kopito, 2000; Rubinsztein, 2006). To investigate the role of these pathways in the cellular response to VAPB-P56S aggregates, we assessed the distribution of their markers, such as Hsp-70, ubiquitin, vimentin, Atg8/LC3, and Atg12 in HeLa cells transfected with HA-VAPB-P56S. In contrast to the aggregates induced by mutant Htt fragments [GFP-Htt(Q74)] (Waelter et al., 2001), used as a positive control in our experiments, VAPB-P56S aggregates did not colocalize with any of the above-mentioned markers (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (data not shown). Consistent with these data, the treatment of HA-VAPB-P56S-expressing cells with proteasome inhibitor MG132 for 4 or 24 h did not significantly influence aggregate formation or change the number of VAPB-P56S clusters (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (data not shown). However, when we tested the solubility of VAPB-P56S in cells treated with 10 μm MG132 for 24 h, the VAPB-P56S levels increased in the soluble fraction whereas the insoluble fraction was unaffected (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), suggesting that solely nonaggregated VAPB-P56S is significantly degraded by the proteasome.
Several studies demonstrated that increased expression of chaperones, such Hsp70 and Hsp104, suppresses aggregate formation (Muchowski et al., 2000; Vacher et al., 2005). We cotransfected HeLa cells with HA-VAPB-P56S and Hsp70 or Hsp104; in contrast to mutant Htt-expressing cells, we found no colocalization of the chaperones with VAPB-P56S and no significant difference in the size and number of VAPB-P56S clusters (data not shown). Furthermore, we observed no upregulation of the ER stress-response markers such as the chaperone Grp78/BiP and p-EIF2α in VAPB-P56S-expressing cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these data indicate that VAPB-P56S aggregates do not seem to recruit protein quality control and degradation machinery in contrast to many well characterized aggregation-prone proteins.
VAPB-P56S recruits wild-type VAPA and VAPB via the TMD
In vitro GST pull-down experiments suggested that VAPB-P56S associates with exogenous wild-type VAPB (Kanekura et al., 2006). We investigated the effect of VAPB-P56S on endogenous VAPA and VAPB proteins in our cellular model and examined the specific VAP domains involved in the binding. In COS-1 cells transfected with the wild-type HA-VAPB, endogenous VAPB is present in both the soluble and insoluble fraction, as in nontransfected cells (Fig. 5E) (data not shown). Expression of HA-VAPB-P56S, however, reduced the amount of endogenous VAPB in the soluble fraction and increased the amount of insoluble endogenous VAPB (Fig. 5E), suggesting that VAPB-P56S traps endogenous VAPB in the aggregates. Indeed, HeLa cells expressing VAPB-P56S recruited a considerable amount of endogenous VAPA to mutant VAP aggregates, whereas VAPA-P56S clusters accumulated endogenous VAPB (Fig. 5A,B). Colocalization and coimmunoprecipitation experiments confirmed that both wild-type VAPA and VAPB bind to VAPB-P56S (Fig. 5C,D,F). Together, these data indicate that mutant VAPB-P56S is capable of recruiting both wild-type VAPA and VAPB, providing a possible explanation for the dominant effect of the P56S mutation in VAPB-linked ALS patients.

VAPB-P56S recruits both wild-type VAPA and VAPB. A, B, HeLa cells transfected with HA-VAPB-P56S (A) or HA-VAPA-P56S (B) and stained with anti-HA (green) and anti-VAPA (red; A) or anti-VAPB (red; B) antibodies. Endog., Endogenous. C, D, HeLa cells double transfected with HA-VAPB-P56S and myc-VAPB-wt (C) or myc-VAPA-wt (D), fixed and stained with anti-HA (green) and anti-Myc antibodies (red) to visualize the expressing VAP protein. E, Lysates (L) of COS-1 cells transfected with HA-VAPB-wt and HA-VAPB-P56S were solubilized with Triton X-100, fractionated in supernatant (S) and pellet (P), and analyzed by immunoblotting using anti-VAPB (#1006-03) antibodies. F, COS-1 cells cotransfected with HA-VAPB-P56S and myc-VAPA-wt or myc-VAPB-wt were immunoprecipitated with control IgG or HA antibodies. The cells were incubated for 2 h at 20°C before lysis to solubilize VAPB-P56S. Each immunoprecipitation reaction is shown in three lanes: I, input to IP reaction; S, supernatant remaining after IP; P, precipitated pellet. The L, I, S, and P samples were immunoblotted for the indicated proteins. G, COS-1 cells cotransfected with HA-VAPB-P56S and GFP-VAPB-wt-N or GFP-VAPB-TMD were immunoprecipitated and immunoblotted as in F. H–K, HeLa cells single or double transfected with GFP-VAPB-TMD (H, J) or GFP-VAPB-wt-N (I, K) and HA-VAPB-P56S (J, K) and stained with anti-calreticulin to reveal the ER (H, I), or anti-HA (J, K) antibodies to visualize the expressing VAP protein. L–N, HeLa cells single or double transfected with GFP-VAPB-P56S-N and HA-VAPB-wt (M) or HA-VAPB-P56S (N) and stained with anti-calreticulin (L) or anti-HA (M, N) antibodies. Scale bar, 10 μm.
Structural studies have suggested that VAPs form dimers via the central coiled-coil domain in the cytosolic tail and the GxxxG-motif in the TMD (Russ and Engelman, 2000; Kaiser et al., 2005). In contrast to the Caenorhabditis elegans MSP protein itself (Haaf et al., 1996; Smith and Ward, 1998), the N-terminal MSP homology domain in VAP is monomeric in solution and does not dimerize in vitro (Kaiser et al., 2005). To determine which part of wild-type VAPB is responsible for the observed interaction with VAPB-P56S, we generated GFP-tagged constructs encoding the transmembrane portion of VAPB (VAPB-TMD) and the cytosolic part of VAPB (VAPB-wt-N) (Fig. 4A). As expected, GFP-VAPB-TMD in HeLa cells showed ER localization (Fig. 5H), whereas GFP-VAPB-wt-N revealed a diffuse cytosolic and nuclear distribution (Fig. 5I). Coexpression experiments demonstrated that VAPB-TMD was recruited to the mutant aggregates (Fig. 5J,K) and coprecipitated with the full-length VAPB-P56S (Fig. 5G), whereas VAPB-wt-N showed no interaction with the mutant VAPB (Fig. 5K,G), indicating that wild-type VAPs bind to VAPB-P56S aggregates via the TMD.
Next, we examined the effect of the P56S mutation on the formation of the cytosolic aggregates. HeLa cells expressing GFP-VAPB-P56S-N form compact cytosolic aggregates (Fig. 5L), which are larger but less abundant compared with the VAPB-P56S clusters (∼4 ± 1 clusters per cell with a diameter of ∼3.2 μm). Coexpression of GFP-VAPB-P56S-N and HA-VAPB-wt or HA-VAPB-P56S showed that, in contrast to VAPB-wt, VAPB-P56S strongly coclusters with mutant cytosolic aggregates (Fig. 5M,N). Similar results were obtained using GFP-tagged constructs containing mutant and wild-type MSP domains (data not shown). We conclude that the P56S mutation dramatically enhances the binding between the cytosolic VAPB portions, providing a likely explanation for the observed VAP-P56S aggregation.
The P56S mutation in VAPB perturbs the binding to FFAT motif-containing proteins
To examine whether P56S influences the interaction of VAPB with its binding partners, we used pull-down assays combined with mass spectrometry. We generated biotinylation-tagged VAPB-wt (Bio-HA-VAPB-wt) and P56S (Bio-HA-VAPB-P56S) constructs and coexpressed them with the protein-biotin ligase BirA in HeLa cells (Lansbergen et al., 2006). Localization of biotinylated wild-type and mutant VAPB proteins was similar to that of wild-type and mutant HA-VAPB (data not shown). Biotinylated proteins from cells expressing either BirA alone or in combination with wild-type or mutant Bio-HA-VAPB were isolated using streptavidin beads and analyzed on a Coomassie-stained gel by Western blotting (data not shown) and by mass spectrometry. After background correction using the BirA-only control data, the mass spectrometry results from VAPB-wt-expressing HeLa cells revealed several FFAT motif-containing proteins (i.e., Nir2, ORP3, ORP6, and ORP9) (supplemental Table 1, available at www.jneurosci.org as supplemental material), whereas no FFAT-domain proteins were pulled down from Bio-HA-VAPB-P56S-expressing cells, indicating that the P56S mutation inhibits the interaction with FFAT motif-containing proteins. Similar results were obtained using GST-pull down experiments (data not shown).
To confirm this finding, we generated GFP-FFAT reporter constructs containing either the FFAT motif of Nir2 (residues, EFFDAHE) or the permutated version of this sequence (GFP-FFAT-SCR; residues, EDNFAHE). Structural studies of VAPA have shown that two amino acids, K87 and M89, are critical for FFAT binding (Kaiser et al., 2005). We made the double point mutation K87D/M89D in HA-tagged VAPB-construct (VAPB-K87D/M89D) and used it as a negative control. Coexpression of wild-type VAPB and GFP-FFAT in HeLa cells showed their prominent colocalization in cluster-like structures in the ER (Fig. 6A), reminiscent of OSER (Snapp et al., 2003). In contrast, none of the mutated VAPB constructs induced ER-clustering or colocalization with GFP-FFAT (Fig. 6B,C). Coimmunoprecipitation experiments confirmed that wild-type VAPB and not VAPB-K87D/M89D or VAPB-P56S were pulled down by GFP-FFAT (Fig. 6G,H). Consistently, GST-pull down experiments showed that wild-type VAPB (full length and cytosolic domain) associated with GFP-FFAT, whereas the VAPB-P56S mutant forms were unable to bind to GFP-FFAT (Fig. 6I). The VAP–FFAT interaction was specific for the FFAT domain, because GFP-FFAT-SCR did not interact with VAPB-wt or the mutated VAPB proteins (Fig. 6H,I).

VAPB-P56S disturbs binding to FFAT motif-containing proteins. A–C, HeLa cells double transfected with GFP-FFAT plus HA-VAPB constructs and stained with anti-HA (red) antibodies to visualize the expressing VAP protein. D–F, HeLa cells double transfected with ORP3-GFP plus HA-VAPB constructs and stained with as in A–C. G, COS-1 cells cotransfected with GFP-FFAT and VAPB constructs were immunoprecipitated with GFP antibodies. Each IP reaction is shown in three lanes: I, input to IP reaction; S, supernatant remaining after IP; P, precipitated pellet. The I, S, and P samples were immunoblotted for the indicated proteins. H, COS-1 cells cotransfected with GFP-FFAT and HA-VAPB-wt were immunoprecipitated and immunoblotted as in G. I, Coomassie-stained gel of purified GST-VAPB proteins (top). The GST fusion proteins were loaded with HeLa-cell extracts transfected with GFP-FFAT and GFP-FFAT-SCR, precipitated, and immunoblotted (bottom two panels). Twenty percent of the input (I) is loaded on the gel. J, COS-1 cells cotransfected with ORP3-GFP and HA-VAPB constructs were immunoprecipitated with control IgG and GFP antibodies and immunoblotted as in G. Scale bar, 10 μm.
Mass spectrometry results were further confirmed by coexpression experiments using the FFAT motif-containing protein ORP3. Whereas the wild-type VAPB and ORP3-GFP prominently colocalized in cluster-like structures in the ER (Fig. 6D), none of the mutated VAPB constructs coincided with ORP3-GFP (Fig. 6E,F). Coimmunoprecipitation experiments showed that only wild-type VAPB and not its mutated versions were pulled down by ORP3-GFP (Fig. 6J). These experiments consistently demonstrate that the P56S mutant VAPB is unable to interact with FFAT motif-containing proteins.
Mutant VAPB and VAP knockdown induce cell death and Golgi fragmentation in primary neurons
As in non-neuronal cells, VAPB-P56S forms cytosolic aggregates in primary neurons, which do not colocalize with ER markers (Fig. 7A,B). To examine whether mutant VAPB leads to an increase in cell death in neuronal cells, we transfected primary cultured rat hippocampal neurons at DIV13 with mutant and wild-type VAPB. Two days after transfection, the number of neurons expressing VAPB-P56S was decreased by ∼60% compared with wild-type VAPB and control neurons (Fig. 7C,E) (data not shown), suggesting that neuronal viability is greatly reduced in cultures transfected with mutant VAPB. Furthermore, neurons expressing VAPB-P56S show aberrant neuronal morphology (Fig. 7A,C). Hence, neuronal cells may be uniquely sensitive to mutant VAPB because the expression of VAPB-P56S does not increase cell death in HeLa or COS-1 cells (data not shown).

VAPB-P56S expression and VAP reduction causes cell death and Golgi fragmentation in primary neurons. A, B, Primary neurons expressing wild-type and mutant HA-tagged VAPA and VAPB were fixed and stained with HA-antibodies (green) and anti-PDI antibodies (red in B). Enlargement of the boxed region containing a single confocal stack (0.8 μm) is shown on the right. C, Primary neurons transfected with wild-type and mutant HA-tagged VAPB and shRNA against VAPA and VAPB (right) at DIV13 for 2 (left, middle) and 6 (right) d and stained with anti-HA (green; left, middle) or anti-β-galactosidase (green, right) and anti-PDI antibodies (red; insets). D, Representative images of primary neurons expressing wild-type (left) and mutant (middle) HA-tagged VAPB and shRNA against VAPA and VAPB (right), fixed and stained with HA-antibodies (green; left, middle) or β-galactosidase (right) and the cis-Golgi marker GM130 (red; insets). E, Quantification of neuronal survival by counting the number of wild-type VAPB-, mutant VAPB-, and β-galactosidase-expressing neurons per coverslip, 2 d (VAPB-WT and mutant) or 6 d (shRNA) after transfection from six and four independent experiments, respectively. F, Quantification of percentage of neurons with dispersed Golgi apparatus of control GFP, wild-type, or mutant VAPB- and shRNA-expressing cells 2 d (VAPB-WT and mutant) or 3 d (shRNA) after transfection. Scale bars: A, 10 μm; C, 100 μm ***p < 0.0005; **p < 0.001; *p < 0.10. Error bars indicate SEM.
Because we showed that VAPB-P56S recruits endogenous VAPA and VAPB into insoluble aggregates and reduces cytosolic VAP levels (Fig. 5A,E), we hypothesized that decreased VAP family protein levels would phenocopy the mutant VAPB effects. To reduce VAP expression in neurons, we used a plasmid (pSuper)-based method for VAP knockdown and designed two rat VAP-shRNAs (VAPA-shRNA4 and VAPB-shRNA1) based on siRNA sequences shown to be effective to decrease VAP expression in HeLa cells (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). RNAi constructs were cotransfected in hippocampal neurons at DIV13 with a plasmid encoding for β-gal, to highlight the transfected neurons and immunostained for VAPA and VAPB 6 d later (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). VAPA-shRNA- or VAPB-shRNA-expressing neurons showed ∼70% reduction in immunostaining for the respective VAP (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), indicating an effective knockdown for both VAP-shRNA constructs.
We next investigated whether knockdown of VAP influences neuronal viability. Although no effect on neuronal survival was observed in VAPB knockdown neurons, the absence of VAPA decreased the number of transfected neurons by ∼80% compared with control neurons (Fig. 7E). A second VAP-shRNA, VAPA-shRNA2, showing ∼60% reduced VAPA immunostaining, confirmed the observed cell death; ∼30% of the transfected neurons survived (data not shown). Similar results are obtained after knockdown of both VAPA and VAPB, where only ∼20% of the neurons survived compared with control cells (Fig. 7E), indicating that VAP family proteins are important for neuronal viability. Because VAPA is the most abundant VAP in the hippocampus (Fig. 1F), it is likely that VAPA is redundant for VAPB function in hippocampal neurons. However, we cannot rule out a specific role for VAPA in neuronal survival.
Because a disrupted Golgi apparatus is an early pathological hallmark of neurodegeneration that is generally caused by disturbed intracellular trafficking (Gonatas et al., 2006), we analyzed the Golgi morphology in neurons expressing VAPB-P56S or VAP-shRNA. Transfection of hippocampal neurons at DIV13 results in significant Golgi fragmentation in ∼15% of the VAPB-P56S-expressing neurons and ∼15% (ranging from 10 to 20%) in VAP knockdown neurons compared with ∼1% (ranging from 0 to 3%) in control neurons (Fig. 7D,F). Overall, these data show that VAPB-P56S expression in neurons has similar effects as knockdown of endogenous VAP levels, suggesting that the dominant effect of VAPB-P56S is caused by a loss-of-function of VAP in these cells.
Motor neurons show reduced VAP immunoreactivity in ALS tissue
To examine whether loss of VAP function might also be involved in the pathogenesis of sporadic ALS and SOD1-linked ALS, we analyzed the VAPA and VAPB expression levels in spinal cord sections from human ALS patients and in ALS-prone transgenic mice that express G93A-mutated SOD1 (Jaarsma et al., 2001). No change in VAP expression was observed in G93A-SOD1 mice before disease onset (10–25 weeks of age) or in aged controls, but symptomatic and end-stage G93A-SOD1 mice (30–40 weeks) showed a reduction of VAPA and VAPB immunoreactivity as determined by immunohistochemistry and Western blot analysis (Fig. 8A,D). This reduced VAP expression could, at least in part, be explained by the loss of motor neurons, because the reduction of VAP expression temporally correlated with the onset of motor neuronal loss. However, calreticulin, a resident ER protein, was significantly less affected in the end-stage G93A-SOD1 mice compared with VAP (Fig. 8D). Also in spinal cord sections from ALS patients (n = 4), VAPB immunoreactivity in motor neurons was reduced compared with controls (n = 3) and significantly lower VAPB expression was seen in the pyramidal tract (Fig. 8E,F). Double labeling showed that, in the spinal cords of SOD1-ALS mice and ALS patients, VAPB was not present in ubiquitinated inclusions, which are a dominant pathological hallmark of ALS (Fig. 8B) (data not shown). However, a reduced VAPB immunoreactivity was primarily observed in motor neurons displaying Golgi fragmentation (Fig. 8C), consistent with the loss-of function data in primary neuron cultures (Fig. 7). Our observations suggest that the reduced expression of VAP is not only coupled to VAPB-linked ALS, but more generally associated with the viability of motor neurons in both sporadic and familiar ALS.

Reduced VAPB expression in ALS patients and SOD1-ALS transgenic mice. A1–C3, Immunoperoxidase (A) and confocal immunofluorescence staining (B, C) of VAPB in cervical spinal cord of SOD1-G93A-transgenic ALS mice before symptom onset (A2) and at end-stage disease (A3). VAPB immunoreactivity (green; B2) is not present in ubiquitinated inclusions (red; B3) in motor neurons of symptomatic SOD1-G93A mice (B). Motor neurons displaying Golgi fragmentation visualized by anti-GM130 antibody (red; C2) show reduced VAPB expression (green; C1). D, Western blot analysis of spinal cord homogenates of young nontransgenic (20 weeks), aged nontransgenic (80 weeks), presymptomatic SOD1-G93A (15 weeks and 20 weeks), and symptomatic SOD1-G93A (30 weeks and end-stage) mice. E, F, VAPB-immunoperoxidase staining in control (E) and human ALS (F) lumbar spinal cord. Compared with motor neurons (E3) and pyramidal tract (E2) in control issue, ALS spinal cord sections show a substantial loss of VAPB immunoreactivity in remaining motor neuronal profiles (F3, F4), and reduced staining of the pyramidal tract (F2). pyr, Pyramidal tract. Scal bars: A1–A3; E3, F3, 100 μm; B1, C1, 20 μm; E1, F1, 800 μm; E2, F2, 50 μm.
Discussion
Characterization of VAPB-P56S aggregates
Our results show that mutant VAPB protein aggregates are different in several aspects from any other described aggregate-prone mutants in neurodegenerative diseases (Taylor et al., 2002; Forman et al., 2004; Selkoe, 2004). First, the accumulation of VAPB-P56S into micrometer-scale aggregates is remarkably efficient. This is different from clustering of other aggregate-prone proteins, such as mutant SOD1 and Htt, where micrometer-scale aggregates form over days at high levels of mutant protein (Waelter et al., 2001; Chai et al., 2002; Kim et al., 2002; Matsumoto et al., 2005). This is consistent with the idea that the P56S mutation creates a high affinity for self-association (discussed below).
Second, VAPB-P56S aggregates do not recruit protein quality control and degradation machinery components. Most aggregate-prone proteins identified in neurodegenerative diseases recruit molecular chaperones or proteasome components (Kopito, 2000; Taylor et al., 2002; Rubinsztein, 2006). Furthermore, aggregation of mutant Htt and SOD1 is enhanced in response to proteasome inhibition (Waelter et al., 2001; Iwata et al., 2005). We do not observe these phenomena in cells expressing VAPB-P56S, and this could explain the efficient aggregation of the mutant VAP protein.
Third, mutant VAPB clusters are ultrastructurally distinct from other micrometer scale aggregates that are usually composed of a network of fibrillar polymers (Taylor et al., 2002; Ross and Poirier, 2005). Instead, the VAPB-P56S aggregates consist of tubules, which can be continuous with the ER, suggesting that they are derived from it. Still, VAPB aggregates are clearly distinct from the ER as they lack ER markers, and tubule borders within the aggregates show an electron-dense appearance suggestive of the presence of high levels of protein. Furthermore, photobleaching experiments show that the VAPB-P56S proteins are mostly immobile, similar to the aggregated SOD1 and Htt mutants (Chai et al., 2002; Kim et al., 2002), but different from ER clusters that can be obtained by overexpressing ER proteins such as cytochrome b(5) (Snapp et al., 2003). Occasionally, the VAPB-P56S tubular aggregates are continuous with the outer membranes of mitochondria, which may be explained by the close association of some ER compartments with mitochondria (Goetz and Nabi, 2006).
Mutant VAPB induces membrane-protein aggregates
How does mutant VAPB form aggregates? Structural analysis indicates that wild-type VAP proteins form dimeric complexes where both the coiled-coil and the C-terminal transmembrane domains induce VAP dimerization whereas two FFAT motifs bind the MSP homology domains in a 2:2 complex (Russ and Engelman, 2000; Kaiser et al., 2005). In VAPB-wt, the P56 residue lies within the loop connecting the d1- and d2-strands within the MSP homology domain (Kaiser et al., 2005) (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). The characteristic S-shape of this loop is maintained by the cis-peptide bond conformation stabilized by the proline residue. This conformation seems required to establish the hydrogen-bonding network that maintains the precise register of the d1 strand with respect to the e strand. The side chain of P56 and the d1 strand pack against hydrophobic core residues of the MSP homology domain (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). We show that the mutant and not the wild-type MSP homology domains are recruited to the aggregates. The most likely explanation for the clustering of mutant VAPB is that the serine substitution breaks the cis-conformation of the peptide bond to assume the energetically much more favorable trans-conformation. This structural change, together with the placement of a polar side chain in close proximity to a hydrophobic environment, is expected to destabilize the hydrogen-bonding interaction network established by the d1–d2 loop and d1 strand (Nishimura et al., 2004), consequently exposing hydrophobic residues to the solvent (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). The aberrant VAPB structure may induce self-association of dimeric VAPB with neighboring mutant VAPB molecules, whereby VAPB complexes are still tethered to the ER membrane by their TMDs (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). Interactions between the TMDs of the mutant and wild-type VAPB proteins cause entrapment of the latter in the aggregates.
Mutant VAPB recruits wild-type VAP, disrupts FFAT binding, and causes cell death
Why does mutant VAPB and reduced VAP expression leads to death of neuronal cells? VAP proteins are adaptors for recruiting FFAT motif-containing lipid-binding proteins to ER membranes, and disruption of VAP induces the mislocalization of these proteins (Loewen et al., 2003). Knockdown experiments and analysis of FFAT-motif ceramide transporter CERT mutants revealed that VAPs are implicated in ceramide transport and sphingomyelin synthesis in cultured mammalian cells, but most likely are involved in several other lipid-trafficking pathways (Kawano et al., 2006; Perry and Ridgway, 2006). In Drosophila, the FFAT motif-containing protein rdgB (retinal degeneration B) is implicated in phospholipid transport and metabolism and is essential for photoreceptor viability (Lev, 2004).
Close examination of the high-resolution crystal structure of the complex formed between the MSP domain of VAPA and a FFAT motif-containing peptide originating from ORP1 shows that P56, and in particular N57, is positioned next to the FFAT peptide binding site (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). As discussed above, the local conformational change induced by the serine substitution is expected to impair the hydrogen-bonding network formed between VAPB MSP and the C-terminal moiety of the bound FFAT peptide. Here, we show that the interaction with the FFAT motif is disrupted by the P56S mutation. Although the disruption of FFAT binding alone cannot explain the effects of the dominant VAPB mutation, we believe that repressed FFAT binding, in addition to the recruitment of wild-type VAP to mutant VAPB aggregates, further impairs normal VAP function and accelerates the reduced ER anchoring of FFAT motif-containing lipid-binding proteins. We hypothesize that reduced levels of VAP similar to its yeast homolog Scs2 (Kagiwada et al., 1998) disturbs lipid homeostasis. Evidence supporting this model comes from the observed Golgi dispersion in primary neurons expressing VAP-P56S and shRNA-VAP constructs. Disruption of intracellular transport and abnormal lipid metabolism has been shown to lead to Golgi disassembly (Fukunaga et al., 2000; Gonatas et al., 2006).
Model of VAP leading to motor neuron degeneration
Expression analyses show that VAPs are ubiquitously expressed in all tissues and throughout the CNS, raising the following question: why does its mutation in vivo preferentially affects motor neurons? We show that VAPB is highly abundant in motor neurons compared with other cells in the CNS. Because of their exceptionally large size and complex morphology, motor neurons are likely to require a high rate of lipid metabolism and well organized lipid transport mechanisms, which may be linked to the high level of VAPB expression and explain the preferential vulnerability of motor neurons to VAPB mutations.
Our data point to several potential deleterious effects by which VAPB-P56S can cause degeneration and death of neurons. VAPB-P56S forms stable aggregates that are continuous with the ER, mitochondria, and possibly other membranous organelles, recruits wild-type VAPs, and impairs normal VAP function. This, in combination with the disrupted binding to FFAT motifs by mutant VAPB, may result in abnormal lipid transport and biosynthesis and induce slow degeneration of neurons (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). Loss of DVAP-33 in Drosophila results in motor abnormalities, defects at the neuromuscular junctions, and death at the early larval stage (Pennetta et al., 2002), indicating that in flies deleterious consequences of the loss of VAP are restricted to motor neurons. We show that VAP expression is reduced in motor neurons from sporadic ALS patients and SOD1-ALS mice, suggesting that the decreased expression of VAPB might be a common feature associated with motor neuron degeneration. We propose that VAPB mutation or deficiency results in an abnormal lipid metabolism in motor neurons. Interestingly, a number of observations have linked defects in lipid homeostasis to ALS pathogenesis and motor neuron degeneration. Altered levels of sphingomyelin, ceramides, and cholesterol esters, and altered expression of genes involved in the control of lipid metabolism have been identified in the spinal cord of ALS patients and transgenic SOD1-ALS mice (Malaspina et al., 2001; Cutler et al., 2002). In addition, mice with a targeted disruption in the liver X receptor β, a ubiquitous sterol-activated nuclear receptor involved in cholesterol and sterol metabolism, exhibit degenerative changes in motor neurons and muscle atrophy reminiscent of ALS (Andersson et al., 2005; Xie et al., 2005). Furthermore, plant and bacterial sterol derivatives have been shown to be neurotoxic, and have been linked to the pathogenesis of the Guam ALS-parkinsonism dementia complex (Ly et al., 2007)
In sum, we hypothesize that the combination of loss of function (disrupted FFAT-motif binding), dominant-negative effects (wild-type VAP recruitment), and gain of toxic function (disrupted membrane trafficking) may lead to VAPB-linked motor neuron disease. The dominant-negative VAPB-P56S effect correlates well with the VAP knockdown phenotype and reduced expression of VAP in sporadic and familial ALS, indicating the reduction of VAP family proteins is a likely factor contributing to motor neuron degeneration.
Animal models will provide additional insight into the cellular mechanisms by which a reduction in ER-resident membrane VAP proteins can result in specific degeneration of motor neurons in vivo and reveal the particular lipid-trafficking routes affected by mutant VAP aggregates.
Footnotes
-
This work was supported by the Netherlands Organization for Scientific Research (NWO-VIDI), European Science Foundation (European Young Investigators Award), and the ALS Association. E.T. was supported by the Prinses Beatrix Fonds from C.C.H. and D.J. We thank Dr. K. Simons for tsVSVG-YFP, Dr. V. Ollkonen for ORP3-GFP, Dr. H. Kampinga for Htt(Q74)-GFP and GFP-Hsp-70, Dr. S. Lindquist for Hsp-104, Dr. R. Kopito for Atg8/LC3 and Atg12 antibodies, M. Schlager for providing cell line extracts, S. Spangler and N. Keijzer for preparing primary neuronal cultures, Dr. D. Troost and M. Ramkema for providing human ALS and control spinal cord sections, Karel Bezstarosti for help with mass spectrometry analyses, and Dr. W. van Cappellen for assistance with live cell imaging.
- Correspondence should be addressed to either Casper C. Hoogenraad or Dick Jaarsma, Department of Neuroscience, Erasmus Medical Center, P.O. Box 2040, 3000CA, Rotterdam, The Netherlands. c.hoogenraad{at}erasmusmc.nl or d.jaarsma{at}erasmusmc.nl





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}