
Abstract
Elevated brain glutamate with activation of neuronal glutamate receptors accompanies neurological disorders, such as epilepsy and brain trauma. However, the mechanisms by which excitotoxicity triggers neuronal injury are not fully understood. We have studied the glutamate receptor agonist kainic acid (KA) inducing seizures and excitotoxic cell death. KA caused the disintegration of the endoplasmic reticulum (ER) membrane in hippocampal neurons and ER stress with the activation of the ER proteins Bip, Chop, and caspase-12. Salubrinal, inhibiting eIF2α (eukaryotic translation initiation factor 2 subunit α) dephosphorylation, significantly reduced KA-induced ER stress and neuronal death in vivo and in vitro. KA-induced rise in intracellular calcium was not affected by Salubrinal. The results show that ER responses are essential parts of excitotoxicity mediated by glutamate receptor activation and that Salubrinal decreases neuronal death in vivo. Inhibition of ER stress by small molecular compounds may be beneficial for treatment of various neuronal injuries and brain disorders.
- kainic acid
- hippocampus
- salubrinal
- PERK
- eIF2α
- caspase-12
Introduction
Neuronal degeneration and dysfunction occur in many human neurological diseases leading to severe incapabilities and increased suffering of patients. The causes of cell loss in these disorders are not fully understood but involve both caspase-dependent and -independent pathways (Leist and Jäättelä, 2001; Yuan et al., 2003). Cell death in neurons is controlled by the activity of signaling pathways and proteins with a cross talk between various organelles (Ferri and Kroemer, 2001). Mitochondria are crucially involved in cell death control and dysfunctions as shown also in many brain diseases (Lindholm et al., 2004). Recently, other organelles, such as the endoplasmic reticulum (ER), mediating cell stress responses have been linked to human diseases such as diabetes, Parkinson's disease, and amyotrophic lateral sclerosis (Wootz et al., 2004; Lindholm et al., 2006; Wu and Kaufman, 2006).
ER is an important cell organelle that is responsible among others for correct folding and sorting of proteins (Boyse and Yuan, 2006). ER functions can be disturbed by different insults such as accumulation of unfolded proteins and changes in calcium homeostasis (Verkhratsky, 2005; Boyse and Yuan, 2006). Disturbed ER functions induce expression of chaperones, attenuate protein translation, and activate ER-associated degradation (Bertolotti et al., 2000; Boyse and Yuan, 2006). This occurs by the activation of ER sensor proteins controlled by the chaperone Bip/Grp78 (Bip), which is localized in the ER (Bertolotti et al., 2000). ER stress leads to activation of PRK (RNA-dependent protein kinase)-like ER protein kinase (PERK)/pancreatic eukaryotic translation initiation factor 2 subunit α (eIF2α) kinase, activating transcription factor-6 (ATF6), and the inositol-requiring enzyme 1 (IRE1), which in turn activate distinct signaling cascades mediating the ER stress response (Lee et al., 2002; Ma et al., 2002; Liu et al., 2003). ER stress has been widely studied for its role in the unfolded protein response (UPR), in cellular homeostasis and in calcium regulation (Verkhratsky, 2005; Boyse and Yuan, 2006). Apart from the UPR that is mainly adaptive and restorative in function, prolonged ER stress can trigger mitochondria-dependent and -independent forms of cell death (Breckenbridge et al., 2003; Rao et al., 2004; Hetz et al., 2006). However, little is known about the role of ER responses in mediating pathophysiological reactions in acute brain injuries. Here we have studied excitotoxic brain injury induced by kainic acid (KA) and ER stress and its inhibition for neuroprotection against cell degeneration caused by glutamate receptor overstimulation occurring in different brain disorders.
Materials and Methods
Animals
Experiments were approved by the ethical committee and performed in accordance with the European Communities Council Directive (86/609/EEC). Every attempt was made to reduce the number and to minimize pain and suffering of animals. Adult male Wistar rats (200–300 g; B&K, Hull, UK) were given injections of KA (Calbiochem, Espoo, Finland) into the lateral ventricle (0.35 μg/μl) in a volume of 0.5 μl per side, as described previously (Mudo et al., 1995), and killed at different time points from 3 to 48 h. Controls received an equal volume of saline. Salubrinal (Sal; Calbiochem) was dissolved in dimethylsulfoxide (Sigma, Helsinki, Finland) and further diluted with saline. Rats were given injections of Sal 30 min before KA either intracerebroventricularly (1 μl) using a 75 μm solution or intraperitoneally (0.1 ml) with 1 mg/kg. Rats were decapitated in deep anesthesia, and brains were rapidly dissected, frozen in isopentane, cooled in liquid nitrogen, and stored at −70°C until analysis. The right side of the brain was used for histology and the left for Western blot analyses.
Neuronal cultures
Hippocampal neurons prepared from embryonic day 17 rats (Harlan, Horst, The Netherlands) were cultured for 7 d on poly-ornithine (Sigma)-coated dishes in Neurobasal medium with B27 supplement (Invitrogen, Espoo, Finland) as described previously (Korhonen et al., 2001, 2003; Sokka et al., 2005). Different concentrations of KA (Calbiochem) or glutamate (Sigma) were added to the cells for various periods of time. In some experiments, 50 μm Sal, 1 mm kynurenic acid (Sigma), and 10 μm MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate; Sigma] were added 30 min before KA. Concentrations of 50 and 20 μm BAPTA-AM [1,2-bis(o-aminophenoxy) ethane-N,N,N',N'-tetraacetic acid; Sigma] were added 2 h and 30 min, respectively, before KA to lower increases in cell calcium (Zafra et al., 1991). Neuronal survival was determined by counting nuclei with fragmented/condensed nuclei using Hoescht blue (Sigma) or using staining with propidium iodine (Sigma), which is excluded from viable cells (Sundberg et al., 2006). In some experiments, we also used the MTT (Sigma) assay for cell viability (Korhonen et al., 2001, 2004).
Western blotting
Hippocampal tissue and neurons were lysed using ice-cold radioimmunoprecipitation assay buffer (150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1% SDS, 50 mm Tris-HCl, pH 8.0) supplemented with protease inhibitor mixture (Roche, Espoo, Finland). Equal amounts of protein were subjected to SDS-PAGE and blotted onto nitrocellulose filters (Amersham Biosciences, Helsinki, Finland). These were first incubated for 1 h in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20, and 5% skimmed milk and then overnight at 4°C with primary antibodies: anti-Bip (1:1000; BD Biosciences, Helsinki, Finland); anti-Chop (1:250; Santa Cruz Biotechnology, Heidelberg, Germany); anti-caspase-12 (1:2000; Chemicon, Helsinki, Finland); anti-phosphorylated (p)-PERK (1:500) and anti-PERK (1:500; both from Santa Cruz Biotechnology); anti-p-eIF2 (1:1000) and anti-eIF2 (1:1000; both from Cell Signaling, Espoo, Finland); and anti-actin (1:5000; Sigma). After washing, the filter was incubated with horseradish peroxidase-conjugated secondary antibodies (1:2500; Jackson ImmunoResearch, Espoo, Finland), followed by detection using enhanced chemiluminescence (Pierce, Helsinki, Finland). Quantification was performed using GelDoc (Bio-Rad, Espoo, Finland).
Immunochemistry
In vivo. Freshly frozen 10 μm rat brain sections were cut on a cryostat, mounted onto Superfrost slides (Metzel-Gläser, Braunschweig, Germany), and fixed for 10 min at −20°C using acetone–methanol (1:1). Slides were first incubated for 1 h with 5% bovine serum albumin (BSA: Sigma)/PBS/0.1% Triton X-100 at room temperature and then overnight at +4°C with anti-Bip (1:100), anti-Chop (1:100), anti-p-eIF2α (1:200), anti-p-PERK (1:100), anti-active caspase-3 (1:200; Cell Signaling), anti-caspase-12 (1:200), anti-ATF6 (1:100; Pierce), and anti-p-c-jun (1:200; Santa Cruz Biotechnology) antibodies. After washing with PBS/0.05% Tween 20, secondary Cy3- or Cy2-conjugated anti-mouse antibody (1:300; Jackson ImmunoResearch) was added for 1 h, and the slides were washed and mounted using gel-mounting medium (Sigma). Controls without a primary antibody showed no staining.
In vitro. Neurons were fixed for 20 min using 4% paraformaldehyde, incubated for 1 h using 5% BSA/0.1% Triton X-100, and stained as above.
Fluoro-Jade C and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining
Frozen 10 μm sections were immersed for 5 min in 1% sodium hydroxide/80% ethanol, rinsed for 2 min in 70% ethanol, and then rinsed in distilled water. Potassium permanganate solution (0.06%) was added for 10 min, followed by a 10 min incubation in 0.0001% Fluoro-Jade C (Chemicon) (Korhonen et al., 2001; Schmued et al., 2005). After washing, the sections were air dried for 10 min at 50°C, cleared in xylene, and mounted in dibutyl phthalate xylene (Sigma). Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining (Roche) was done as described previously (Korhonen et al., 2001, 2003). The number of Fluoro-Jade- and TUNEL-positive cells was counted at the same bregma level in control and treated rats (n = 4–8).
Analysis of ER fragmentation
Hippocampal neurons were loaded for 30 min with 100 nm Blue-white ER-tracker (Invitrogen, Espoo, Finland), and different concentrations of KA or glutamate were added for 60 min after changing the medium. Cells were observed under a fluorescent microscope (Leica, Espoo, Finland) using UV filters. To visualize ER membranes, cells were also stained using an anti-inositol-3-phosphate receptor (IP3-R) antibody (1:100; Chemicon).
Ca2+ imaging
Conventional wide-field fluorescence microscopy integrated in CellR Olympus (Tokyo, Japan) microscopy station was used to monitor Ca2+ transient responses. Dishes were pretreated with vehicle (1% dimethylsulfoxide) or 50 μm Sal for 30 min (short-term treatment) or for 20–24 h (long-term treatment). Cultures were washed and incubated for 40 min at room temperature with fura-2 AM dye (2 μm; Sigma). A concentration of 50 μm KA or 50 μm glutamate was applied for 1 min through a bath perfusion system. Ca2+ transient responses were measured after background subtraction as the ratio between the fluorescence intensity at 340 and 380 nm, respectively (Grynkiewicz et al., 1985). Plots were made using Microcal (Northampton, MA) Origin 6.0 software. Peak amplitude of Ca2+ responses was calculated as mean amplitude of the five time points after agonist application. Statistical differences were estimated using the t test.
Quantitative PCR
RNA was prepared from hippocampal neurons and cDNA synthesized using 50 U of SuperScript II reverse transcriptase and components given by the vendor (Invitrogen). Quantitative PCR was performed using LightCycler (Roche) and the following primers: BiP: forward 5′-AAGGTGAACGACCCCTAACAAA-3′, reverse 5′-GTCACTCGGAGAATACCATTAACATCT-3′; Chop: forward 5′-GCCTTTCGCCTTTGAGACAGT-3′, reverse 5′-TGAGATATAGGTGCCCCCAATT-3′; activating transcription factor-4 (ATF4), forward 5′-CTACTAGGTACCGCCAGAAG-3′, reverse 5′-GCCTTACGGACCTCTTCTAT-3′; β-actin: forward 5′-CACACTGTGCCCATCTATGA-3′, reverse 5′-CCATCTCTTGCTCGAAGTCT-3′. Amplification was performed using an initial 10 min step at 95°C, followed by 50 cycles with 15 s at 95°C, 5 s at 60°C, and 4–12 s at 72°C, and with a final extension for 10 min at 72°C. Specificity of the product was confirmed by melting-curve analysis. Quantification was done from data on the reaction kinetics, and expression levels were related to β-actin. To study splicing of XBP, reverse transcription-PCR and the following primers were used: forward 5′-AGAGTAGCAGCACAGACTGCGCG-3′; reverse 5′-GGAACTGG-GTCCTTCTGGGTA-3′.
Quantification and statistics
Statistical comparisons were performed using one-way ANOVA, followed by Student's t test. For each time point in vivo, four to eight rats were used, and the in vitro assays were repeated more than three times. p ≤ 0.05 was considered significant.
Results
Activation of KA receptors induces ER responses in hippocampal neurons
To study the role of ER in excitotoxic neuronal response, we used hippocampal neurons that abundantly express KA glutamate receptors (Olney et al., 1974; Nadler et al., 1978). The addition of 100 μm KA rapidly induced the fragmentation of the ER membrane shown by the ER-tracker dye and by staining with antibodies against IP3-R (Fig. 1). This concentration has been shown to induce a delayed degeneration of the neurons (Korhonen et al., 2001). To show that the neurons do not undergo a rapid necrotic response after KA, we studied lower concentrations of the compound (25 μm) that also induced ER fragmentation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Treatment of cells for 1 h using 25–200 μm KA did not cause any damage to the neuronal plasma membrane as shown by propidium iodine exclusion, in contrast to 300 μm glutamate that did so to some extent (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results showed that the ER fragmentation produced by KA is not secondary to necrosis that may follow excessive glutamate receptor activation.

KA induces ER membrane changes in hippocampal neurons. A concentration of 100 μm KA was added to the hippocampal neurons cultured as described in Materials and Methods. The integrity of ER was analyzed by the ER-tracker and by immunostaining for IP3-Rs. Arrows show disintegration of ER membranes 30 and 60 min after KA. Scale bar: top panels, 25 μm; middle and bottom panels, 10 μm.
We then studied whether KA also triggers ER stress as evident from alterations in ER stress proteins. As shown before, KA administration in vivo induces cell death particularly in the hippocampal CA3 region at 24 h (Fig. 2a) (Korhonen et al., 2001). The ER chaperone Bip was elevated in the hippocampus at 12 h with a maximum at 24 h (Fig. 2b). Similarly, the ER stress-inducible leucine zipper-containing transcription factor Chop/Gadd153 (Chop) was increased at 24 h after KA (Fig. 2b). Bip- and Chop-positive cells increased particularly in CA3 neurons after KA, with an accumulation of Chop within neuronal nuclei (Fig. 2c). In KA-treated hippocampal neurons, Bip and Chop mRNA increased approximately twofold (Fig. 2d). ATF6 that controls the expression of ER sensors was also activated by KA, as shown both in vivo and in vitro (Fig. 2e).

KA induces nerve cell death and ER stress in hippocampal neurons. a, Sections were prepared from rat hippocampus after KA injections. Cell degeneration was analyzed by Fluoro-Jade C staining. Arrows show neuronal loss in the CA3 region at 24 h. DG, Dentate gyrus. b, Bip and Chop in hippocampus at different times after KA as detected by immunoblotting and specific antibodies. β-Actin was used as the control. p < 0.01 for control versus 24 h KA (n = 3). c, Immunostaining of Bip and Chop in the hippocampal CA3 region after KA. d, Bip and Chop mRNA levels in cultured neurons treated with 100 μm KA analyzed using quantitative PCR as described in Materials and Methods. Bip, p < 0.05 for control versus 24 h KA (n = 5); Chop, p < 0.05 control versus 24 h KA (n = 5). e, Immunostaining for ATF6 in the rat hippocampal CA3 area (top panels) and in cultured neurons (bottom panels). ATF6 is present in nuclei after KA. Scale bars: a, 500 μm; c, d, 25 μm. Error bars indicate SEM. C, Control.
KA induces PERK and eIF2α phosphorylation in hippocampal neurons
To study which other ER pathways are activated by KA in the neurons, we used antibodies against phosphorylated forms of PERK and its downstream target, eIF2α. A concentration of 100 μm KA within 30 min increased the levels of p-PERK and p-eIF2α in the cultured neurons (Fig. 3a,b), whereas total levels of the proteins did not change (data not shown). An increased number of p-PERK- and p-eIF2α-immunoreactive CA3 neurons was also evident in vivo after KA (Fig. 3c). Increases in p-PERK and p-eIF2α by 100 μm KA were transient both in cultures (Fig. 3a) and in vivo (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Studies of the dose–response revealed that 25 μm KA increased eIF2α and Bip in the hippocampal neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This was also observed using 75 μm glutamate to stimulate the neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Treatment with KA also induced the expression of ATF4 that is downstream of PERK (Fig. 3d). Splicing of XBP1 mRNA, activated via the IRE1 pathway did not occur significantly after KA (Fig. 3e), whereas increased p-c-Jun was readily observed (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Data showed that the effect of KA on p-eIF2α was inhibited by kynurenic acid (Fig. 3f), blocking glutamate receptors (Zafra et al., 1991). These results demonstrate that KA through glutamate receptor activation induces ER stress with the activation of sensor proteins and downstream signaling in the hippocampal neurons.
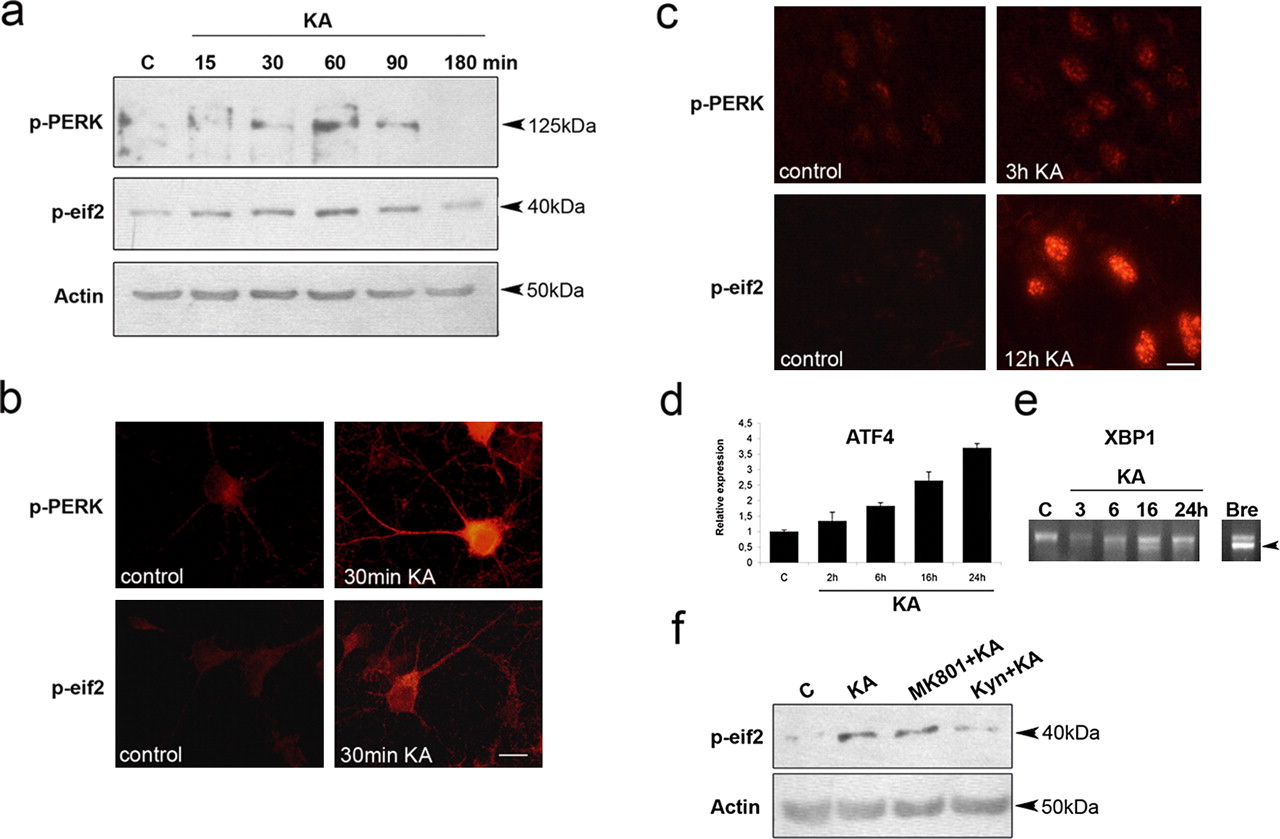
KA induces PERK and eIF2α phosphorylation in hippocampal neurons. a, p-PERK and p-eIF2α was studied in hippocampal neurons treated with KA using immunoblotting and specific antibodies. b, Immunostaining of p-PERK and p-eIF2α in cultured neurons. Note the increased staining 30 min after KA. c, Immunostaining of p-eIF2α and p-PERK in CA3 in hippocampal neurons in vivo after KA. d, ATF4 mRNA levels in cultured neurons treated with 100 μm KA and analyzed by quantitative PCR. Note the increase in expression from 6 h onward. p < 0.01 for control versus 16 and 24 h KA (n = 5). e, Splicing accompanying XBP1 activation was analyzed in the neurons using reverse transcription-PCR. BrefeldinA (Bre; 0.5 μg/ml) as a positive control induced XBP1 activation but not KA. f, Hippocampal neurons treated for 1.5 h with 100 μm KA alone or in the presence of 10 μm MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] blocking NMDA receptors and 1 mm kynurenic acid (Kyn) inhibiting glutamate receptors. Scale bars: b, 25 μm; c, 10 μm. Error bars indicate SEM. C, Control.
KA increased cleavage of caspase-12 in hippocampal neurons
Among cell-death proteins, caspase-12 resides in the ER membrane and can be cleaved by ER stress (Nakagawa et al., 2000; Nakagawa and Yuan, 2000). In rat hippocampus, caspase-12 was cleaved 12 h after KA (Fig. 4a), with the immunoreactivity present within neuronal nuclei in the CA3 region (Fig. 4b). In cultured neurons, the cleavage of caspase-12 occurred rapidly after KA addition, showing the existence of a fast signal from the receptor (Fig. 4c). Incubation of the neurons in the presence of BAPTA-AM to chelate cell calcium (Zafra et al., 1991) decreased caspase-12 cleavage (Fig. 4d). This shows that elevation in calcium occurring after KA (see supplemental Fig. 4, available at www.jneurosci.org as supplemental material) and the stimulation of glutamate receptors causes caspase-12 cleavage at the ER. Apart from calcium, KA is known to activate other signaling cascades downstream of the receptor (Lerma, 2003).

Cleavage of caspase-12 after KA in hippocampal neurons. a, Caspase-12 in rat hippocampus at different times after KA injections as detected by immunoblotting. β-Actin was used as the control. b, Immunostaining in CA3 neurons in vivo 12 h after KA showed nuclear localization of caspase-12. Scale bar, 25 μm. c, Caspase-12 in cultured hippocampal neurons was cleaved 15 min after 100 μm KA. d, Lowering of intracellular calcium by 50 μm BAPTA-AM inhibited the effect of 100 μm KA on caspase-12. C, Control; B, BAPTA.
Sal inhibits KA-induced neuronal death in vivo
Sal is an inhibitor of eIF2α dephosphorylation that was recently shown to counteract ER stress-induced cell degeneration in culture (Boyce et al., 2005). To study the effect of Sal on hippocampal neurons, we added Sal in combination with KA. Sal decreased the number of dying neurons with condensed nuclei by ∼70% (Fig. 5a) (p < 0.01), as well as increased neuronal survival shown by the MTT assay (data not shown). Sal also reduced increases in Bip and the caspase-12 cleavage because of KA (Fig. 5b) and decreased the number of active caspase-3-positive neurons in KA-treated cultures (Fig. 5c). To study whether Sal may influence the calcium signal in neurons induced by KA, we measured increases in calcium in control and Sal-treated cells. As shown in experiments using fura-2, neither short-term nor long-term treatment with Sal affected the calcium elevation in neurons after stimulations by KA and glutamate (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). This shows that Sal does not act upstream of ER in reducing calcium rise induced by KA treatments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sal inhibits KA-induced neuronal death in vivo. a, Number of degenerating hippocampal neurons in culture treated for 24 h with 100 μm KA alone or in the presence of 50 μm Sal. Insets show cells stained with Hoescht blue to reveal nuclear morphology. Sal protected against KA-induced cell death. p < 0.05; n = 6. b, Immunoblots of Bip and caspase-12 in cultured neurons treated with KA and Sal. Note the inhibition of caspase-12 cleavage and reduced Bip in KA-treated cells by Sal. c, Immunostaining for active caspase-3 was done in control and 100 μm KA and 50 μm Sal-treated neurons. Insets show immunopositive neurons. p < 0.05 for control versus KA and for Sal plus KA versus KA (n = 6). d, Sal was injected intracerebroventricularly or intraperitoneally 30 min before KA. The number of degenerating neurons was assayed by Fluoro-Jade C staining in the hippocampal CA3 region at 24 h. Note the reduced cell degeneration by Sal treatments. p < 0.01, control versus KA and for Sal plus KA versus KA (n = 6–8). e, Immunostaining for active caspase-3 and TUNEL in the CA3 region after KA and Sal treatments. f, Sal induces eIF2α phosphorylation in hippocampal CA3 neurons in vivo. Sal was given to rats 1 μl of 75 μm intracerebroventricularly or 1 mg/kg intraperitoneally, and the hippocampus was analyzed 24 h later. Scale bars: a, 25 μm; c, 50 μm; d, 500 μm; f, 150 μm. Error bars indicate SEM. C, Control.
As in cultures, Sal administrated in vivo was able to protect against excitotoxic neuronal death in the hippocampus. Thus, the number of degenerating CA3 neurons was reduced by 75% in Sal-treated rats compared with controls after intracerebroventricular injections (p < 0.01; n = 8) and by 60% after intraperitoneal injections of Sal (Fig. 5d) (p < 0.01; n = 6). Sal also decreased by ∼70% the number of active caspase-3- and TUNEL-positive CA3 neurons in the rat hippocampus (Fig. 5e). The levels of p-eIF2α were increased by Sal in the rat hippocampus, in line with eIF2α as a target for its action (Fig. 5f). These results show that Sal is able to penetrate into brain tissue in vivo and thereby affords significant neuroprotection against excitotoxic cell degeneration in brain.
Discussion
KA is known to activate non-NMDA glutamate receptors and trigger a delayed type of excitotoxic cell death in vulnerable brain regions, such as in the hippocampus and amygdala (Olney et al., 1974; Nadler et al., 1978; Korhonen et al., 2001). The mode of cell death caused by KA includes activation of caspases as well as changes in expression of various pro- and anti-apoptotic molecules (Korhonen et al., 2001, 2003; Sokka et al., 2005). Members of the Bcl-2 and IAP (inhibitor of apoptosis protein) families have been shown to modulate the KA-mediated cell death (Lopez et al., 1999; Korhonen et al., 2003; Sokka et al., 2005). Caspase inhibitors can afford partial protection against KA-induced neuronal death, indicating that these enzymes are involved in excitotoxic injury (Kondratyev and Gale, 2000; Korhonen et al., 2001). However, the precise signals and pathways by which glutamate receptor activation triggers neuronal death have remained elusive.
In this study, we show that neuronal degeneration mediated by KA involves ER stress with the activation of ER sensors such as the PERK, ATF6, and IRE1 pathways. Preceding these, KA induced fragmentation of the ER membrane in neurons. Studies using specific blockers showed that the effects of KA on ER are attributable to activation of glutamate receptors. KA also induced cleavage of caspase-12 that occurred rapidly with the presence of the protein in neuronal nuclei. The translocation of caspase-12 into cell nuclei has been reported before in cultured myoblasts after ER stress and may be related to the ensuing cell death (Fujita et al., 2002). Activation of KA receptors is known to induce different signaling pathways and ion fluxes including elevation of calcium in neurons (Lerma, 2003). We observed that blocking calcium by BAPTA inhibited caspase-12 cleavage, demonstrating that increased calcium can trigger the effect of KA on this ER caspase.
Previously, caspase-12 cleavage has been observed in hippocampal neurons lacking the calcium-binding protein hippocalcin (Korhonen et al., 2005). In addition, cells lacking caspase-12 are resistant against degeneration induced by the amyloid β peptide and by tunicamycin causing ER stress (Nakagawa et al., 2000; Nakagawa and Yuan, 2000). The precise role of caspase-12 in ER stress-mediated cell death is not clear, however, because the human gene shows large deletions (Obeng and Boise, 2005). Data showed that Sal treatment also reduced caspase-3 activation with an increase in neuronal viability. This shows that other caspases than caspase-12 are activated by KA and ER stress and contribute to the excitotoxic neuronal injury.
Apart from caspases, KA induced the transcription factor Chop in the hippocampal neurons. Chop has been linked to the ER stress-induced cell death and may act partly by inhibiting Bcl-2 (Urano et al., 2000; McCullough et al., 2001). ER stress can also, via the IRE1 kinase, activate the JNK pathway involved in cell-death control. Previously, JNK-3 was shown to be crucial for excitotoxic damage in the brain (Yang et al., 1997), and we also observed an increase in downstream p-c-Jun levels in KA-treated hippocampal neurons. It has also been shown that Grp78/Bip that suppress ER stress protects neurons against glutamate-induced excitotoxicity (Yu et al., 1999). It is likely that the different signals activated downstream of ER stress interact with and facilitate each other in inducing neuronal damage after KA treatment.
Small molecular compounds acting on intracellular pathways have emerged as promising cytoprotective agents with the capacity to inhibit cell death and restore function. Sal significantly reduced KA-induced neuronal degeneration both in cultured neurons and in hippocampal neurons in vivo. The effect of Sal was mediated via the inhibition of ER stress as shown by effects on caspase-12, caspase-3, and Bip. Previously, Sal was shown to inhibit dephosphorylation of eIF2α and to counteract ER stress in cultured cells (Boyce et al., 2005). As shown by our data, the protective effect of Sal in vivo was robust, although not complete. This suggests that other pathways not blocked by Sal are also involved in neuronal death after the brain insult. It is known that ER responses interact with other cell organelles, including mitochondria. Dysregulated mitochondrial functions with disturbed calcium homeostasis have been considered to underlie excitotoxic and other brain injuries (Ankarcrona et al., 1995). The present results also demonstrate an unprecedented involvement of ER stress responses in excitotoxic neuronal damage induced by KA. The effect of on ER occurred rapidly and was related to an increase in calcium and the induction of different signaling pathways from the ER after KA. Data obtained with Sal showed that the inhibition of ER stress is also neuroprotective in vivo. Sal and other compounds preventing ER stress may therefore be of value in novel therapies for excitotoxic and other brain disorders.
Footnotes
- Received October 2, 2006.
- Revision received November 27, 2006.
- Accepted December 15, 2006.
-
This work was supported by the Academy of Finland, the Sigrid Juselius Foundation, the Arvo and Lea Ylppö Foundation, Liv och Hälsa, the Magnus Ehrnrooth Foundation, the Maud Kuistila Foundation, Fondi di Ateneo at University of Palermo, Uppsala University, and the Minerva Foundation. A.-L.S. received a scholarship from von Frenckell's Foundation, and S.R. received a scholarship from the Finnish Medical Society (Finska Läkaresällskapet). We thank E. Lehto and J. Mäkelä for excellent technical assistance and T. Nyman and S. Laitinen for help with PCR.
- Correspondence should be addressed to Dr. Laura Korhonen, Minerva Medical Research Institute, Biomedicum Helsinki, Haartmaninkatu 8, FIN-00290 Helsinki, Finland. Laura.T.Korhonen{at}helsinki.fi
- Copyright © 2007 Society for Neuroscience 0270-6474/07/270901-08$15.00/0