

Abstract
The AMPA receptor subunit glutamate receptor 1 (GluR1 or GluR-A) contributes to amygdala-dependent emotional learning. It remains unclear, however, to what extent different amygdala pathways depend on GluR1, or other AMPA receptor subunits, for proper synaptic transmission and plasticity, and whether GluR1-dependent long-term potentiation (LTP) is necessary for auditory and contextual fear conditioning. Here, we dissected the role of GluR1 and GluR3 (GluR-C) subunits in AMPA receptor-dependent amygdala LTP and fear conditioning using knock-out mice (GluR1−/− and GluR3−/−). We found that, whereas LTP at thalamic inputs to lateral amygdala (LA) projection neurons and at glutamatergic synapses in the basal amygdala was completely absent in GluR1−/− mice, both GluR1 and GluR3 contributed to LTP in the cortico-LA pathway. Because both auditory and contextual fear conditioning were selectively impaired in GluR1−/− but not GluR3−/− mice, we conclude that GluR1-dependent synaptic plasticity is the dominant form of LTP underlying the acquisition of auditory and contextual fear conditioning, and that plasticity in distinct amygdala pathways differentially contributes to aversive conditioning.
Introduction
One basic mechanism that is thought to be involved in memory formation is synaptic plasticity mediated by AMPA receptors containing the glutamate receptor 1 (GluR1) subunit (Zamanillo et al., 1999; Reisel et al., 2002). Mice lacking the GluR1 subunit both fail to express hippocampal CA3-to-CA1 long-term potentiation (LTP) and exhibit impaired spatial working memory. However, both LTP and spatial working memory are restored on selective reexpression of GluR1 in hippocampal pyramidal neurons (Mack et al., 2001; Schmitt et al., 2005). GluR1 is involved in many forms of activity-dependent synaptic plasticity in brain regions other than the hippocampus (Malinow and Malenka, 2002). However, the existence of GluR1-independent forms of LTP (Hoffman et al., 2002; Jensen et al., 2003) indicates that other subunit combinations, such as GluR2/3 (GluR-B/C), may also play an important role (Malinow and Malenka, 2002) (but see Meng et al., 2003).
Here, we studied the function of GluR1 and GluR3 in projection neurons in two nuclei of the basolateral amygdaloid complex (BLA), the lateral (LA) and the basal (BA) amygdala. The BLA plays an important role during the formation of a Pavlovian association between a conditioned stimulus (CS) and an aversive unconditioned stimulus (US) (LeDoux, 2000; Davis and Whalen, 2001; Maren, 2001). Whereas the LA is thought to represent the site of convergence between a discrete auditory CS and the US during auditory fear conditioning (Romanski and LeDoux, 1992; Nader et al., 2001), the BA, by means of its strong anatomical and functional interactions with the hippocampal formation (Maren and Fanselow, 1995; Pitkanen et al., 2000), may constitute an important substrate integrating BLA and hippocampus-dependent context–US associations (Kim and Fanselow, 1992; Phillips and LeDoux, 1992; Calandreau et al., 2005).
In vivo and in vitro electrophysiological studies have identified cellular correlates of auditory fear conditioning, such as NMDA receptor-dependent LTP (Maren and Quirk, 2004; Sigurdsson et al., 2007). The mechanisms underlying amygdala LTP have been studied most extensively in the thalamo- and cortico-LA pathways (Sigurdsson et al., 2007), where both GluR1/2 and GluR2/3 subunit combinations are thought to be expressed (Farb and LeDoux, 1997, 1999; Radley et al., 2007). Whereas in vivo studies indicate that the thalamo-amygdala pathway is rapidly potentiated during the acquisition of conditioned fear (Quirk et al., 1995, 1997), the relative contribution of these two pathways to the acquisition of auditory fear conditioning is still a matter of debate. Furthermore, a recent study has demonstrated, using viral transfection techniques, that disrupting GluR1 trafficking blocks thalamo-amygdala LTP, but only partially reduces the acquisition of auditory fear conditioning (Rumpel et al., 2005). We therefore examined the contribution of GluR1 and GluR3 to synaptic transmission and plasticity in different amygdala pathways. Although our study reveals pathway-specific involvements of GluR1- and GluR3-dependent mechanisms to glutamatergic synaptic transmission and plasticity, the former appears to dominate during auditory and contextual fear conditioning.
Materials and Methods
Gene-targeted mice
The gene-targeted mouse lines for the GluR1 (GluR-A) and GluR3 (GluR-C) knock-out were generated in the laboratory of R. Sprengel and were published and described in detail by Zamanillo et al. (1999) and Sanchis-Segura et al. (2006), respectively. In brief, in both targeting events mouse R1-embryonic stem cells (Nagy et al., 1993) were used to manipulate the Gria1 and Gria3 alleles. R1-embryonic stem cell clones that were identified by Southern blot as being positive for the targeting events were injected into C57BL/6/N blastocysts to produce chimeric animals. The chimeric male mice were then backcrossed for more than six generations with C57BL/6/N animals. Both the GluR1 and the GluR3 knock-out mice are kept as a heterozygous line. For individual experiments, the colonies of wild-type (WT) and knock-out mice were simultaneously produced by mating of heterozygous mice.
Whole-cell recordings
Standard procedures were used to prepare 350- to 400-μm-thick coronal slices from 4- to 5-week-old male wild-type, GluR1−/−, and GluR3−/− mice following a protocol approved by the Veterinary Department of the Canton of Basel-Stadt (Humeau et al., 2003). Briefly, the brain was dissected in ice-cold artificial CSF (ACSF), mounted on an agar block, and sliced with a vibratome (Leica VT 1000; Leica, Wetzlar, Germany) at 4°C. Slices were maintained for 45 min at 35°C in an interface chamber containing ACSF equilibrated with 95% O2/5% CO2 and containing the following (in mm): 124 NaCl, 2.7 KCl, 2 CaCl2, 1.3 MgCl2, 26 NaHCO3, 0.4 NaH2PO4, 18 glucose, 4 ascorbate, and then for at least 45 min at room temperature before being transferred to a superfusing recording chamber. Whole-cell recordings from LA projection neurons were performed at 30–32°C. Neurons were visually identified with infrared videomicroscopy using an upright microscope equipped with a 40× objective (Olympus, Tokyo, Japan). Patch electrodes (3–5 MΩ) were pulled from borosilicate glass tubing and normally filled with a solution containing the following (in mm): 120 K-gluconate, 20 KCl, 10 HEPES, 10 phosphocreatine, 4 Mg-ATP, and 0.3 Na-GTP (pH adjusted to 7.25 with KOH or CsOH, respectively, 295 mOsm). For voltage-clamp experiments, K-gluconate was replaced by equimolar Cs-gluconate. All experiments were performed in the presence of the GABAA receptor blocker picrotoxin (100 μm). In current-clamp recordings, the membrane potential was kept manually at −70 mV. Data were recorded with an Axopatch 200B, filtered at 2 kHz, and digitized at 10 kHz. In all experiments, series resistance was monitored throughout the experiment by applying a hyperpolarizing current or voltage pulse. If the series resistance changed by >15%, the data were not included in the analysis. Data were acquired and analyzed with ClampEx9.2, ClampFit9.2 (Molecular Devices, Palo Alto, CA), and the Mini Analysis Program (Synaptosoft, Decatur, GA). Monosynaptic EPSPs exhibiting constant 10–90% rise times and latencies were elicited by stimulation of afferent fibers with a bipolar twisted platinum/10% iridium wire (25 μm diameter). Although we never observed any antidromic spikes, we cannot exclude the possibility that some fibers originating from LA projection neurons were stimulated. Miniature EPSCs (mEPSCs) were recorded in the presence of the sodium channel blocker TTX (1 μm) at −70 mV and analyzed off-line by a computer program (Mini Analysis Program). mEPSC amplitudes and decay time constants were obtained by monoexponentially fitting the decay of each individual event. To construct cumulative histograms, an equal number of events was randomly selected from each neuron and pooled. To record quantal events from defined afferent pathways, stimulation-evoked EPSCs were desynchronized by replacing extracellular Ca2+ with Sr2+ (2 mm). The threshold for Sr2+-mEPSC detection was set to 5 pA and at least 120 events were analyzed per cell. To compare the AMPA/NMDA ratio of evoked synaptic transmission, the AMPA component was measured as the EPSC peak amplitude at −70 mV; the NMDA component was determined by measuring the current amplitude at 100 ms after EPSC onset at +30 mV. LTP was induced by pairing afferent stimulation (four times 100 Hz; 1 s; 0.1 Hz) with postsynaptic depolarization to −20 mV. LTP was quantified for statistical comparisons by normalizing and averaging EPSP slopes during the last 5 min of experiments relative to the 5 min of baseline before LTP induction. All values are expressed as means ± SEM. Statistical comparisons were done with paired or unpaired Student's t test as appropriate (two-tailed p < 0.05 was considered significant).
Extracellular field recordings
Adult (4–6 months of age) wild-type, GluR1−/−, and GluR3−/− mice were killed with desflurane. The brains were removed and cooled to 0–4°C in ACSF of the following composition (in mm): 124 NaCl, 2 KCl, 1.25 KH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26 NaHCO3, 12 glucose, bubbled with 95% O2–5% CO2, pH 7.4. All experiments were conducted according to the Norwegian Animal Welfare Act and the European Union's Directive 86/609/EEC. Coronal slices (400 μm) containing the amygdala were cut with a vibroslicer in 4°C O2/CO2-bubbled ACSF. Slices were placed in an interface chamber exposed to humidified gas at 28–32°C and perfused with ACSF. Orthodromic synaptic stimuli (50 μs; < 300 μA; 0.1 Hz) were delivered alternately through two tungsten electrodes: one placed in the internal capsule, close to the medial border of the lateral nucleus of the amygdala (thalamic pathway), and the other in the capsula externa (cortical pathway). Extracellular synaptic responses were monitored by two glass electrodes (filled with ACSF), placed in the LA. After stable synaptic responses had been obtained in both pathways for at least 15 min, one pathway was tetanized (100 Hz for 1 s repeated four times at 5 min intervals). The second pathway served as a control pathway and never exhibited any significant changes. In a subset of experiments, one of the stimulation electrodes was placed at the medial border of the BA and a recording electrode within the BA. Six consecutive responses (1 min) were averaged and normalized to the mean value recorded 1–4 min before the first tetanization. Data from the different pathways were pooled across animals of the same genotype and are presented as mean ± SEM. Statistical significance was evaluated using a paired two-tailed t test, whereas LTP levels between genotypes were statistically compared by linear mixed-model analysis.
Behavior
Subjects.
All experiments were conducted in two replications with 10- to 12-month-old male GluR1−/− (n = 9), GluR3−/− (n = 9), and wild-type littermate (n = 13) mice. The wild-type group consisted of six littermates of the GluR1−/− mice and seven littermates of the GluR3−/− mice. There was no significant difference between the two groups of wild-type mice originating from the two colonies (GluR1 and GluR3) in any of the behavioral measures. Subjects were housed individually in a holding room with a 12 h light/dark cycle (lights on from 7:00 A.M. to 7:00 P.M.), with food and water ad libitum. All experiments were conducted under the auspices of the United Kingdom Home Office Project and personal licenses held by the authors.
Apparatus.
Behavioral procedures were performed in two identical standard operant chambers (internal dimensions, 12.5 cm wide by 12.5 cm deep by 30 cm high; Coulbourn Instruments, Allentown, PA), housed in sound attenuating boxes. Each chamber had two aluminum walls, a transparent Perspex wall and a Perspex door that served as a fourth wall. The two aluminum walls were divided into three sections, and the ceiling was also aluminum and contained an infrared detector measuring the animal's locomotor activity. The chambers received ambient illumination from a house light operated at 24 V located on the middle section at the top of the right side wall. A speaker was mounted at the top of the left side aluminum wall located in the middle section, through which a computer generated tone (80 dB) was emitted. The subject's behavior was recorded using a digital camera directed at the transparent wall and attached to a VCR.
Fear conditioning.
Mice were transferred to the operant chamber and, after an initial acclimatization period of 6 min, were presented with three pairings of the auditory conditioning stimulus with footshock (0.4 mA; 2 s). The cue was presented for 30 s, and the shock was administered for the last 2 s, coterminating with the auditory cue. Pairings were separated by 2 min, and mice were removed from the chamber 30 s after the last shock presentation. Twenty-four hours after training, mice were tested for CS-induced conditioned responses. For each chamber, the sections of the side aluminum walls were replaced with sections containing checkerboard patterns, which were also attached to each Perspex wall, and additionally fresh sawdust was scattered on the floor of each chamber. After an initial acclimatization period of 6 min, the CS was presented for 8 min (CS test). Twenty-four hours later, an additional test was performed for context conditioning. The chambers were altered to the original configuration used during conditioning. Subjects were placed in the chamber for 8 min (context test).
Scoring.
During each stage of fear conditioning, the mouse's tendency to freeze was scored. Observation was performed using a time-sampling procedure. Every 5 s, each mouse was judged as either freezing or active. Freezing was defined as the absence of visible movement, except for respiration (Blanchard and Blanchard, 1969). Scoring began ∼10 s after the mouse was placed in the chamber and continued until the session had finished. From this observation, a percentage freezing score was calculated by dividing the number of intervals the subject was judged to be freezing by the total number of observations. All scoring was conducted by observers who were “blind” with respect to the critical aspects of the manipulation; that is, they were unaware of the genotype of the mice and their conditioning history. The interrater reliability between observers who used this scoring procedure was ∼95%. To provide an independent assessment of changes in locomotor activity, we also recorded the activity of the mice using an infrared movement detector (Coulbourn Instruments; model H24-61MC; set to mouse sensitivity) mounted (22 cm above the floor) in the ceiling of the apparatus. This device calculated the animal's movement in “movement units”; each unit corresponding to whether movement was detected during a 20 ms period (maximum movement unit pulses was 50 pulses per second).
Reagents
AP5 was from Tocris-Cookson (Bristol, UK). TTX was from Latoxan (Valence, France). All other drugs were from Fluka/Sigma (Buchs, Switzerland).
Results
Reduction of AMPA receptor-mediated synaptic transmission at thalamo- and cortico-LA synapses in GluR1−/− mice
We first analyzed the GluR1 and GluR3 contribution to AMPA receptor-mediated synaptic transmission by recording mEPSCs in LA projection neurons in the presence of TTX (Fig. 1). GluR1- and GluR3-deficient mice (Zamanillo et al., 1999; Sanchis-Segura et al., 2006) exhibited a similar decrease in mEPSC amplitude and mEPSC frequency indicating that GluR1 and GluR3 AMPA receptor subtypes contribute to excitatory transmission (Fig. 1) (wild type: −12.4 ± 0.9 pA, n = 20; GluR1−/−: −8.1 ± 0.4 pA, n = 14, p < 0.01; GluR3−/−: −9.1 ± 0.5 pA, n = 6, p < 0.01; wild type: 5.8 ± 1.5 Hz, n = 20; GluR1−/−: 2.8 ± 0.9 Hz, n = 14, p < 0.05; GluR3−/−: 2.1 ± 0.5 Hz, n = 6, p < 0.05). In addition, mEPSCs exhibited a slower decay time constant with no difference in the 10–90% rise time in GluR3−/− mice (Fig. 1c) (wild type: 5.0 ± 0.3 ms, n = 20; GluR1−/−: 5.4 ± 0.1 ms, n = 14, NS; GluR3−/−: 6.1 ± 0.2 ms, n = 6, p < 0.05).

Reduced mEPSC amplitude and frequency in GluR1−/− and GluR3−/− mice. a, Representative sample traces from WT, GluR1−/−, and GluR3−/− mice. Calibration: 12 pA, 350 ms. b, Histograms illustrating the relative reduction in large amplitude events in GluR1−/− and GluR3−/− animals relative to wild-type controls. Histograms are normalized to the total number of events recorded in each genotype. The gray line indicates mEPSC amplitude distribution in wild-type mice. The traces represent averaged mEPSC waveforms obtained from all events. Calibration: 7 pA, 5 ms. c, Cumulative distributions of mEPSC amplitude, interevent intervals, and τdecay for all genotypes containing 300 randomly selected events from each cell (wild type, n = 20 cells; GluR1−/−, n = 14; GluR3−/−, n = 6). The insets show mean ± SEM for mEPSC amplitude, frequency, and τdecay. The traces illustrate increased decay in GluR3−/− mice (traces are peak-scaled averages obtained from all cells). Calibration, 5 ms. *p < 0.05; **p < 0.01.
The comparable overall reduction in AMPA receptor currents at synapses on LA projection neurons in GluR1−/− and GluR3−/− mice appeared, however, to be input-specific, because AMPA receptor-mediated synaptic transmission at thalamo- and cortico-LA synapses was affected only in GluR1−/− mice. Whereas GluR1−/− mice showed a dramatic reduction in the AMPA/NMDA receptor ratio at both thalamo- and cortico-LA synapses, in GluR3−/− mice the AMPA/NMDA ratio was not different from wild-type mice in either pathway (Fig. 2) (thalamic: wild type, 6.5 ± 1.6, n = 8; GluR1−/−, 2.7 ± 0.3, n = 9, p < 0.05; GluR3−/−, 8.4 ± 1.2, n = 13, NS; cortical: wild type, 7.6 ± 1.9, n = 8; GluR1−/−, 2.4 ± 0.3, n = 9, p < 0.05; GluR3−/−, 8.0 ± 0.9, n = 13, NS). Because the mEPSC amplitude and frequency were equally affected in both knock-out mice, the regular AMPA/NMDA ratio in GluR3−/− mice indicates that either a different population of synapses (other than thalamo- and cortico-LA synapses) contributed to the loss of mEPSCs in absence of GluR3 or, alternatively, that there was a concomitant reduction in NMDA receptor-mediated transmission in GluR3−/− mice.
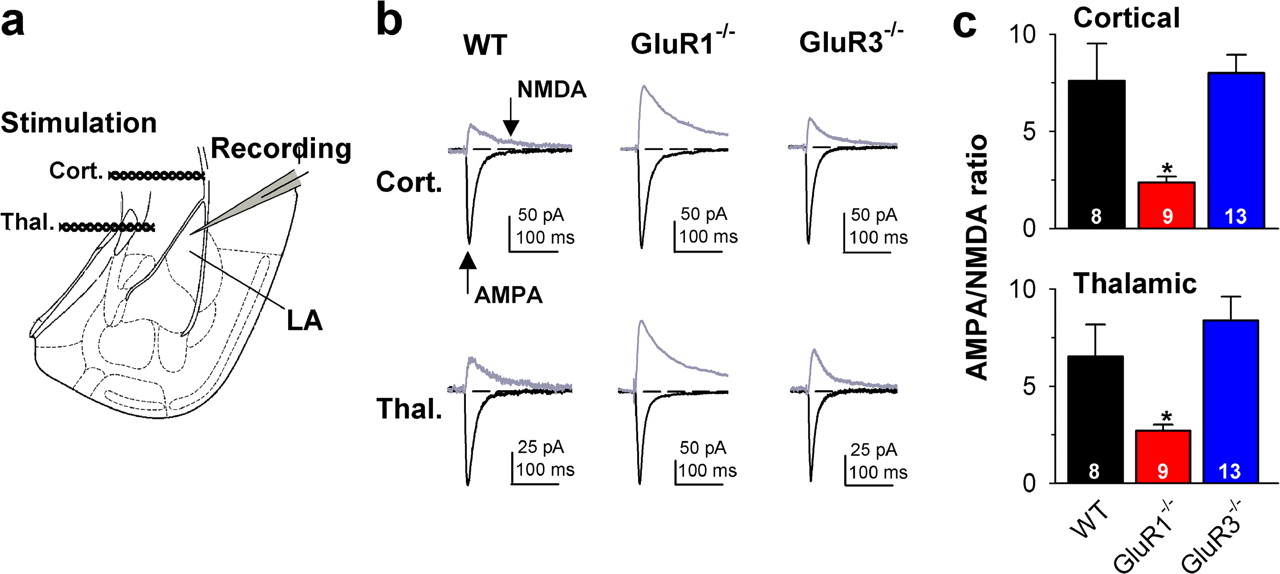
GluR1−/− mice exhibit a decreased AMPA/NMDA ratio of evoked synaptic transmission at thalamo- and cortico-LA synapses. a, Placement of stimulation and recording electrodes. b, Sample traces depicting evoked EPSC waveforms at thalamo- and cortico-LA synapses recorded at −70 mV and at +30 mV in wild-type, GluR1−/−, and GluR3−/− mice. The AMPA component was obtained by measuring the EPSC peak amplitude at −70 mV; the NMDA component was determined by measuring the current amplitude at 100 ms after EPSC onset at +30 mV (arrows). c, Averaged data illustrating the significant (p < 0.05) reduction in the AMPA/NMDA ratio at cortical and thalamic afferents in GluR1−/− mice. Number of experiments is indicated on bars. *p < 0.05. Error bars indicate SEM.
To exclude a reduction in the NMDA receptor component, we examined synaptic efficacy in terms of quantal amplitude of AMPA receptor-mediated transmission at thalamo- and cortico-LA synapses. We recorded stimulation-induced desynchronized mEPSCs (in the presence of extracellular Sr2+ instead of Ca2+). Consistent with the reduced TTX–mEPSC amplitude and the marked reduction in the AMPA/NMDA ratio, stimulation-induced mEPSCs at thalamo- and cortico-LA synapses were significantly smaller in GluR1−/− mice compared with wild-type mice (Fig. 3a–c) (thalamic: wild type, −14.0 ± 0.2 pA, n = 4; GluR1−/−, −8.2 ± 1.8 pA, n = 5, p < 0.05; cortical: wild type, −16.9 ± 0.4 pA, n = 4; GluR1−/−, −9.9 ± 1.1 pA, n = 5, p < 0.01). In GluR3−/− mice, however, quantal amplitudes were not different from wild-type animals (thalamic: −13.8 ± 1.9 pA, n = 5, NS; cortical: −16.4 ± 1.7 pA, n = 5, NS) (Fig. 3a–c). There was no difference in the decay kinetics between groups (Fig. 3d). Thus, at thalamic and cortical inputs to LA projection neurons, the lack of GluR1 leads to a pronounced reduction in the efficacy of AMPA receptor-mediated synaptic transmission, which also might include a reduction in the number of functional synapses. For GluR3−/− mice, we observed that the amplitude of Sr2+ mEPSCs was unchanged compared with control animals, suggesting that pathways other than the thalamo- and cortico-LA pathways are affected in the absence of GluR3.

GluR1−/− mice exhibit a reduction in quantal amplitude at thalamo- and cortico-LA synapses. a, Sample traces of evoked EPSCs in the presence of Ca2+ or Sr2+. In the presence of Sr2+, asynchronous quantal events are detectable. Calibration: 20 pA, 50 ms; insets, 10 pA, 25 ms. b, Normalized histograms illustrating the selective reduction in mEPSC amplitude in the two pathways in GluR1−/− mice (wild type, n = 691 events from 5 cells; GluR1−/−, n = 985 events from 5 cells; GluR3−/−, n = 713 events from 5 cells). Fits indicate mEPSC amplitude distribution in wild-type animals. c, Same data plotted as cumulative probability distributions and averaged means ± SEM (insets). d, GluR1−/− and GluR3−/− exhibited no significant difference in mEPSC kinetics (wild type, n = 5; GluR1−/−, n = 5; GluR3−/−, n = 5; p > 0.05). Superimposed traces represent amplitude-scaled mEPSC waveforms obtained by averaging the scaled mean mEPSC waveforms from each cell. Calibration, 4 ms. *p < 0.05; **p < 0.01.
LTP in the thalamo-amygdala pathway depends on GluR1
We analyzed whether deletion of either the GluR1 or the GluR3 subunit affected LTP in the neural pathways associated with auditory cued fear conditioning (Sigurdsson et al., 2007). In slices from wild-type mice, the average field EPSP (fEPSP) slope in the thalamic pathway was significantly increased 40–45 min after the last tetanization when compared with the pretetanic control value (117 ± 5%; n = 22; p < 0.01) (Fig. 4a). In GluR1−/− mice, however, we failed to observe a persistent potentiation (102 ± 5%; n = 27; p = 0.77), whereas the amount of LTP in GluR3−/− mice was similar to wild-type animals (112 ± 6%; n = 17; p = 0.64) (Fig. 4a).

Selective absence of thalamo-LA LTP in GluR1−/− but not GluR3−/−mice. a, Time course of the fEPSP slope at thalamic afferents in wild-type (n = 22), GluR1−/− (n = 27), and GluR3−/− (n = 17) mice. Whereas wild-type and GluR3−/− animals exhibited significant LTP (p < 0.01), LTP could not be induced in GluR1−/− animals (p = 0.77). LTP was induced by tetanic stimulation of thalamic afferents (100 Hz; 1 s; repeated 4 times with 5 min interval). The depicted traces show averaged fEPSPs before and 45 min after the last tetanization. Calibration: 0.5 mV, 5 ms. b, Pairing-induced LTP at thalamic afferents is selectively abolished in GluR1−/− but not GluR3−/− mice. Time course of the EPSP slope at thalamo-LA synapses in wild-type (n = 12), GluR1−/− (n = 9), and GluR3−/− (n = 5) mice. LTP was induced by pairing afferent stimulation (4 times; 1 s, 100 Hz; arrow) with postsynaptic depolarization to −20 mV. The depicted traces show averaged EPSPs for 2 min of baseline and 2 min of LTP (25–30 min after pairing). Calibration: 2 mV, 10 ms.
In GluR1−/− mice, the failure to observe tetanization-induced LTP in the thalamo-amygdala pathway could be explained by an insufficient depolarization during the induction caused by the reduced AMPA receptor-mediated responses. We therefore used whole-cell recordings to compensate for a possible deficit in postsynaptic depolarization and induced LTP by pairing a similar presynaptic stimulation pattern (1 s; 100 Hz; four times; 0.1 Hz) with postsynaptic depolarization to −20 mV. In addition, we blocked GABAA receptor-mediated inhibition, thereby facilitating the induction of LTP (Wigström and Gustafsson, 1985; Bissière et al., 2003). In general, thalamo-LA LTP induced by pairing presynaptic stimulation with postsynaptic depolarization in the presence of a GABAA receptor antagonist (Fig. 4b) appeared to be more stable than LTP induced by tetanic stimulation alone (Fig. 4a). This may indicate that the extent of postsynaptic depolarization during LTP induction at thalamo-LA synapses has an impact on LTP stability or, alternatively, that thalamo-LA fEPSPs contain an additional nonsynaptic component that decays over time. Similar to the extracellular results, however, whole-cell recordings revealed an almost complete lack of LTP in the thalamo-amygdala pathway in GluR1−/− mice (GluR1−/−: 114 ± 8% of baseline, n = 9, NS), whereas the magnitude of LTP was of similar size in wild-type and GluR3−/− mice (wild type: 159 ± 21% of baseline, n = 12, p < 0.05; GluR3−/−: 149 ± 9% of baseline, n = 5, p < 0.05) (Fig. 4b). Thus, LTP expression at thalamo-amygdala synapses depends on the presence of the GluR1 subunit.
LTP in the cortico-amygdala pathway depends on GluR1 and GluR3
Next, we examined whether LTP in the cortico-amygdala pathway was similarly GluR1 dependent. Cortical afferents to the LA were stimulated by placing a stimulation electrode in the external capsule above the LA (Humeau et al., 2003). Whereas LTP was readily induced in wild-type animals (125 ± 3%; n = 34; p < 0.01), we failed to obtain LTP in both GluR1−/− and GluR3−/− mice (GluR1−/−: 104 ± 3%, n = 33, NS; GluR3−/−: 105 ± 4%, n = 26, NS) (Fig. 5a). To exclude a failure in LTP induction, we used the same pairing procedure as in the thalamo-amygdala pathway. Although LTP was not completely abolished, we observed a substantial reduction in this pathway both in GluR1- and GluR3-deficient mice (wild type: 195 ± 28%, n = 14, p < 0.01; GluR1−/−: 137 ± 13%, n = 7, p < 0.05; GluR3−/−: 142 ± 5%, n = 5, p < 0.01) (Fig. 5b). This suggests that both GluR1 and GluR3 subunits contribute to LTP expression at cortico-LA synapses. Moreover, because quantal amplitude at cortico-LA synapses was reduced in GluR1- but not in GluR3-deficient mice, a reduced synaptic efficacy and/or density in this pathway can hardly account for the similar reduction in LTP in the two genotypes. Thus, in contrast to thalamo-amygdala LTP, LTP in the cortico-amygdala pathway does not solely depend on the GluR1 subunit.

Equal reduction of LTP at cortico-LA synapses in GluR1−/− and GluR3−/− mice. a, Time course of the fEPSP slope in the cortico-LA pathway in wild-type (n = 34), GluR1−/− (n = 33), and GluR3−/− (n = 26) mice. LTP in both GluR1−/− and GluR3−/− animals was absent 40–45 min after induction (p > 0.05). LTP was induced by tetanic stimulation of cortical afferents (4 times; 1 s, 100 Hz). The depicted traces show averaged fEPSPs before and 45 min after the last tetanization. Calibration: 0.5 mV, 5 ms. b, Pairing-induced LTP at cortico-LA synapses is equally reduced in GluR1−/− and GluR3−/− mice. Time course of the EPSP slope in the cortico-LA pathway in wild-type (n = 14), GluR1−/− (n = 7), and GluR3−/− (n = 5) mice. LTP was induced by pairing afferent stimulation (4 times; 1 s; 100 Hz; arrow) with postsynaptic depolarization to −20 mV. The depicted traces show averaged EPSPs for 2 min of baseline and 2 min of LTP (25–30 min after pairing). Calibration: 2 mV, 10 ms.
LTP within the BA is GluR1 dependent
Based on previous studies indicating a role for the BA in both contextual and cued fear conditioning (Anglada-Figueroa and Quirk, 2005; Calandreau et al., 2005; Goosens and Maren, 2006), we next analyzed whether deletion of either the GluR1 or the GluR3 subunit affected LTP within the BA. As at thalamo-LA synapses, we found that LTP in the BA was normal in GluR3−/− mice, but completely absent in GluR1−/− mice (wild type: 125 ± 4%, n = 12, p < 0.05; GluR1−/−: 103 ± 3%, n = 16, p = 0.23; GluR3−/−: 124 ± 8%, n = 13, p < 0.05) (Fig. 6a,b).

Selective absence of LTP in the BA in GluR1−/− but not GluR3−/− mice. a, Placement of stimulation and recording electrodes. b, Time course of the fEPSP slope in the basal amygdala of wild-type (n = 12) and GluR1−/− (n = 16) mice. Whereas wild-type animals exhibit robust LTP (p < 0.05), LTP is completely abolished in GluR1−/− mice (p > 0.05). c, GluR3−/− animals (n = 13) exhibit normal LTP (p < 0.05). LTP was induced by tetanic stimulation of local afferents (100 Hz; 1 s; repeated 4 times with 5 min interval). The depicted traces show averaged fEPSPs before and 45 min after the last tetanization. Calibration: 0.5 mV, 2.5 ms.
GluR1−/− mice exhibit complete absence of freezing behavior during fear conditioning
To study whether GluR1- and/or GluR3-containing AMPA receptors participate in emotional learning, we analyzed the acquisition and retention of auditory cued and contextual fear conditioning. Before CS–US pairings, all genotypes displayed similar levels of baseline activity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Conversely, after the first CS–US pairing, activity levels decreased as freezing responses increased for GluR3−/− and wild-type mice. In contrast, GluR1−/− mice showed similar activity levels throughout the conditioning session, indicating major impairments during the acquisition phase of fear conditioning in the absence of GluR1 (Fig. 7a,b). This is substantiated by the detailed analysis of the activity levels and freezing responses during the three intertrial intervals (ITIs) preceding CS1, CS2, and CS3, respectively (Fig. 7c,d). Main effects analysis of mean activity levels revealed no effect of genotype during the first ITI period (F < 1). However, a significant effect of genotype was revealed during the second and third ITI time bins (smallest F(2,32) = 8.97; p = 0.001) (Fig. 7c). Multiple post hoc Newman–Keuls comparisons supported this, showing that during the second ITI, wild-type mice differed from both GluR1−/− (p < 0.0001) and GluR3−/− mice (p < 0.05). However, during the third ITI, GluR3−/− and wild-type mice showed similar levels of activity suppression, suggesting that acquisition of fear conditioning may be delayed in GluR3−/− mice. In contrast, GluR1−/− mice exhibited no change in activity across the ITI period (p > 0.05) (Fig. 7c). The analysis of the freezing data suggested a similar pattern of behavior (Fig. 7d). Thus, whereas GluR3−/− mice exhibited delayed, but mostly normal levels of activity suppression and freezing behavior, GluR1−/− animals showed a complete absence of any CS- or context-induced fear behavior during the acquisition phase of fear conditioning.

GluR1−/− mice exhibit a complete lack of CS- and context-induced fear behavior during conditioning. a, Mean infrared activity (IR) levels during the three presentations of the 30 s auditory CS. Whereas WT (n = 13; p < 0.05) and GluR3−/− (n = 9; p < 0.05) mice exhibit a significant reduction in activity levels during conditioning, activity levels in GluR1−/− mice (n = 9) remain unaffected (p > 0.05). Number of CS is indicated on the x-axis. A two-way ANOVA revealed no main effect on genotype on baseline activity before CS–US pairing time bin; largest F(11,308) = 1.20, p > 0.20. Analysis of the activity scores during the three CS presentations revealed a main effect of genotype (F(2,28) = 6.68; p < 0.01), and CS presentation (F(2,28) = 34.96; p < 0.0001). There was a significant genotype by CS presentation interaction (F(4,56) = 7.43; p < 0.0001). Analysis of the simple main effects followed by post hoc Newman–Keuls comparisons revealed a significant effect of genotype during the second and third CS presentation (smallest F(2,56) = 3.18; p < 0.05) with GluR1−/− mice differing from GluR3−/− and wild-type groups (p < 0.05). b, Percentage of freezing responses during the three presentations of the 30 s auditory CS. Only WT (n = 13; p < 0.05) and GluR3−/− (n = 9; p < 0.05) animals show a significant increase in freezing levels. Freezing levels of GluR1−/− mice do not increase during conditioning (n = 9; p > 0.05). The number of CS is indicated on the x-axis. There is a main effect of genotype (F(2,28) = 61.30; p < 0.0001) and of CS presentation (F(2,28) = 122.87; p < 0.0001) and a significant interaction between these two factors (F(4,56) = 30.43; p < 0.0001). Simple main effects analysis followed by post hoc Newman–Keuls comparisons revealed a significant effect of genotype during the second and third CS presentations (smallest F(2,56) = 38.57; p < 0.001), with GluR1−/− mice differing from all other genotypes (p < 0.05). c, Mean IR activity levels during the ITIs. Number of ITIs preceding CS1, CS2, and CS3 are indicated on the x-axis. During the ITIs, GluR1−/− mice do not exhibit any reduction in locomotor activity with conditioning (n = 9; p > 0.05), whereas activity levels are significantly reduced in WT (n = 13; p < 0.05) and GluR3−/− (n = 9; p < 0.05) mice. A two-way mixed ANOVA of the activity data revealed a main effect of genotype (F(2,28) = 6.47; p < 0.01) and of activity time bin (F(2,56) = 59.38; p < 0.0001) and a genotype by ITI time bin interaction (F(4,56) = 22.07; p < 0.0001). d, Percentage of freezing responses during the ITIs does not increase in GluR1−/− mice (n = 9; p > 0.05), whereas WT (n = 13; p < 0.05) and GluR3−/− (n = 9; p < 0.05) mice exhibit a significant increase in freezing levels with a main effects of genotype (F(2,28) = 147.07; p < 0.0001) and of ITI time bin (F(2,56) = 219.52; p < 0.0001) and an interaction between the two factors (F(4,56) = 62.47; p < 0.0001). Simple main effects analysis followed by multiple post hoc Newman–Keuls comparisons revealed again a significant effect of genotype during the second and third ITI bins (smallest F(2,84) = 92.93; p < 0.0001), with wild-type mice differing from both knock-out groups during the second ITI bin, and GluR1−/− differing from GluR3−/− and wild-type mice during the third ITI bin (p < 0.05). Error bars indicate SD.
The impaired fear responses of GluR1−/− mice during the acquisition phase of fear conditioning were not caused by a deficit in detecting novel auditory stimuli or insensitivity to the footshook as revealed by the unconditioned suppression of activity elicited by the novel tone and by the initial agitation induced by the footshock (Bouton and Bolles, 1980) (Fig. 8a). The locomotor activity was scored in 0.5 s time bins for the last 4 s of the first ITI and the first 4 s of the first CS tone presentation. An ANOVA confirmed that there was a main effect of phase (ITI vs CS), which did not interact with genotype, and a significant interaction between phase and time bin (F(3,84) = 3.54; p < 0.05). The test of simple main effects revealed a significant effect of time bin only during the CS period (F(3,84) = 7.83; p < 0.001) that reflected a gradual rise in locomotor activity during the CS for all genotypes suggesting that all genotypes were able to detect the presentation of the tone. Similarly, the unconditioned reaction to footshock measured by mean locomotor activity responses, for the first 1.25 s after presentation of the footshock was similar for all genotypes (Fig. 8b), indicating that all mice were able to detect the US.

Comparable CS and US reactivity in wild-type, GluR1−/−, and GluR3−/− mice. a, Mean infrared activity (IR) levels for the 4 s period before presentation of the first CS during conditioning, and the 4 s period of CS presentation does not differ between WT, GluR1−/−, and GluR3−/− mice. Activity scores averaged across 0.5 s time bins (WT, n = 13; GluR1−/−, n = 9; GluR3−/−, n = 9; p > 0.05). A two-way ANOVA of the activity data revealed no main effect of genotype (F(2,28) = 2.52; p > 0.09). There was a main effect of phase (ITI vs CS; F(1,28) = 13.58; p < 0.01), main effect of time bin (F(1,28) = 14.16; p < 0.01), and a significant interaction between these factors (F(3,84) = 3.54; p < 0.05), but no interactions involving genotype. The test of simple main effects revealed a significant effect of time bin only during the CS period (F(3,84) = 7.83; p < 0.001) that reflected a gradual rise in locomotor activity during the CS for all genotypes, suggesting that all genotypes were able to detect the presentation of the tone. b, Comparable activity levels of wild-type, GluR1−/−, and GluR3−/− animals for the 1.25 s period after presentation of the first shock. Activity scores averaged across 0.25 s time bins (WT, n = 13; GluR1−/−, n = 9; GluR3−/−, n = 9; p > 0.05). A two-way ANOVA with genotype and time bin as factors revealed no main effect of genotype (F(2,28) = 2.36; p = 0.1121). However, there was a main effect of time bin, which reflected a reduction in locomotor activity after offset of the shock (F(4,112) = 10.01; p < 0.0001).
Absence of long-term memory of conditioned fear in GluR1−/− mice
Fear memory was tested by assessing retention of conditioned responses 24 h after conditioning by measuring locomotor activity and freezing responses before and after the CS presentation in a novel context (Fig. 9a,b). Before the presentation of the auditory CS, both freezing and activity levels were comparable for all groups. On presentation of the auditory CS, both GluR3−/− and wild-type mice exhibited freezing responses, indicating a memory for the CS. GluR1−/− mice, however, showed no change in activity or freezing responses throughout the duration of the CS presentation, demonstrating that the GluR1 deletion results in a complete failure to acquire and/or retain memory for cued auditory fear conditioning (Fig. 9a). This pattern of results was also evident in the analysis of the locomotor activity data (Fig. 9b). Before the presentation of the cue, there was an apparent trend for GluR1−/− mice to show higher baseline levels of activity in the novel context. However, this difference was not significant. In contrast, there was a significant difference between the genotypes in locomotor activity during the tone presentation. Both the WT and GluR3−/− mice showed lower levels of locomotor activity than GluR1−/− mice on presentation of the CS.

GluR1−/− but not GluR3−/− mice exhibit severe memory deficits in cued and contextual fear conditioning. Retention test. a, b, The percentage observations of a freezing response (a) and the mean IR locomotor activity responses (b) during the baseline period before the CS and during the CS presentation in WT (n = 13), GluR1−/−, and GluR3−/− mice (n = 9 each). No differences in baseline freezing (F(2,28) < 1; p > 0.10; two-way ANOVA) or activity (F(2, 28) = 1.52; p > 0.10; two-way ANOVA) levels was observed. Two-way ANOVAs were conducted on the freezing response and activity data obtained during presentation of the CS. Analysis of the freezing data revealed a main effect of genotype (F(2,28) = 33.42; p < 0.0001), of time bin (F(15,420) = 23.79; p < 0.0001), and an interaction between these two factors (F(30,420) = 6.69; p < 0.0001). Analysis of the simple main effects followed by post hoc Newman–Keuls comparisons revealed a main effect of genotype during freezing bins 13–23 (smallest F(2,205) = 3.56; p < 0.05) with GluR1−/− differing from GluR3−/− and WT groups (p values < 0.05). Analysis of the infrared activity data, similarly, revealed a main effect of genotype (F(2,28) = 6.03; p < 0.01), and of activity time bin (F(15,420) = 17.01; p <. 00001), and a genotype by activity time bin interaction (F(30,420) = 1.781; p < 0.01). Simple main effects analysis revealed a main effect of genotype during activity bins 13–19 and 21–23 (smallest F(2,66) = 4.37; p < 0.05). Post hoc Newman–Keuls comparisons revealed differences between GluR1−/− mice and the remaining groups (p < 0.005). c, d, Context extinction test: the percentage observations of a freezing response (c) and the mean IR locomotor activity responses conditioning context (d) of WT (n = 13), GluR1−/−, and GluR3−/− mice (n = 9 each). WT and GluR3−/− mice showed a steady suppression of locomotor activity. GluR1−/− mice showed considerably reduced freezing responses and higher levels of locomotor activity relative to GluR3−/− and WT mice. The analysis of the freezing response data revealed a main effect of genotype (F(2,28) = 17.57; p < 0.0001) and of time bin (F(15,420) = 4.91; p < 0.0001), and an interaction between the two factors (F(30,420) = 2.92; p < 0.001). Simple main effects analysis revealed a main effect of genotype at bins 4–6 and 8–16 (smallest F(2,154) = 4.54; p < 0.05). Post hoc Newman–Keuls comparisons identified differences between GluR1−/− mice and all other groups (p values < 0.05). A two-way ANOVA analysis of the activity data revealed a main effect of genotype (F(2,28) = 6.65; p < 0.01), of activity time bin (F(15,420) = 2.21; p < 0.01), and a genotype by activity time bin interaction (F(30,420) = 3.004; p < 0.0001). Analysis of the simple main effects followed by post hoc Newman–Keuls comparisons revealed a main effect of genotype at bins 4–6 and 8–16 (smallest F(2,59) = 3.43; p < 0.05), with GluR1−/− mice differing from all other groups (p values < 0.05).
Twenty-four hours later, retention of contextual fear conditioning was assessed. The experimental chambers were altered back to the original configuration used during conditioning. Subjects were placed in the chamber for 8 min. GluR3−/− and wild-type mice showed a steady suppression of locomotor activity, as a consequence of the freezing elicited by the context (Fig. 9c,d). In contrast, GluR1−/− mice failed to show enhanced freezing responses and exhibited correspondingly higher levels of locomotor activity relative to GluR3−/− and wild-type control mice (Fig. 9c,d). In conclusion, our findings demonstrate that GluR1−/−, but not GluR3−/− mice, are impaired in the formation of conditioned fear memories for both contextual and auditory cues.
Discussion
Our electrophysiological analysis revealed a striking difference between GluR1 and GluR3 function in terms of AMPA receptor-mediated synaptic transmission and LTP at thalamic and cortical synapses onto LA projection neurons. In the absence of GluR1, the postsynaptic efficacy and possibly the number of thalamo- and cortico-amygdala synapses were strongly reduced. Although AMPA receptor-mediated synaptic transmission at thalamo- and cortico-LA synapses appeared normal when GluR3 is missing, we found a significant reduction in the amplitude and frequency of mEPSCs recorded in TTX. In principle, this reduction in mEPSC frequency can be caused by presynaptic and/or postsynaptic mechanisms (e.g., by a reduction in the release probability or by removal of postsynaptic AMPA receptors at a subset of synapses). However, the finding that the postsynaptic quantal amplitude at thalamo- and cortico-LA inputs was normal, whereas the overall quantal amplitude of mEPSCs measured in TTX was markedly reduced, cannot be explained by a presynaptic mechanism. This indicates that in LA projection neurons postsynaptic GluR3-containing AMPA receptors play a more prominent role at other glutamatergic inputs, such as for example at intra-amygdala connections. Thus, different AMPA receptor subunits participate in a pathway-specific manner to synaptic transmission in LA projection neurons and the GluR3 content is low or absent at thalamic and cortical inputs under baseline conditions.
Whereas LTP at thalamo-amygdala synapses and fear memory appeared to be normal in the GluR3−/− mice, LTP and fear memory were severely impaired in the GluR1−/− mice. These results are in agreement with previous work indicating that the thalamo-amygdala pathway is rapidly potentiated during auditory fear conditioning (Quirk et al., 1995, 1997), that local prevention of GluR1 trafficking in the LA interferes with thalamo-amygdala LTP and auditory fear conditioning (Rumpel et al., 2005), and that fear conditioning induces rapid expression of GluR1 (Yeh et al., 2006). Together with previous studies (Humeau et al., 2005; Rumpel et al., 2005), our findings strongly support a postsynaptic expression mechanism for thalamo-amygdala LTP (but see Apergis-Schoute et al., 2005).
Although baseline synaptic transmission at cortico-LA synapses was impaired in GluR1−/−, but not GluR3−/− mice, LTP expression was equally reduced in the two genotypes. We and others have shown that cortico-LA LTP can be induced presynaptically and postsynaptically depending on the induction protocol (Huang and Kandel, 1998; Tsvetkov et al., 2002, 2004; Humeau et al., 2003, 2005; Shaban et al., 2006; Humeau and Lüthi, 2007). Together with our previous studies (Humeau et al., 2003), the present results indicate that presynaptic and postsynaptic expression mechanisms may coexist at cortico-LA synapses.
The loss of synaptic plasticity in the BA in GluR1−/− mice is consistent with the observed impairment in contextual fear conditioning, thereby extending the parallels between the behavioral effects of GluR1 deletion and lesions of the LA and/or BA, which also disrupt conditioned freezing to discrete cues and to context (Phillips and LeDoux, 1992; Nader et al., 2001; Calandreau et al., 2005; Anglada-Figueroa and Quirk, 2005; Goosens and Maren, 2006). Our results suggest that GluR1-dependent synaptic plasticity may play a general role at different synapses within the amygdala during the acquisition of both cued and contextual fear conditioning.
GluR1−/− mice displayed a robust deficit in the acquisition of conditioned freezing, both to a discrete CS and to the experimental context. The deficit in fear conditioning was unlikely to be attributable to differences in baseline activity levels or to differences in CS or US detection, because unconditioned CS and US responses were normal. In contrast, GluR3−/− mice revealed a delayed acquisition of conditioned freezing behavior. However, after the third CS–US pairing and during CS and context memory tests, freezing behavior of GluR3−/− mice was equal to wild-type animals.
The relatively normal conditioned freezing displayed by the GluR3−/− mice, which exhibit deficient LTP at the cortico-LA pathway, suggests that AMPA receptor plasticity in this pathway is not essential for conditioned freezing. It could be argued that the delayed acquisition of conditioned freezing observed after the first tone–footshock pairing may reflect a modulatory role for GluR3-dependent plasticity in the cortico-LA pathway. This dissociation in electrophysiological phenotypes between the two mouse mutants may therefore explain the different behavior, suggesting that the deficits in conditioned freezing observed in GluR1−/− mice are attributable to the reduction of AMPA receptor-dependent synaptic transmission and/or the absence of GluR1-dependent synaptic plasticity at thalamo-LA synapses (Rumpel et al., 2005).
There are a number of important caveats, however. First, relatively normal conditioned freezing in GluR3−/− mice does not completely preclude a role for plasticity at cortico-LA synapses in supporting this behavior in normal wild-type mice. The LA receives auditory inputs from thalamus and cortex (Carlsen and Heimer, 1988; Farb and LeDoux, 1997, 1999; McDonald, 1998; Smith et al., 2000), and conditioned freezing to a simple auditory CS can be mediated by either of these pathways (Romanski and LeDoux, 1992). It may be the case therefore that plasticity in both cortical and thalamic pathways can contribute to conditioned freezing in normal animals. It is also possible that the intact LTP at thalamo-LA synapses in GluR3−/− mice can compensate and support the behavior in these mutant mice, and that plasticity in both thalamic and cortical inputs must be impaired before a behavioral deficit becomes evident (as in the GluR1−/− mice). Alternatively, it has been argued that the cortico-LA pathway is important for more complex auditory stimuli or for stimulus discriminations (Thompson, 1962; Jarrell et al., 1987; Shaban et al., 2006) (but see Armony et al., 1997), and therefore the thalamic pathway is more likely to support conditioning to a simple tone CS. Indeed, the present data suggest that synaptic plasticity in the cortico-LA pathway has a more subtle and limited role in this particular form of conditioned freezing, thus potentially reflecting the relatively simple nature of the tone CS used in the present study.
Second, it is also possible that the deficits in conditioned freezing in the GluR1−/− mice, in particular to the experimental context, could be explained by a deficit in hippocampal synaptic plasticity (Zamanillo et al., 1999). Previous studies have shown that GluR1−/− mice are impaired on hippocampus-dependent tasks that require the rapid encoding and/or expression of trial-specific memories (Reisel et al., 2002; Schmitt et al., 2003, 2004). Thus, it is possible that a GluR1-dependent, hippocampal memory could play a role in fear conditioning tasks such as that used in the present study in which acquisition is rapid, and occurs in just one or two trials. Against this, it could be argued that because GluR1 gene inactivation disrupts conditioned freezing not only to context but also to a discrete CS, then the latter, at least, is more likely to be the result of an amygdala dysfunction. This is based on several lesion studies that have shown that hippocampal damage produces deficits in conditioned freezing to context but not to a discrete CS (Kim and Fanselow, 1992; Phillips and LeDoux, 1992; Maren et al., 1997) (but see Richmond et al., 1999). In contrast, lesions of the BLA reliably disrupt freezing responses to both kinds of cues (Phillips and LeDoux, 1992). Targeted lesions of the BA, the main amygdala target of hippocampal afferents and origin of projections to the hippocampus (Pitkanen et al., 2000), revealed a selective impairment of contextual fear conditioning (Calandreau et al., 2005) (but see Goosens and Maren, 2006). In accordance with a role of the BA in contextual conditioning, we found that GluR1−/− but not GluR3−/− mice are deficient in BA LTP.
In summary, we conclude that activity-dependent synaptic plasticity in distinct amygdala pathways depends on different AMPA receptor subunit combinations. The absence of conditioned freezing to the auditory CS and the experimental context in GluR1−/− mice suggests a dominant role for GluR1-dependent synaptic plasticity in amygdala-dependent emotional learning.
Footnotes
-
This work was supported by the Volkswagen Foundation, Centre National de la Recherche Scientifique, and the Novartis Research Foundation. We thank G. Casassus for helpful discussions and comments on this manuscript, and Peter H. Seeburg for his generous support.
- Correspondence should be addressed to Andreas Lüthi, Friedrich Miescher Institute for Biomedical Research, Maulbeerstrasse 66, CH-4058 Basel, Switzerland. andreas.luthi{at}fmi.ch





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}