

Abstract
The mammalian neurotrophins (NTs) NGF, BDNF, NT-3, and NT-4 constitute a family of secreted neuronal growth factors. In addition, NTs are implicated in several forms of activity-dependent synaptic plasticity. Although synaptic secretion of NTs has been described, the intracellular signaling cascades that regulate synaptic secretion of NTs are far from being understood. Analysis of NT secretion at the subcellular level is thus required to resolve the role of presynaptic and postsynaptic NT secretion for synaptic plasticity. Here, we transfected cultures of dissociated rat hippocampal neurons with green fluorescent protein-tagged versions of BDNF and NT-3, respectively, and identified NT vesicles at glutamatergic synapses by colocalization with the cotransfected postsynaptic marker PSD-95 (postsynaptic density-95)-DsRed. Depolarization-induced secretion of BDNF and NT-3 was monitored with live cell imaging. Direct postsynaptic depolarization with elevated K+ in the presence of blockers of synaptic transmission allowed us to investigate the signaling cascades that are involved in the postsynaptic NT vesicle secretion process. We show that depolarization-induced postsynaptic NT secretion is elicited by Ca2+ influx, either via L-type voltage-gated calcium channels or via NMDA receptors. Subsequent release of Ca2+ from internal stores via ryanodine receptors is required for the secretion process. Postsynaptic NT secretion is inhibited in the presence of KN-62 ([4(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl] phenyl isoquinolinesulfonic acid ester) and KN-93 (N-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide), indicating a critical dependence on the activation of α-calcium–calmodulin-dependent protein kinase II (CaMKII). The cAMP/protein kinase A (PKA) signaling inhibitor Rp-cAMP-S impaired NT secretion, whereas elevation of intracellular cAMP levels was without effect. Using the Trk inhibitor k252a, we show that NT-induced NT secretion does not contribute to the NT release process at synapses, and BDNF does not induce its own secretion at postsynaptic sites. Release experiments in the presence of the fluorescence quencher bromphenol blue provide evidence for asynchronous and prolonged fusion pore opening of NT vesicles during secretion. Because fusion pore opening is fast compared with compound release, the speed of NT release seems to be limited by diffusion of NTs out of the vesicle. Together, our results reveal a strong dependence of activity-dependent postsynaptic NT secretion on Ca2+ influx, Ca2+ release from internal stores, activation of CaMKII, and intact PKA signaling, whereas Trk signaling and activation of Na+ channels is not required.
Introduction
Brain-derived neurotrophic factor (BDNF) and other members of the protein family of neurotrophins (NTs) have been implicated in a multitude of processes that are important for synaptic development and synaptic plasticity in the rodent CNS (Lu, 2004; Poo, 2001). In comparison, our knowledge regarding the processes that govern synaptic secretion of the NTs is scarce.
In developing CNS neurons, acute application of BDNF is known to enhance primarily glutamatergic synaptic transmission (Lessmann et al., 1994; Kang and Schuman, 1995; Levine et al., 1995) and to inhibit GABAergic synaptic currents (Tanaka et al., 1997; Frerking et al., 1998; Brunig et al., 2001). The enhancing effect of acutely added BDNF on glutamatergic synapses is mostly presynaptic in origin (Carmignoto et al., 1997; Lessmann and Heumann, 1998; Li et al., 1998). In addition, postsynaptic NMDA receptor gating has been described to be regulated by BDNF signaling (Levine et al., 1995, 1998). Interestingly, acute application of BDNF to the dentate gyrus in vivo can cause long-term synaptic plasticity of glutamatergic synapses, involving mechanisms requiring new protein synthesis (Messaoudi et al., 1998; Bramham and Messaoudi, 2005). For GABAergic synapses, acute application of BDNF to hippocampal slices and dissociated cultures is known to reduce postsynaptic activation of GABAA receptors (Tanaka et al., 1997; Brunig et al., 2001). An additional transient and presynaptic enhancement of GABAergic transmission has been described as well in cultured hippocampal neurons (Wardle and Poo, 2003; Jovanovic et al., 2004).
Long-term incubation of synaptic networks of dissociated hippocampal and neocortical neurons with BDNF enhances the development of both glutamatergic and GABAergic synaptic transmission (Rutherford et al., 1997; Vicario-Abejon et al., 1998; Bolton et al., 2000; Paul et al., 2001; Baldelli et al., 2002; Yamada et al., 2002). Likewise, BDNF contributes to homeostatic plasticity of glutamatergic and GABAergic synapses in developing neocortical and hippocampal cultures, scaling up glutamatergic synapses under conditions when excitatory drive in the synaptic network is low, and scaling up GABAergic synapses when the excitatory drive is high (Turrigiano and Nelson, 2000).
The importance of BDNF for long-term potentiation (LTP) is evident from two different experimental approaches. First, in the visual cortex, exogenous supply of BDNF can increase the likelihood to observe LTP under conditions of suboptimal LTP stimulation (Akaneya et al., 1997; Huber et al., 1998; Kinoshita et al., 1999). Second, using BDNF knock-out mouse models, the availability of endogenous BDNF has been shown to be required for the proper establishment of LTP in the hippocampus (Korte et al., 1995; Patterson et al., 1996), somatosensory cortex (Itami et al., 2003), and visual cortex (Bartoletti et al., 2002; Abidin et al., 2006) and for the maintenance of late LTP (Korte et al., 1998; Patterson et al., 2001, Bramham and Messaoudi, 2005). This requirement for BDNF in LTP can be explained by at least two different mechanisms: first, BDNF has a permissive effect on LTP by supporting high-frequency transmitter release at a level that is sufficient for LTP induction (Figurov et al., 1996; Gottschalk et al., 1998; Pozzo-Miller et al., 1999; Abidin et al., 2006). Second, BDNF can have an instructive effect for expression of LTP in the hippocampus and in the somatosensory cortex (e.g., by allowing membrane insertion of new AMPA receptors) (Kossel et al., 2001; Itami et al., 2003). An important function of acutely released BDNF for synaptic plasticity in the hippocampus is most evident from experiments that show impaired synaptic plasticity when extracellular BDNF is scavenged during high-frequency synaptic stimulation (Kang et al., 1997; Chen et al., 1999; Kossel et al., 2001; Walz et al., 2006). These latter data support a requirement for acute (i.e., within minutes of synaptic activation) activity-dependent synaptic secretion of BDNF to mediate plasticity.
Despite the plethora of the above-mentioned synaptic actions of the neurotrophins, our knowledge regarding the mechanisms of synaptic secretion of NTs is still modest (von Bartheld et al., 2001; Lessmann et al., 2003). Several studies have addressed the mechanisms of neuronal secretion of NGF and BDNF using ELISA measurements of neurotrophin content in the extracellular medium (Blochl and Thoenen, 1995; Canossa et al., 1997). These studies revealed an important role of patterned high-frequency electrical stimulation for the release of BDNF (Balkowiec and Katz, 2000; Lever et al., 2001; Gartner and Staiger 2002) and an involvement of intracellular Ca2+ stores in the release process (Blochl and Thoenen, 1996; Griesbeck et al., 1999; Canossa et al., 2001; Balkowiec and Katz, 2002; Wang et al., 2002). Using green fluorescent protein (GFP) fusion proteins of neurotrophins, it recently became possible to investigate synaptic secretion of NTs directly, at high spatial and temporal resolution (Hartmann et al., 2001; Kohara et al., 2001; Kojima et al., 2001). These studies revealed postsynaptic secretion of BDNF and of other neurotrophins from cultured hippocampal neurons, in response to tetanic synaptic or depolarizing stimulation (Hartmann et al., 2001; Kojima et al., 2001) that proceeds one order of magnitude more slowly than the release of neurotransmitters (Brigadski et al., 2005). On a longer time scale (i.e., in the range of hours), axonal release of BDNF from cultured cortical neurons was reported as well (Kohara et al., 2001).
Previous studies using ELISA measurements that addressed the intracellular signaling cascades involved in the NT release process could not distinguish between synaptic network effects (e.g., favoring overall excitation, thus indirectly supporting NT secretion) and direct actions on the NT release process. Our study is now aimed at elucidating the postsynaptic signaling cascades that directly regulate the synaptic secretion of NTs.
The results show that postsynaptic secretion of BDNF and NT-3 relies on the influx of extracellular Ca2+, either via postsynaptic NMDA receptors or L-type voltage-gated Ca2+ channels (VGCCs). The release process requires the subsequent amplification of the initial Ca2+ influx via thapsigargine-sensitive internal Ca2+ stores and involves the activation of ryanodine receptors. This synaptic NT secretion is critically dependent on the activation of postsynaptic α-calcium–calmodulin-dependent protein kinase II (CaMKII) and requires basal levels of protein kinase A signaling. Neither action potential firing nor postsynaptic tyrosine receptor kinase B (TrkB) or TrkC receptor activation contribute to the postsynaptic secretion of NTs under these circumstances. Finally, experiments with the fusion pore permeable dye bromphenol blue (BPB) suggest that postsynaptic NT secretion proceeds via prolonged and/or repetitive fusion pore openings during depolarization.
Materials and Methods
Cell culture.
Dissociated postnatal rat hippocampal microcultures were prepared essentially as described previously (Lessmann and Heumann, 1998; Brigadski et al., 2005): primary rat postnatal [postnatal day 0 (P0) to P2] neocortical astrocytes were isolated and cultured for 2–4 weeks in DMEM medium, containing 10% FCS until expanded to confluence. Astrocytes were passaged and seeded on glass coverslips at a density of 80,000 cells per 3.5 cm dish in DMEM/10% FCS to yield astrocyte islands of 100–300 μm in diameter after 7–14 d in vitro (DIV). (β-d-Arabinofuranosyl)cytosine (5 μm) was added 5 d after seeding of astrocytes to avoid further growth of astrocyte islands. Dissociated postnatal rat (P0–P2) hippocampal neurons were plated in DMEM/10% FCS at a density of 1–10 neurons per astrocyte island onto the astrocyte coverslips. After 20 h, the plating medium was exchanged to serum-free medium (Neurobasal with 2% B27 supplement; Invitrogen, San Diego, CA).
Transfection.
Rat hippocampal microcultures were transfected with expression vectors coding for C-terminal GFP-tagged rBDNF or hNT-3, respectively (Haubensak et al., 1998; Brigadski et al., 2005.) These cytomegalovirus promoter driven constructs yield an approximately fivefold overexpression of BDNF-GFP and NT-3-GFP in our neurons (compare supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The respective expression plasmids were used for transfection at 8–9 DIV, using the Ca2+ phosphate precipitation method as decribed previously (Haubensak et al., 1998). During incubation (1.5–3 h), 10 μm DNQX and 50 μm d,l-APV were added to reduce excitotoxicity. The transfection medium was replaced, cultures were washed three times with warmed HEPES buffered saline (HBS), and the cultures were kept in neuron-conditioned Neurobasal/B27 medium thereafter. The cells were used for time-lapse imaging experiments 1–3 d after transfection.
FM 4-64 release assay.
Activity-dependent labeling of presynaptic terminals was performed as described previously (Klau et al., 2001). Briefly, coverslips with hippocampal neurons were incubated at 10–12 DIV for 2 min at room temperature (RT) in HBS containing 50 mm K+ (replacing an equal amount of Na+), 2 mm Ca2+, 1 mm Mg2+, and 10 μm FM 4-64 (Invitrogen, Eugene, OR). Cells were washed four times in dye-free HBS with low K+ (i.e., 5 mm), 0 mm Ca2+, and 3 mm Mg2+ and were transferred into the recording chamber of an inverted microscope for recordings. Imaging was performed as described below for NT secretion.
Cotransfection with synaptic marker proteins.
The construction and use of a PSD-95 (postsynaptic density-95)-DsRed expression plasmid was described previously (Brigadski et al., 2005). Intact synaptic targeting of the resulting protein was confirmed by colocalization of PSD-95-DsRed with the green fluorescent activity-dependent label of active synapses, FM 1-43 (data not shown). To identify postsynaptic structures, in BDNF-GFP and NT-3-GFP expressing hippocampal neurons, cells were cotransfected with the respective NT-GFP construct together with PSD-95-DsRed (DNA ratio, GFP:DsRed, 1:1; 3 μg per dish overall).
Fluorescence imaging.
Visualization of epifluorescence signals was performed as described previously (Hartmann et al., 2001). Briefly, coverslips with transfected cells were transferred into Petriperm dishes (Vivascience-Sartorius, Hannover, Germay) with folio bottom and inspected with an inverted microscope (Olympus IX70; Olympus Optical, Tokyo, Japan) using 40× [numerical aperture (NA), 1.0] and 100× (NA, 1.35) oil immersion objectives. Image capture was performed using a cooled CCD camera (Sensys 1401E; 12 bit dynamic range; Photometrics, Huntington Beach, CA), controlled by MetaVue software (Molecular Devices, Menlo Park, CA). The exposure times for time-lapse recordings (between 0.3 and 1.5 s per image when using the 40× objective) were adjusted for every cell such that no area of the CCD chip was driven into saturation. Processing of images was performed by MetaVue and Adobe Photoshop software (Adobe Systems, San Jose, CA) without compromising the evident primary image information. However, in the figures, fluorescence in the soma and the proximal dendrites is often enhanced close to saturation to make clearly visible single vesicles also in distal neurites.
Confocal imaging.
In some experiments, colocalization of BDNF-GFP and NT-3-GFP with PSD-95-DsRed was evaluated with a Nipkow spinning disk confocal system (Visitech, Sunderland, UK), attached to a conventional fluorescence microscope (Olympus BX51 WI) equipped with a high-aperture water immersion objective (60×; NA, 0.9). Full-frame image capture was performed with a cooled CCD camera (CoolSnap HQ; 12 bit dynamic range; Roper Scientific, Trenton, NJ). Green and red fluorescence was excited with the 488 and 568 nm lines, respectively, of a Kr/Ar laser (Laser Physics, West Jordan, UT).
Neurotrophin release.
Real-time imaging of synaptic secretion of GFP-tagged NTs was performed as described previously (Hartmann et al., 2001): coverslips of transfected hippocampal neurons were mounted in the perfusion chamber of an inverted fluorescence microscope. Cells with synaptically localized NT-GFP were locally superfused with HBS (containing 2 mm Ca2+ and 1 mm Mg2+) throughout the experiment, and images were captured at 5 or 10 s intervals, respectively. After 5 min control period (i.e., superfusion with HBS), cells were depolarized for 1–5 min by superfusing 50 mm K+ containing (replacing an equal amount of Na+) HBS at RT. If not stated otherwise, the elevated K+ solution contained DNQX (10 μm), d,l-APV (50 μm), and gabazine (50 μm) to inhibit postsynaptic glutamate and GABA receptors. Regions containing postsynaptic clusters of NT-GFP were analyzed over time for changes in intracellular fluorescence intensity. Background fluorescence levels of a void region in the same field of view was recorded in parallel and subtracted. Fluorescence decrease caused by photobleaching was corrected for by extrapolation of the fluorescence decrease during an initial 5 min control period over the whole recording time. Original fluorescence values at a given time point were normalized to the corresponding value of this extrapolated bleaching curve, thus providing a measure of the fluorescence decrease because of release. This fluorescence decrease has been shown previously to reflect tetanus toxin and Ca2+-sensitive release of NTs (Hartmann et al., 2001).
Reagents.
Tetrodotoxin (TTX), k252a, and human recombinant BDNF were purchased from Alamone Labs (Jerusalem, Israel). Nifedipine (Nif), 8Br-cAMP, DNQX, gabazine, NMDA, d,l-APV, Bafilomycine A1, KN-92 (2-[N-(4′-methoxybenzenesulfonyl)]amino-N-(4′-chlorophenyl)-2-propenyl-N-methylbenzylamine phosphate), and KN-93 (N-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide) were obtained from Sigma (St. Louis, MO). Thapsigargine, KN-62 (4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl] phenyl isoquinolinesulfonic acid ester), and Rp-cAMPs were purchased from Tocris Bioscience (Ballwin, MO). Ryanodine and cyclopiazonic acid (CPA) were obtained from Alexis Biochemicals (San Diego, CA).
Results
Measuring postsynaptic secretion of neurotrophins
To focus on synaptic secretion of BDNF and NT-3 at glutamatergic synapses, cultured hippocampal neurons were cotransfected at 8–9 DIV with PSD-95-DsRed (as a postsynaptic marker of glutamatergic synapses) and BDNF-GFP or NT-3-GFP, respectively. Two days after transfection, postsynaptic accumulations of NT vesicles were identified by colocalization of the NT-GFP signal with PSD-95-DsRed (Fig. 1 A,B) (as indicated by immunocytochemistry, this paradigm yielded an approximately fivefold overexpression of the GFP-tagged NTs, compared with endogenous levels) (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Secretion of BDNF or NT-3 from these postsynaptic sites was recorded using time-lapse video microscopy (see Materials and Methods) and was evident as a decrease in intracellular GFP fluorescence intensity after synaptic depolarization (Fig. 1 C,D). To obtain a reproducible depolarization of the entire cell membrane (including all postsynaptic structures) and a comparable degree of depolarization between experiments, we chose to directly depolarize the cell membrane with elevated extracellular K+ (50 mm; 5 min). This solution was supplemented with blockers of synaptic transmission (50 μm gabazine, 50 μm d,l-APV, 10 μm DNQX) to exclude indirect effects of presynaptically released glutamate and GABA via postsynaptic ionotropic receptors. As shown previously, metabotropic glutamate receptors do not mediate NT secretion under these conditions (Hartmann et al., 2001). Using this paradigm, synaptic network effects are excluded, and all pharmacological effects in this study can be directly related to the postsynaptic release process of NTs itself, rather than to changed excitatory drive or altered synaptic efficacy in our cultures.

Investigation of postsynaptic secretion of BDNF and NT-3. A , Hippocampal neurons were cotransfected at 8 DIV with BDNF-GFP and PSD-95-DsRed. Colocalization of both proteins were monitored at 10–11 DIV. B , Boxed area in A shown at higher magnification. Postsynaptic vesicle clusters of BDNF (green) were identified by colocalization with PSD-95 (red). C , Time course of BDNF release in response to depolarization with elevated K+ (50 mm; 300 s). Pictures show postsynaptic BDNF-GFP vesicle clusters at indicated time points (0 s, start of depolarization; compare D ). Note the gradual decrease of intracellular BDNF-GFP after the start of depolarization (+150 s; +300 s). D , Time course of intracellular fluorescence of regions of interest in C . Note the decrease in intracellular fluorescence during stimulation (bar). E , Postsynaptic BDNF secretion in response to different depolarization paradigms (red, 6 × 10 s elevated K+ at 10 s intervals; green, 300 s K+). Comparable degrees of NT secretion after 120 s are evident.
On average, this depolarizing stimulation led to a release of ∼10% of stored NTs (i.e., remaining intracellular fluorescence for BDNF, 90.6 ± 1.3%, n = 20 cells; NT-3, 89.6 ± 1.3%, n = 17 cells), 300 s after starting the depolarization, compared with unstimulated negative controls (remaining fluorescence, 99.6 ± 0.9%; n = 9 cells). The average time constants (monoexponential fit) of secretion for BDNF (173 ± 33 s) and NT-3 (238 ± 25 s) were similar to previously published values (Brigadski et al., 2005). Other patterns of postsynaptic depolarization were also effective to induce significant postsynaptic NT secretion. Thus, six times interspersed 10 s applications of elevated K+ also induced a significant loss of postsynaptic NT-GFP fluorescence. At 120 s (a common time point of stimulation for both paradigms), the remaining fluorescence intensity amounted to 95.2 ± 2.2% (6× 10 s stimulation), compared with 93.7 ± 2.3% (300 s stimulation; both significantly different from negative control with p < 0.05). This indicates detectable postsynaptic secretion of NTs, also when using less robust depolarization paradigms (Fig. 1 E). Although the initial release time course was comparable, the larger amount of secreted BDNF after 5 min depolarization (remaining fluorescence, 87.8 ± 4.3%) allowed us to investigate the postsynaptic signaling cascades involved in NT secretion with higher sensitivity. Thus, all subsequent experiments were performed using the 5 min depolarization protocol. Because the efficiency of release varied between different preparations (range, 8–15% release), matched positive controls (i.e., 300 s elevated K+ plus gabazine/APV/DNQX) were included in each set of experiments.
Postsynaptic NT secretion proceeds via prolonged fusion pore opening
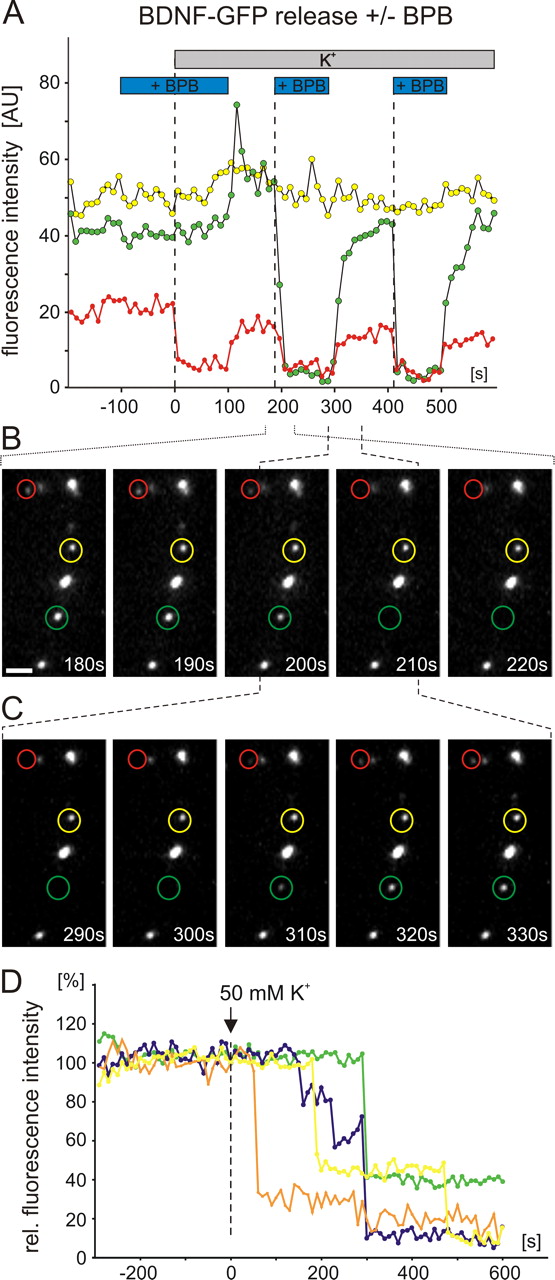
To investigate whether postsynaptic secretion of NTs proceeds via full collapse vesicle fusion or via graded release of NTs through prolonged or repetitive opening of vesicle fusion pores, we made use of the recently developed technique to visualize the kinetics of first fusion pore openings (Harata et al., 2006). For this purpose, the extracellular solution was supplemented with the fluorescence quencher BPB (2 mm), which completely eliminates vesicular GFP fluorescence after first fusion pore opening (Harata et al., 2006). Under these conditions, the elevated K+-induced fluorescence decrease of BDNF-GFP vesicles started considerably faster and was more efficient (remaining fluorescence after 300 s, 79.8 ± 3.1%; n = 6 cells) than under control conditions (90.6 ± 1.3%; n = 20 cells) (Fig. 2 A). This enhanced decay in vesicular BDNF-GFP fluorescence was accompanied by a sudden and complete loss of fluorescence of single vesicles after the onset of stimulation: as shown in Figure 2 B (regarding the evidence for single vesicle fluorescence in these experiments) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), in the presence of BPB, single BDNF-GFP vesicles now were often observed to lose their entire fluorescence between just two image captures (interval, 10 s). This observation is in contrast with the graded loss of single-vesicle BDNF-GFP fluorescence even over hundreds of seconds, that was commonly observed in the absence of BPB (compare Fig. 2 C). This suggests that BPB passed the fusion pore of BDNF-GFP vesicles after first opening and effectively quenched the entire GFP fluorescence in these vesicles (cf. Harata et al., 2006). The quenching strongly accelerated the observed overall fluorescence decrease (compare Fig. 2 A). As shown in Figure 3 A–C, the quenching effect of BPB was reversible and could be observed repetitively for the same vesicle, indicating prolonged and/or repetitive fusion pore opening of vesicles that stayed at the same release site during ongoing depolarization. As shown in Figure 3 D, the delay between the start of the depolarization and first fusion pore opening varies greatly for different vesicles of the same cell. This is an indication for asynchronous release of NTs from different release sites. The more graded loss of BDNF-GFP fluorescence under control conditions (i.e., in the absence of BPB) (Fig. 2 C) indicates that BDNF vesicles fuse with the membrane to slowly release their contents via repetitive and/or prolonged fusion pore opening.

Bromphenol blue quenching of GFP fluorescence reveals fast initial fusion pore opening of BDNF-GFP vesicles. Hippocampal neurons were transfected with GFP-tagged BDNF, and depolarization-induced (50 mm K+, 300 s; postsynaptic receptors blocked with 10 μm DNQX, 50 μm d,l-APV, and 50 μm gabazine) decrease in intracellular GFP fluorescence was monitored. A , Average time course of fluorescence decay in the absence/presence of the green fluorescence quencher BPB (2 mm in all superfusion solutions). In the absence of BPB, the fluorescence decay reflects release of BDNF-GFP, but in the presence of BPB, the decay represents the kinetics of first fusion pore openings of the BDNF-GFP vesicles (compare Results). Note the fast onset of first fusion pore openings compared with the delayed and less pronounced release of BDNF-GFP. B , Two representative series of pictures (dashed blue box in A ) for a cell measured in the presence of BPB. Note the sudden and complete loss of fluorescence of a single vesicle (blue arrows) within a 10 s imaging interval. C , Two representative series of pictures (dashed red box in A ) for another cell measured in the absence of BPB. Note the graded fluorescence decay of single vesicles (red arrows) even during a 200 s imaging interval. The sudden BPB-dependent quenching of intravesicular BDNF-GFP fluorescence explains the faster and more complete decrease of the average data for BPB cells in A . Scale bars, 2 μm.

Prolonged or repetitive fusion pore opening of BDNF-GFP vesicles. All experimental details are as in Figure 2. A , Fluorescence intensity of single BDNF vesicles in the color-coded circles in B and C . Horizontal bars indicate superfusion conditions. B , C , Two image sequences (time windows indicated by stippled brackets) of the vesicles analyzed in A . The repeated disappearance/reappearance of the vesicles marked in red and green, after wash-in/washout of BPB, indicates prolonged and/or repetitive fusion pore opening during the depolarization. The transient increase in fluorescence intensity of the “green” vesicle at +100 s is caused by reduced quenching of GFP fluorescence after neutralization of intragranular pH through the fusion pore (Brigadski et al., 2005). The unchanged fluorescence of the “yellow” vesicle reflects absence of fusion pore opening for this vesicle. D , Recording as in A of single vesicle and vesicle cluster fluorescence in response to elevated K+ solution in the presence of BPB. Note the large scatter in delays until first fusion pore opening for individual vesicles from the same dendrite. Scale bar, 2 μm.
NT secretion depends on activity of postsynaptic L-type VGCC
Postsynaptic secretion of BDNF has been shown previously to depend on Ca2+ influx (Hartmann et al., 2001). This strict dependence of the postsynaptic release of NTs on Ca2+ influx was confirmed for the current recording conditions: whereas elevated K+-induced depolarization in Ca2+ free solution was ineffective, subsequent perfusion of 2 mm Ca2+ in depolarizing solution immediately provoked NT secretion (Fig. 4 A). This suggests that postsynaptic Ca2+ channels were gated by the elevated K+-induced depolarization and now permitted Ca2+ entry to activate the secretion process. Because of the known dendritic and postsynaptic localization of L-type voltage-gated Ca2+ channels (Hell et al., 1993), we investigated whether these channels contribute to the postsynaptic Ca2+ influx that induced NT secretion. Indeed, preincubation of our neurons with the selective L-type Ca2+ channel blocker nifedipine (1, 10, or 50 μm) strongly inhibited secretion of BDNF-GFP and NT-3-GFP (BDNF plus 50 μm Nif, 97.6 ± 1.3%; NT-3 plus 50 μm Nif, 97.7 ± 0.8%; BDNF control, 91.8 ± 1.8%; NT-3 control, 88.3 ± 1.0%; BDNF plus 10 μm Nif, 100.5 ± 1.8%; BDNF plus 1 μm Nif, 98.4 ± 0.8%) (Fig. 4 B,C). These data indicate that, under our conditions, the postsynaptic depolarization-induced Ca2+ influx that triggers NT secretion is mediated by L-type Ca2+ channels.

Depolarization-induced postsynaptic NT secretion depends on Ca2+ influx. Hippocampal neurons were transfected with GFP-tagged BDNF and NT-3, and depolarization-induced (50 mm K+, 300 s; postsynaptic receptors blocked with 10 μm DNQX, 50 μm d,l-APV, and 50 μm gabazine) postsynaptic secretion of NTs was measured as the decrease in intracellular fluorescence intensity. A , Change in BDNF-GFP secretion after consecutive depolarizations of the same cell in the absence/presence of extracellular Ca2+ (2 mm). B , Time course of secretion of BDNF-GFP and NT-3-GFP. Preincubation with the L-type Ca2+ channel antagonist nifedipine (50 μm; 10 min) significantly inhibited (p < 0.0001, at t = 300 s) secretion of both NTs. C , Residual intracellular fluorescence after 300 s stimulation for conditions as indicated. **p < 0.0001 compared with negative control (neg. con). Note the strong dependence of BDNF and NT-3 secretion on the activity of L-type VGCCs. Error bars represent SEM.
Because BDNF and NT-3 were equally dependent on Ca2+ influx and their secretion showed similar efficiencies and time courses, data for both NTs were pooled in all subsequent experiments.
Activation of postsynaptic NMDA receptors can elicit NT secretion, but activation of voltage-dependent Na+ channels is not required
We next tested, whether Ca2+ influx via NMDA receptors can elicit postsynaptic secretion of NTs as well. Therefore, neurons were stimulated with 300 μm NMDA (in Mg2+-free solution to remove NMDA receptor inhibition). To assure that direct postsynaptic NMDA receptor (NMDAR) activation was the only possible release stimulus, these experiments were performed in the presence of nifedipine (10 μm), DNQX (10 μm), and gabazine (50 μm). Indeed, this stimulation was also able to induce efficient release of BDNF-GFP. Nevertheless, the release occurred slightly delayed and was less pronounced (remaining fluorescence after 300 s, 93.9 ± 1.1%; significantly different from negative control with p < 0.001) compared with elevated K+ stimulation (90.2 ± 1.4%) (Fig. 5 A). Together, these results suggest that Ca2+ influx via postsynaptic NMDA receptors is sufficient to initiate NT secretion.

Activation of NMDA receptors can trigger postsynaptic secretion of BDNF-GFP. Experimental details are as in Figure 4. A , Time course of secretion of BDNF-GFP after application of 300 μm NMDA in saline containing 2 mm Ca2+ and 0 mm Mg2+. These experiments were performed in the presence of DNQX (10 μm), gabazine (50 μm), and nifedipine (10 μm) to allow for selective observation of NMDAR-mediated release. B , Time course of elevated K+ induced secretion of BDNF in the absence or presence of tetrodotoxin (TTX; 0.8 μm). Error bars represent SEM. neg. control, Negative control.
Activation of TTX-sensitive voltage-gated Na+ channels has been shown in different preparations to be a prerequisite for elevated K+-induced NT secretion (Blochl and Thoenen, 1995; Androutsellis-Theotokis et al., 1996; Kojima et al., 2001). We thus tested whether the postsynaptic secretion process itself requires gating of TTX-sensitive Na+ channels. As is evident from Fig. 5 B, elevated K+-induced NT secretion did not depend on the activation of voltage-dependent Na+ channels, stressing that, under our recording conditions, postsynaptic secretion of NTs occurs independent of action potential firing and action potential driven synaptic network activity.
Synaptic NT secretion is dependent on Ca2+ release from internal stores and CaMKII activity
Release of NGF and BDNF from neuronal networks has been shown previously to involve activation of intracellular Ca2+ stores (Blochl and Thoenen, 1995; Griesbeck et al., 1999). To determine whether endoplasmic reticulum resident Ca2+ stores can directly regulate postsynaptic NT secretion, we performed experiments in which intracellular calcium stores were depleted by preincubation with the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) inhibitor thapsigargine (10 μm; 10 min) before depolarizing the neurons with elevated K+. Interestingly, postsynaptic NT secretion was completely inhibited under these conditions (BDNF-GFP plus thapsigargine, 100.3 ± 1.1%; BDNF-GFP control, 91.2 ± 1.2%) (Fig. 6 A,B). Similarly, preincubation of the cells with another SERCA inhibitor (CPA, 30 μm) also significantly (p < 0.01) inhibited secretion of BDNF-GFP (BDNF-GFP plus CPA, 97.7 ± 1.1%). These data stress the importance of intracellular Ca2+stores for the secretion process, although the primary influx of Ca2+ for depolarization-induced secretion is inevitable (compare Fig. 4).

Dependence of NT secretion on intracellular Ca2+ stores and CaMKII. Hippocampal neurons were transfected with BDNF-GFP (BDNF) and monitored for postsynaptic neurotrophin secretion as in Figure 5. A , C , Time course of depolarization-induced (50 mm K+) release of BDNF-GFP. B , D , Average residual BDNF-GFP fluorescence after 300 s depolarization. B , Preincubation with thapsigargine (10 μm; 10 min) or CPA (3 μm, 30 min) to deplete internal Ca2+ stores or with ryanodine (80 μm; 30 min) to block Ca2+-induced Ca2+ release from internal stores significantly inhibited NT secretion (*p < 0.0001). D , Preincubation with 10 μm KN-62 or 10 μm KN-93 to inhibit CaMKII significantly inhibited NT secretion. For KN-62, data from BDNF and NT-3 release were pooled (*p < 0.01). neg. control, Negative control; rel., relative.
Given the combined dependence of the release process on Ca2+ influx and internal Ca2+ stores, we argued that Ca2+-induced Ca2+ release via ryanodine receptors in the ER might be instrumental. Accordingly, preincubation of cells with ryanodine (80 μm) blocked NT secretion (BDNF-GFP plus ryanodine, 102.4 ± 2.3%; significantly different from BDNF-GFP control with p < 0.0005).
Postsynaptic activation of CaMKII as well as synaptic BDNF secretion are known to be important regulators of synaptic plasticity. To identify a possible link between both molecules, we inhibited CaMKII by preincubating (30 min) the neurons with either KN-62 (10 μm) or KN-93 (10 μm), respectively (the inactive isomer KN-92 served as a negative control). Indeed, both CaMKII inhibitors strongly reduced BDNF-GFP secretion (remaining fluorescence after 300 s for BDNF-GFP plus KN-62, 96.2 ± 1.2%; BDNF-GFP plus KN-93, 97.8 ± 1.6%; BDNF-GFP control, 89.0 ± 1.1%; BDNF-GFP plus KN-92, 88.5 ± 1.8%) (Fig. 6 C,D), indicating a pivotal role for CaMKII activity in postsynaptic secretion of NTs. Similar results were obtained with NT-3-GFP (data not shown).
Presynaptic neurotransmitter secretion is unaffected by the various Ca2+ signaling inhibitors
To confirm that the diverse Ca2+ signaling inhibitors used in this study did not interfere with other release processes, we investigated whether they affected secretion of neurotransmitters under identical recording conditions: transmitter vesicles of hippocampal neurons were filled with the lipophilic dye FM 4-64 (see Materials and Methods), and release of the FM dye was taken as an indicator for transmitter release (Fig. 7 A–D) (cf. Cochilla et al., 1999). As evident from Figure 7 C, preincubation of the neurons with 50 μm nifedipine did not significantly change either the FM 4-64 release time course (exponential time constant 11.8 ± 3.3 s; n = 6) or the efficiency of FM 4-64 secretion, which is indicated by the average remaining fluorescence after 40 s of depolarization (Fig. 7 D). Similarly, no significant changes in either the release time course (data not shown) or the release efficiency were observed after treatment of the neurons with thapsigargine (10 μm), KN-62 (10 μm), or 8Br-cAMP (100 μm). These results suggest that the Ca2+ and cAMP signaling modulators used in this study did not interfere with neurotransmitter release, thus highlighting their specific action on postsynaptic NT secretion.
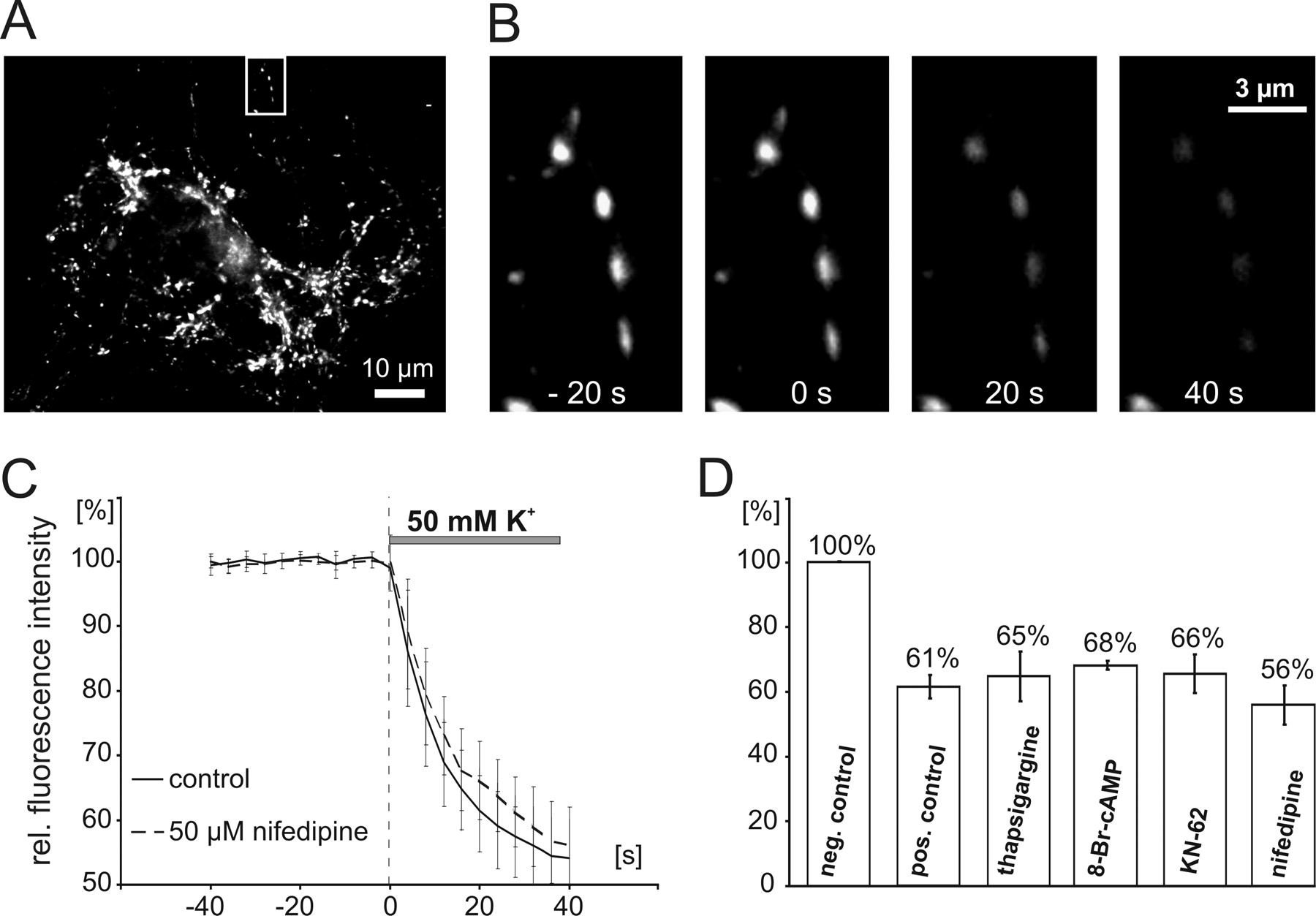
Unaltered presynaptic transmitter secretion during pharmacological treatments. A , Presynaptic terminals of hippocampal neurons were loaded with FM 4-64 at 10 DIV. Presynaptic secretion of FM 4-64 was elicited with elevated K+ (50 mm; 40 s). B , Boxed area in A shown at higher magnification. Pictures show FM 4-64 fluorescence in presynaptic terminals at indicated time points (0 s, start of depolarization; compare C ). C , Average time course of FM release in untreated (control) and treated neurons (50 μm nifedipine). Note the similar time courses of FM secretion, indicating unaltered presynaptic secretion after the drug treatment also used in NT secretion experiments. D , Average residual FM 4-64 fluorescence after 40 s depolarization for different treatments as indicated. All fluorescence values are given relative to the fluorescence levels at the start of the depolarization. Note that the different drug treatments did not significantly change presynaptic release of transmitters, indicating intact synaptic transmitter release under our recording conditions. neg. control, Negative control; rel., relative.
Gating of postsynaptic NT secretion by basal levels of protein kinase A activity
Activation of protein kinase A (PKA) has been suggested previously to regulate BDNF signaling in synaptic plasticity in different neuronal preparations (Ghosh et al., 1994; Meyer-Franke et al., 1995; Boulanger and Poo, 1999), including hippocampal neurons (Patterson et al., 2001). To investigate whether postsynaptic PKA signaling can affect NT secretion, we first chose to investigate NT release in response to acute superfusion with the membrane permeable cAMP analog 8-Br-cAMP (100 μm; 10 min). This time of incubation is sufficient to elicit robust cAMP-dependent signaling in our cultures (Lessmann and Heumann, 1997). However, 8-Br-cAMP alone did not evoke any detectable secretion of BDNF or NT-3 under these conditions (Fig. 8 B). After this preincubation, depolarization-induced postsynaptic secretion was monitored in the same cells: as shown in Figure 8, A and B, the elevation of intracellular cAMP did not change the time course or the efficiency of NT secretion (BDNF-GFP or NT-3-GFP plus 8-Br-cAMP, 93.0 ± 1.6% residual fluorescence, n = 10 cells; BDNF-GFP or NT-3-GFP control, 93.3 ± 1.5%, n = 8 cells).

Postsynaptic cAMP/PKA activity gates secretion of neurotrophins. Hippocampal neurons were transfected with BDNF-GFP (BDNF) and monitored for neurotrophin secretion as described previously. A , C , Averaged depolarization-induced (50 mm K+) release of BDNF versus negative control. B , D , Mean residual fluorescence 300 s after stimulation. A , Preincubation with the PKA activator 8-Br-cAMP (100 μm; 5 min) did not change depolarization-induced BDNF secretion. B , Application of 8-Br-cAMP (100 μm) neither induced BDNF secretion (left) nor changed the amount of released BDNF in response to depolarization (right) (*p < 0.005). C , D , Preincubation with the PKA inhibitor Rp-cAMP-S (100 μm; 5 min) significantly inhibited and delayed BDNF secretion. Experiments were performed in the presence of 10 μm DNQX, 50 μm d,l-APV, and 50 μm gabazine. Note that inhibition of basal levels of PKA signaling delays secretion of BDNF-GFP. neg. control, Negative control; rel., relative.
Interestingly, interference with the basal activity of PKA by preincubating cells with the hydrolysation resistant, membrane permeable, cAMP antagonist Rp-cAMP-S (100 μm; 5 min) significantly delayed the onset (Fig. 8 C) and also inhibited the efficiency of depolarization-induced postsynaptic secretion of NTs (residual fluorescence at 300 s for BDNF-GFP or NT-3-GFP plus Rp-cAMP-S, 95.6 ± 1.0%; BDNF-GFP or NT-3-GFP control, 90.3 ± 1.5%; significantly different with p < 0.02). Together, both sets of data suggest that endogenous levels of intracellular cAMP in our neurons are required and sufficient to gate (cf. Blitzer et al., 1995) the postsynaptic release of NTs via activation of PKA.
NT signaling is not involved in the postsynaptic secretion process
Finally, we aimed to determine whether postsynaptic TrkB or TrkC receptors are involved in the postsynaptic secretion of NTs. First, we applied saturating concentrations of BDNF (100 ng/ml; 5 min) to postsynaptic structures. These experiments were performed in the presence of various transmitter receptor inhibitors (DNQX, APV, GBZ) and nifedipine (10 μm) to avoid any secondary effects of BDNF via modulation of the synaptic network. Under these conditions, BDNF was unable to elicit NT secretion (compare Fig. 9 A).

Absence of NT-induced NT secretion at postsynaptic sites of hippocampal synapses. Hippocampal neurons were transfected with BDNF-GFP (BDNF) and monitored for neurotrophin secretion as described above. A , Averaged depolarization-induced (50 mm K+) or BDNF-induced (100 ng/ml) release of BDNF-GFP versus negative control. All recordings were performed in the presence of 10 μm DNQX, 50 μm d,l-APV, and 50 μm gabazine to block postsynaptic ionotropic receptors. When BDNF was applied, the extracellular solution contained nifedipine (10 μm) to abolish indirect effects of BDNF on L-type Ca2+ channels and to monitor selectively release that was elicited by BDNF-induced release of Ca2+ from internal stores. B , C , Average depolarization-induced (50 mm K+) residual fluorescence, 300 s after start of depolarization, of GFP-tagged BDNF or NT-3 versus control. Preincubation with the Trk kinase inhibitor k252a (200 nm; 30 min) had no significant effect (p > 0.05) on depolarization-induced NT secretion.
Second, we determined whether newly secreted BDNF-GFP and NT-3-GFP (which are known to retain full biological activity) (Brigadski et al., 2005) can sustain their own secretion via autocrine activation of postsynaptic TrkB and TrkC receptors (Canossa et al., 1997; cf. Kruttgen et al., 1998). We thus incubated our neurons with 200 nm k252a (30 min) to inhibit TrkB and TrkC signaling. To distinguish between synaptic signaling of TrkB and TrkC, BDNF-GFP and NT-3-GFP were analyzed separately here. This paradigm failed to reveal a significant reduction of either BDNF-GFP or NT-3-GFP secretion after blocking TrkB/TrkC signaling (BDNF-GFP plus k252a, 91.5 ± 2.7%; BDNF-GFP control, 89.8 ± 1.6%; NT-3-GFP plus k252a, 91.8 ± 1.1%; NT-3-GFP control, 87.9 ± 2.0%; K252a vs control not significantly different with p > 0.05) (Fig. 9 B,C). Overall, these data suggest that any direct BDNF-induced BDNF secretion or NT-3-induced NT-3 secretion at postsynaptic sites is absent.
Discussion
Using recording conditions that allowed us to specifically investigate time-resolved postsynaptic secretion of NTs, our results provide evidence for the following mechanism of depolarization-induced NT secretion (compare Fig. 10): postsynaptic influx of extracellular Ca2+ via NMDARs or via L-type VGCCs, and subsequent amplification of this Ca2+ elevation via thapsigargine-sensitive internal Ca2+ stores, are required for postsynaptic activity-dependent secretion of BDNF and NT-3. Neither action potential firing nor TrkB or TrkC signaling are necessary for this postsynaptic NT secretion. In addition, our data suggest that the NT secretion process proceeds via prolonged and/or repetitive fusion pore openings of NT vesicles.
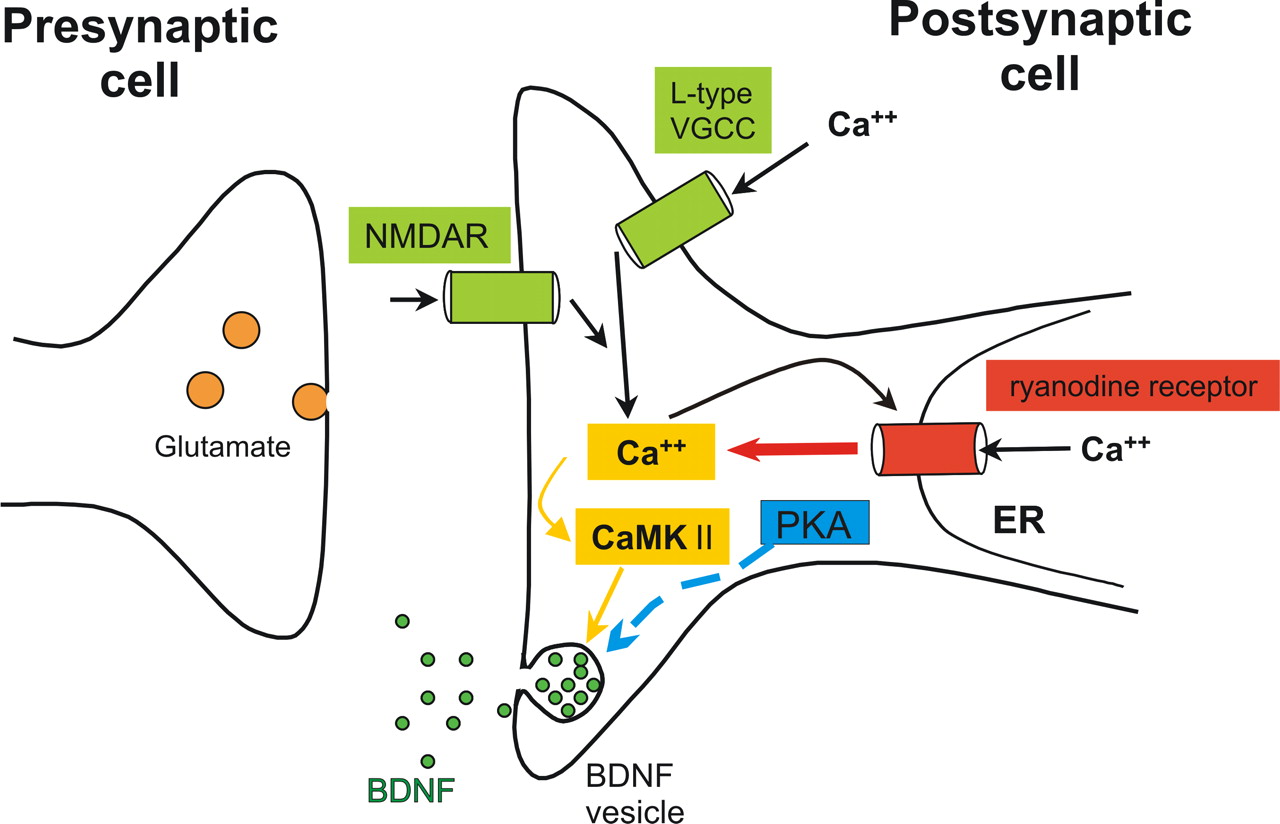
Scheme of the signaling cascades involved in postsynaptic secretion of NTs. Influx of extracellular Ca2+, either via L-type VGCCs or via NMDA receptors, is required for depolarization induced secretion. Amplification of the initial Ca2+ influx through Ca2+-induced Ca2+ secretion via ryanodine receptors is necessary to observe NT secretion. CaMKII signaling is pivotal for the induction of NT secretion. Basal levels of PKA activation “gate” NT secretion. Activation of voltage-gated Na+ currents is not necessary to observe NT release (data not shown). NTs do not sustain their own release from postsynaptic sites (data not shown).
The NT release process is critically dependent on the activation of postsynaptic CaMKII and requires intact cAMP/PKA signaling in the releasing postsynaptic neuron. Thus, our data provide the first direct experimental link between postsynaptic CaMKII and PKA activity and postsynaptic secretion of BDNF, respectively. This link might be instrumental to understand the interplay of these molecules in coregulating synaptic plasticity at glutamatergic synapses.
Requirement to investigate mechanisms of NT secretion at the synaptic level
The intracellular signaling cascades involved in regulating secretion of overexpressed NT-3 and BDNF (Canossa et al., 1997, 2001; Griesbeck et al., 1999) or endogenous BDNF (Balkowiec et al., 2002) have been investigated previously using ELISA procedures. These studies revealed a crucial role of intracellular Ca2+ stores for the release process (Griesbeck et al., 1999) and an important contribution of N-type Ca2+ channels for activity-dependent BDNF secretion (Balkowiec and Katz, 2002). However, most of these studies investigated BDNF bulk secretion from at least 10,000 synaptically connected neurons (but see Balkowiec and Katz, 2002). Despite the valuable insights from these studies, all pharmacological manipulations in these synaptic networks control the overall excitatory drive of the network and thus can have numerous indirect effects on NT secretion, without having any direct influence on the NT secretion process itself. Whereas in the study by Balkowiec and Katz (2002), glutamatergic network effects on the release of endogenous BDNF were excluded their hippocampal cultures were too young to allow addressing synaptic secretion of BDNF.
Selective measurement of synaptic secretion of NTs at the single-cell level has just recently become possible by time-lapse imaging of overexpressed GFP-tagged NTs in central neurons (Haubensak et al., 1998; Kojima et al., 2001). This approach enables investigation of NT secretion from subcellular compartments and with high time resolution (Hartmann et al., 2001; Brigadski et al., 2005) and thus is ideally suited to investigate the intracellular signaling cascades controlling presynaptic versus postsynaptic NT release. Direct membrane depolarization with elevated K+, in the presence of inhibitors of postsynaptic transmitter receptors, was used in this study to assure identical levels of postsynaptic excitation in all experiments and ruling out any synaptic network effects. Imaging of dendritic NT vesicle clusters that colocalized with coexpressed PSD-95-DsRed assured that we analyzed postsynaptic NT secretion from glutamatergic synapses.
Slow secretion of NTs despite fast fusion pore opening: evidence for slow diffusion of NTs from their vesicles
Exocytosis of neurotransmitter and neuropeptide vesicles can, depending on cell context and recording conditions (i.e., extracellular Ca2+ concentration, frequency of stimulation), proceed via full collapse fusion or via a kiss and run mechanism (for review, see An and Zenisek, 2004). In the case of neuropeptides, which are densely packed in quasi-crystalline structures inside a vesicle, opening and dilation of small fusion pores leads to delayed diffusion of peptides from the vesicle (Angleson et al., 1999; Barg et al., 2002). However, although the release time course of NTs from hippocampal neurons was described recently to occur one order of magnitude more slowly than release of small neurotransmitters (Brigadski et al., 2005), the type of vesicle fusion for neuronal release of NTs remained elusive.
The use of the small fluorescent quencher BPB has recently enabled Harata et al. (2006) to distinguish full collapse fusion from kiss and run mechanism in imaging experiments: the kinetics of first fusion pore opening of vesicles with a GFP-tagged cargo can be recorded as a sudden complete drop in vesicular fluorescence in the presence of saturating concentrations of BPB (2 mm) (Harata et al., 2006). If diffusion of the cargo from the vesicle is slow compared with the fusion pore opening/life time, a faster time course and a more pronounced fluorescence decay is expected in the presence of BPB. In fact, our data reveal a strong difference in the fluorescence decay in the absence versus the presence of BPB (Fig. 2), indicating either slow diffusion of BDNF-GFP through a narrow fusion pore or, alternatively, no diffusion of NTs at all through the fusion pore, but onset of release only if subsequent full collapse fusion appears at later time points. Interestingly, the vesicle fluorescence reappeared after washout of BPB during ongoing depolarization (compare Fig. 3 A). In conjunction with the more graded loss of GFP fluorescence and the remaining fluorescence intensity in single vesicles, which undergo release of neurotrophin under control conditions, this indicates the existence of a vesicle pool, which releases NTs via prolonged and/or repetitive fusion pore openings during depolarization. The large scatter in the onset of fusion pore opening for individual vesicles from the same cell (Fig. 3 D) indicates that many of the NT vesicles are not prepared for release (i.e., docked and primed) before stimulation. A similar asynchronous release from individual vesicles has been described for other neuropeptides and seems to be an intrinsic feature of peptide secretion from secretory granules (Han et al., 1999; Barg et al., 2002).
The observed fast (compared with cargo release) onset of NT vesicle fusion pore opening that is evident in the presence of BPB (Fig. 2 A) indicates that the bottleneck for the speed of NT release, in addition to cargo dissolution (cf. Brigadski et al., 2005), is either fusion pore dilation or diffusion of the cargo from the vesicle rather than the onset of the exocytotic event itself.
The role of Ca2+ influx and intracellular Ca2+ stores in NT secretion
As expected for a direct postsynaptic depolarization, NT secretion was critically dependent on Ca2+ influx via VGCCs. Given the high prevalence of L-type VGCCs in the soma and dendrites (Hell et al., 1993), the important role of L-type channels for NT secretion is not surprising.
In contrast, two previous studies that addressed axonal release of endogenous BDNF and NT-3 reported a requirement for activation of N-type Ca2+ channels, whereas inhibition of L-type channels did not affect axonal NT secretion (Balkowiec and Katz, 2002; Wang et al., 2002). This differential dependence on Ca2+ channels likely reflects the different predominance of both types of VGCCs in axons [N-type (Westenbroek et al., 1992, 1995)] versus dendrites [L-type (Hell et al., 1993)] and supports the view that Ca2+ influx is an efficient trigger for NT secretion at either site.
More unexpectedly, however, we observed that a subsequent Ca2+-induced Ca2+ release from internal stores was inevitable for any postsynaptic secretion of NTs to occur, and influx of extracellular Ca2+ alone, even during sustained depolarization, was unable to initiate any NT secretion (Fig. 6). A similar requirement for Ca2+-induced Ca2+ release from internal stores was recently described for bulk secretion of endogenous BDNF from axons of hippocampal neurons (Balkowiec et al., 2002). Interestingly, depolarization-induced bulk secretion of endocytosed BDNF from striatal slices was also described to depend on both Ca2+ influx and Ca2+ release from internal stores (Goggi et al., 2003). Our data now stress a mechanistic role of intracellular Ca2+ stores directly for the Ca2+ influx-induced release process of NTs from postsynaptic structures.
Other studies that addressed elevated K+-induced bulk secretion of NTs from hippocampal neurons described a strict dependence on Ca2+ release from intracellular Ca2+ stores, whereas Ca2+ influx was dispensable (Blochl and Thoenen, 1995; Griesbeck et al., 1999). This independence from Ca2+ influx likely reflected that synaptic networks can bypass Ca2+ influx as a trigger for NT secretion, provided that sufficient amounts of glutamate and acetylcholine are released by elevated K+ to activate the respective metabotropic receptors and subsequent Ca2+ release from internal stores. This is in line with the observed efficient bulk secretion of NTs in response to stimulation of metabotropic acetylcholine and glutamate receptors (Blochl and Thoenen, 1996; Canossa et al., 2001; Balkowiec and Katz, 2002), which activate Ca2+ release from internal stores via IP3 receptors. This release mechanism is clearly distinct from the pathways that elicit synaptic NT secretion in our preparation, which proceeds independent of activation of metabotropic glutamate receptors (Hartmann et al., 2001).
Overall, these data from different groups suggest that a sustained rise in intracellular Ca2+ that is shaped in amplitude and duration by the release of Ca2+ from internal stores is an unequivocal requirement for the NT secretion process. The routes of activation of intracellular Ca2+ stores, however, can be different and depend on the stimulation conditions.
Intracellular Ca2+ stores are well known to participate in the mechanism of dendritic release of neuropeptides (for review, see Ludwig and Pittman, 2003): release of Ca2+ from intracellular stores can either prime the dendritic release machinery for efficient and long-lasting secretion of neuropeptides, or it can cause by itself a sufficient Ca2+ elevation that initiates release (Ludwig et al., 2002; Ludwig and Pittman, 2003). However, a similar obligatory role of intracellular Ca2+ stores in amplifying the initial Ca2+ influx to obtain a sufficient Ca2+ signal for dendritic release (Fig. 6) has not been described, thus far, for other neuropeptides. This release mechanism appears to be unique for neurotrophins and seems to operate for axonal (Balkowiec and Katz, 2002) as well as for dendritic secretion (this study). It is also mechanistically different from the bulk release of neuropeptides/neurotrophins after mobilization of Ca2+ from IP3-dependent internal stores, which is observed after activation of metabotropic acetylcholine and glutamate receptors (see above). The functional significance of the obligatory involvement of internal Ca2+ stores in the release process of NTs remains to be elucidated.
An obvious difference between our results and previous studies lies in the independence of the postsynaptic NT secretion from activation of TTX-sensitive sodium channels (cf. Blochl and Thoenen, 1995; Androutsellis-Theotokis et al., 1996; Kojima et al., 2001). However, this was an expected finding, because in these previous studies, any manipulation that inhibited the overall excitatory drive in the synaptic network (e.g., application of TTX, ceasing glutamatergic transmission) was likely to decrease the bulk NT secretion from the network, which was assayed in these studies. In contrast, in the study by Balkowiec and Katz (2002), which focused on electrical stimulation of very young hippocampal cultures, successful depolarization was dependent on the activation of likely axonal voltage-gated Na+ channels, explaining the TTX sensitivity of BDNF secretion in their study. Our study, however, focused on the postsynaptic secretion process at identified glutamatergic synapses, and our depolarization paradigm (i.e., elevated K+) does not rely on action potential firing but triggers secretion of BDNF-GFP via depolarization induced gating of L-type VGCCs.
After electrical stimulation of glutamatergic synapses, activation of AMPA and/or NMDA receptors can induce the initial Ca2+ influx that triggers the release of NTs (Hartmann et al., 2001). Our new data now go one step further and reveal that postsynaptic NMDAR stimulation is sufficient to elicit postsynaptic secretion of BDNF (Fig. 5 A). This is the first unequivocal demonstration for a direct causal connection between activation of postsynaptic NMDARs and BDNF secretion and suggests that postsynaptic activation of NMDARs during LTP stimulation can be sufficient to evoke BDNF secretion, which then can take part in LTP expression (for review, see Bramham and Messaoudi, 2005). However, our data also reveal that activation of postsynaptic L-type VGCCs can substitute for NMDAR activation in triggering synaptic BDNF secretion, suggesting that both routes of Ca2+ entry into the postsynaptic neuron can initiate BDNF-dependent synaptic plasticity. This is in line with the recently described pivotal role of L-type VGCC in BDNF-dependent LTP in the hippocampal CA1 region (Zakharenko et al., 2003), and our data suggest that activation of L-type VGCCs during induction of this form of LTP could be essential for efficient synaptic BDNF secretion.
Together, Ca2+ influx is essential for triggering NT secretion in response to electrical or synaptic stimulation, whereas the type of Ca2+ channel involved seems to depend on the preponderance of a channel at a given (synaptic) location (compare Fig. 10).
The role of CaMKII in NT secretion
Sustained postsynaptic Ca2+ elevation, causing subsequent activation of CaMKII is a widely accepted key event in LTP induction in CA1 and in hippocampus-dependent memory formation (Silva et al., 1992). Given the importance of long-lasting Ca2+ elevations for NT secretion, we asked whether CaMKII might be directly involved in the postsynaptic secretion of NTs. In fact, postsynaptic release of BDNF and NT-3 was strongly decreased when CaMKII was inhibited (Fig. 6). In our assay system, these data can be reconciled best with a direct action of CaMKII on the postsynaptic NT secretion process (compare Fig. 10). These data are in line with the following: (1) the CaMKII-dependent regulation of catecholamine secretion in PC12 cells (Schweitzer et al., 1995), (2) the CaMKII-dependent regulation of axonal release of NT-3 in the chick optic tectum (Wang et al., 2002), and (3) the CaMKII-controlled, Ca2+-evoked dendritic exocytosis (CEDE) in hippocampal neurons (Maletic-Savatic and Malinow, 1998; Maletic-Savatic et al., 1998). The parallels between this latter study and our own results suggest that the vesicle cargo that was released during CEDE might have been BDNF or NT-3 (cf. Maletic-Savatic et al., 1998). It will be interesting to determine whether these are the same vesicles that deliver new AMPA receptors to potentiated synapses (Shi et al., 1999). Overall, these data provide a possible direct link between postsynaptic activity of CaMKII and BDNF secretion, which could account for, at least in part, the pivotal role of CaMKII in the induction of LTP.
Absence of NT-induced NT secretion from postsynaptic sites
Neurotrophin-induced neurotrophin secretion has been suggested to account for later phases of bulk secretion of BDNF and NT-3 in the hippocampus (Canossa et al., 1997) and in neuroendocrine cells (Kruttgen et al., 1998). Given that binding of neurotrophins to their respective Trk receptors can induce Ca2+ release from IP3-sensitive internal stores, it is not surprising to see that neurotrophins could promote their own secretion, if the relevant Trk receptors are available at the site of secretion. We thus tested whether a similar autocrine loop could account for, at least in part, the postsynaptic NT secretion in our experiments. However, inhibition of Trk signaling altered neither the time course of release nor the amount of secreted BDNF and NT-3, respectively, in our experiments (Fig. 9). Likewise, application of exogenous BDNF under conditions when other sources of Ca2+ elevation were eliminated (i.e., in the presence of nifedipine, DNQX, and APV) also failed to provoke postsynaptic NT secretion (Fig. 9). These data indicate that the time course and the efficiency of NT secretion at postsynaptic sites in our cultures is not shaped by NT-induced NT secretion, not ruling out that Trk-mediated intracellular Ca2+ elevations can become instrumental for NT secretion at other sites of secretion.
Protein kinase A gates synaptic NT secretion
Activation of PKA is involved in presynaptic and postsynaptic forms of activity-dependent synaptic plasticity in the hippocampus, with an important role especially for later stages of LTP (Frey et al., 1993; Huang and Kandel, 1994). At postsynaptic sites, PKA has been described to “gate” (i.e., to facilitate induction of LTP, rather than directly inducing it (Blitzer et al., 1995). A similar gating behavior of PKA for postsynaptic BDNF and NT-3 secretion is indicated by our data: exogenous stimulation of PKA with 8-Br-cAMP did neither induce nor facilitate NT secretion, but depolarization-induced secretion was strongly inhibited when intrinsic cAMP signaling (and thus PKA activity) was reduced (Fig. 8). These results suggest that sustained PKA-dependent phosphorylation of a target protein is required for efficient NT vesicle release, and that the level of basal cAMP in our cultures (cf. Lessmann and Heumann, 1997) is suprathreshold for this gating action of PKA on NT secretion.
At presynaptic sites, PKA has been shown to play an important role in priming transmitter vesicles for release, by regulating the availability of vesicles from the ready releasable pool for exocytosis (Gromada et al., 2001; Nagy et al., 2004). This action is at least in part mediated by PKA-dependent phosphorylation of SNAP-25 (soluble N-ethylmaleimide-sensitive factor attachment protein), which is a component of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex and participates in priming of vesicles (Sutton et al., 1998). It is tempting to speculate that priming of postsynaptic BDNF vesicles can be modulated similarly by PKA.
Interestingly, Patterson et al. (2001) described a PKA-dependent late stage of LTP in the hippocampus that was mediated by downstream activation of the BDNF receptor TrkB. The missing link between PKA and TrkB activation could be the PKA-controlled BDNF release at the stimulated synapses that can be concluded from our data. Such a link is also supported by the modulatory effect of the cAMP/PKA pathway on the secretion of endocytosed BDNF from striatal tissue (Goggi et al., 2003). Together, these data suggest that PKA activation can modulate synaptic plasticity via gating of BDNF release.
In conjunction with our data on the CaMKII dependence of postsynaptic NT secretion (see above), the PKA results suggest that both kinases coregulate (among other processes) postsynaptic secretion of BDNF in certain forms of LTP. A coregulation by PKA and CaMKII of axonal release of NT-3 from retinal ganglion cells in chick tectum has been described previously (Wang et al., 2002), stressing that similar regulatory mechanism could also apply for postsynaptic NT secretion in the hippocampus. It will be challenging to further elucidate the intricate interplay between CaMKII, PKA, and BDNF signaling in synaptic plasticity in the future.
Comparison of presynaptic versus postsynaptic release of NTs
Our study focused on postsynaptic secretion of BDNF and NT-3 at glutamatergic synapse. This approach was chosen to directly relate the activity of postsynaptic signaling mechanisms with the NT secretion process. As also observed in several other studies using overexpressed GFP-tagged NTs, dendritic localization of NT vesicles is, by far, the dominating targeting pattern that is observed in cultured cortical and hippocampal neurons (Hartmann et al., 2001; Kojima et al., 2001; Brigadski et al., 2005). Importantly, dendritic targeting in secretory granules is also observed for endogenous BDNF and NT-3 in hippocampal neurons (Goodman et al., 1996; Swanwick et al., 2004; Brigadski et al., 2005). This rules out that the dendritic targeting of GFP-tagged NTs is an overexpression artifact. This dominating dendritic targeting of NTs in cultured hippocampal neurons, however, does not rule out additional targeting to axons (Hartmann et al., 2001; Kohara et al., 2001). In this study, we concentrated on signaling cascades important for postsynaptic secretion of NTs to account for the importance of this targeting pattern of NTs in the hippocampus.
A previous study that focused on axonal release of NT-3 in the chick optic tectum (Wang et al., 2002) revealed important differences in the secretion process: these authors described a dependence of NT-3 secretion on Ca2+ influx via N-type Ca2+ channels (L-type channels here) and a direct releasing action of PKA (gating of NT secretion here). Nevertheless, important parallels exist, in that also axonal secretion of NT-3 depends, in both cases, on intact intracellular Ca2+ stores and on the activity of CaMKII. It seems important to clearly distinguish between presynaptic and postsynaptic (or axonal vs dendritic) mechanisms of NT secretion, and this might help to avoid any unclear interpretation of NT release data in the future. It is likely that both presynaptic and postsynaptic NT secretion can operate at synapses, but depending on the developmental stage and other specific conditions of a particular synaptic circuit, presynaptic or postsynaptic secretion of NTs could dominate.
Footnotes
-
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grant SFB 553 TP C12, the Stiftung Rheinland-Pfalz für Innovation (V.L.), and DFG Grant GRK 1044 (T.B.). We thank D. Bredt for generously providing PSD-95-GFP, Matthias Hartmann for help with some of the release experiments, Sabine Rickheim-Lowack for excellent technical assistance, and Heiko Luhmann for generous support.
- Correspondence should be addressed to Dr. Volkmar Lessmann, Institute of Physiology, Otto-von-Guericke-University, Leipziger Strasse 44, 39120 Magdeburg, Germany. lessmann{at}med.ovgu.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}