
Abstract
Target-dependent increases in axon growth and varicosities accompany the formation of functional synapses between Aplysia sensory neurons and specific postsynaptic neurons (L7 and not L11). The enhanced growth is regulated in part by a target-dependent increase in the secretion of sensorin, the sensory neuron neuropeptide. We report here that protein kinase C (PKC) activity is required for synapse formation by sensory neurons with L7 and for the target-dependent increases in sensorin synthesis and secretion. Blocking PKC activity reversibly blocked synapse formation and axon growth of sensory neurons contacting L7, but did not affect axon growth of sensory neurons contacting L11 or axon growth of the postsynaptic targets. Blocking PKC activity also blocked the target-induced increase in sensorin synthesis and secretion. Sensorin then activates additional signaling pathways required for synapse maturation and synapse-associated growth. Exogenous anti-sensorin antibody blocked target-induced activation and translocation into sensory neuron nuclei of p42/44 mitogen-activated protein kinase (MAPK), attenuated synapse maturation, and curtailed growth of sensory neurons contacting L7, but not the growth of sensory neurons contacting L11. Inhibitors of MAPK or phosphoinositide 3-kinase also attenuated synapse maturation and curtailed growth and varicosity formation of sensory neurons contacting L7, but not growth of sensory neurons contacting L11. These results suggest that PKC activity regulated by specific cell–cell interactions initiates the formation of specific synapses and the subsequent synthesis and release of a neuropeptide to activate additional signaling pathways required for synapse maturation.
- target selection
- synapse formation
- synapse-associated growth
- sensorin
- MAPK
- PI3K
- Aplysia
Introduction
The formation of specific synapses typically entails the selection of correct partners by interacting neurons, profuse branching of axonal and dendritic arbors, the assembly and apposition of presynaptic and postsynaptic structures, and the stability and maintenance of functional synapses. Several membrane or secretory proteins and their receptors are candidate molecules that might contribute to the formation of specific synapses (Wells et al., 1999; Tao and Poo, 2001; Lee et al., 2003; Blagburn and Bacon, 2004; Graf et al., 2004; McCabe et al., 2004; Shen et al., 2004; Boucard et al., 2005: Chih et al., 2005; Christopherson et al., 2005; Sara et al., 2005; Wang et al., 2007). However, the orchestration of the signaling mechanisms mediating target selection, branching, synapse formation, and maintenance is not well understood.
Neurotrophins and their receptors are one group of molecules that contribute to the formation and maintenance of synapses (Schafer et al., 1983; Wells et al., 1999; Lockhart et al., 2000; Belluardo et al., 2001; Chen et al., 2002; Cohen-Cory, 2002; Vicario-Abejon et al., 2002). Secreted brain-derived neurotrophic factor (BDNF) activates receptors (trkB and P75) to modulate synaptogenesis of both excitatory and inhibitory connections (Martinez et al., 1998; Vicario-Abejon et al., 1998; Alsina et al., 2001; Rico et al., 2002; Aguado et al., 2003; Bamji et al., 2006; Sanchez et al., 2006; Shen et al., 2006) and the fine-tuning of synaptic connections associated with early experience-dependent reorganization in postnatal cerebral cortex (Cabelli et al., 1995, 1997; McAllister et al., 1997; Marshak et al., 2007). Neurotrophin interaction with receptors leads to activation of multiple signaling pathways including mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), and phospholipase C (PLC) [for review, see Huang and Reichardt (2001) and Segal (2003)]. Do neurotrophin secretion, receptor activation, and signaling contribute to the formation of specific synapses?
Aplysia neurons are identified functionally because of their specific connections. In culture Aplysia sensory neurons form specific synapses (Glanzman et al., 1989; Schacher et al., 1999). When contacting L7, sensory neurons extend exuberant axon growth and form numerous varicosities with transmitter release sites compared with growth by sensory neurons contacting L11, which does not induce synapse formation by sensory neurons (Glanzman et al., 1989; Schacher and Montarolo, 1991; Hatada et al., 1999; Kim et al., 2003). Target-induced secretion of sensorin, the sensory neuron-specific neuropeptide (Brunet et al., 1991), contributes to axon growth and maturation of synapses with L7 (Hu et al., 2004b). Sensorin also acts as a neurotrophin-like molecule when mature sensory neuron synapses express long-term facilitation (LTF). Stimuli that produce LTF enhance the local synthesis and secretion of sensorin, which then binds autoreceptors to activate p42/44 MAPK required for LTF and the formation of new synaptic contacts (Hu et al., 2004a, 2006, 2007).
We report here that PKC activity is required to initiate synapse formation and growth by sensory neurons with a specific target and to increase the synthesis and secretion of sensorin required for synapse maturation. The PKC-dependent increase in sensorin synthesis and secretion activates signaling pathways such as MAPK required for axon growth associated with the formation of additional synapses with the appropriate target. Growth of sensory neurons in contact with a nontarget (L11) is unaffected by blocking PKC, the secreted sensorin, or its downstream signaling pathways. Thus the formation and maturation of specific synapses requires the sequential activations of specific signaling pathways linked together by PKC activities and neuropeptide secretion.
Materials and Methods
Cell culture and electrophysiology.
Sensory neurons were isolated from ganglia dissected from adult animals (80–100 g); motor cell L7 and L11 were isolated from juvenile (1–3 g) ganglia and maintained in coculture for up to 42 h as described previously (Rayport and Schacher, 1986; Schacher et al., 1999; Hu et al., 2004b). In most cases, both targets were plated in the same culture dish, and one sensory neuron was plated with each motor neuron target. Standard electrophysiological techniques were used to record the amplitude of the EPSP evoked in L7 or L11 with the stimulation of each sensory neuron (Schacher and Montarolo, 1991; Schacher et al., 1999). The motor targets were impaled with a microelectrode (resistance of 10–15 MΩ) containing 2.0 m K-acetate, 0.5 m KCl, and 10 mm K-HEPES, pH 7.4, and held at −85 mV. Each sensory neuron was stimulated with a brief (0.3–0.5 ms) depolarizing pulse to evoke an action potential using an extracellular electrode placed near the cell body of the sensory neuron.
Imaging sensory neuron growth and varicosities.
After measuring the strength of the synaptic connections, the fluorescent dye 5(6)-carboxy-fluorescein (Molecular Probes, Eugene, OR; 6% in 0.44 m KOH, pH = 7.0) was injected with 0.3–0.5 nA hyperpolarizing current pulses (500 ms at 1 Hz) for 5 min into some of the sensory neurons that were cocultured either with L7 or L11 (Glanzman et al., 1989; Schacher and Montarolo, 1991). Nomarski- or phase-contrast and fluorescent images of the same view areas along the axons of L7 or L11 were taken to map out the location of regenerated sensory neuron neurites and varicosities. Varicosities were defined as swellings along the sensory neuron neurites with diameters ≥1.5 μm. Growth cones at the leading tips of neurites were not counted. Overall growth of neurites along the major processes and the cell bodies of L7 or L11 was used to measure the extent of growth. The cultures were viewed with a Nikon (Tokyo, Japan) Diaphot microscope attached to a silicon-intensified target (SIT, Dage 68; Dage-MTI, Michigan City, IN) video camera, the images were processed by a Dell computer, and images were captured and processed by the microcomputer-controlled imaging device (MCID) software package (Imaging Research, St. Catharines, Ontario, Canada).
Drug treatments.
Cultures were incubated for 8 h or 18–20 h with anti-sensorin antibody (Ab) or with protein A-purified preimmune serum (400 ng/ml) (Hu et al., 2004a,b) starting immediately after plating sensory neurons with their targets. Sensorin peptide (80 ng/ml) was added to cultures either when sensory neurons were first added to cultures or 6 h after plating. PKC inhibitor (PKCI) chelerythrine or bisindolylmaleimide I (2 μm; Calbiochem, La Jolla, CA), PI3K inhibitor LY294002 (2 μm; Calbiochem), or PKA inhibitor KT5720 (5 μm; Calbiochem) was added to some cultures for 16–18 h starting 15 min before sensory neurons were plated with the target motor neuron. To block MAPK activities, cultures were incubated with U0126 (2 μm; Calbiochem) for 16–18 h beginning 15 min before adding sensory neurons. Control cultures were incubated with the compound U0124 (2 μm; Calbiochem).
Immunocytochemistry.
Immunocytochemistry was used to monitor expression of sensorin, total p42/44 MAPK, or phosphorylated p42/44 MAPK (Martin et al., 1997; Liu et al., 2003; Hu et al., 2004a,b). Cultures were rinsed briefly in artificial seawater and fixed in 4% paraformaldehyde and processed as described previously (Martin et al., 1997; Hu et al., 2004a,b). The cells were exposed to rabbit polyclonal antibody specific for sensorin (1:1000), all forms of p42/44 MAPK or phospho-p42/44 MAPK (1:100, Cell Signaling, Beverly, MA) diluted in 2% normal goat serum in 0.01 m PBS with 0.3% Triton X-100 at 4°C for 24 h. The incubated cultures were washed in 0.01 m PBS and incubated in FITC-conjugated goat anti-rabbit IgG (1:200; Sigma, St. Louis, MO) at 4°C for 4 h. After washing in 0.01 m PBS, cultures were imaged directly with filter set for detecting fluorescein signal. To test the specificity of the primary antibody, controls were performed, including the substitution of normal rabbit serum for the primary antibody and omission of the primary antibody. To test specificity of the sensorin antibody, sensorin and its antibody at equimolar concentrations were preincubated for 30 min before their application to fixed cultures for immunostaining (Liu et al., 2003; Hu et al., 2004a,b). All controls showed little immunocytochemical reaction. Illumination for detecting fluorescent signals was maintained at a constant setting for all treatments. Images were taken with a Nikon Diaphot microscope attached to a Dage 68 SIT video camera, processed by a Dell computer with the MCID (7.0) software package from Imaging Systems.
Quantification and data analysis.
All data are expressed as mean ± SEM produced by the indicated treatments. The averages for EPSP amplitude, axon growth, and varicosities are measured in millivolts, micrometers, and number, respectively. The intensity of sensorin immunostaining was tested by measuring average fluorescent intensity in the sensory neuron cell body, the entire main axon, the axon stump, and the neurites and varicosities contacting the major processes of L7 with the MCID (7.0) software package from Imaging Systems. Staining intensities for sensorin for the various experimental treatments were normalized to the intensities measured in each cellular compartment after control treatments (100%). The overall staining intensity of total or phosphorylated MAPK immunofluorescence was determined by averaging intensity for the entire cell body (cytoplasm plus nucleus). Staining in the nucleus or cytoplasm was determined by measuring average intensity over that area. ANOVA and Scheffé's F test were used to gauge significant differences between treatments.
Results
PKC mediates target-dependent increase in sensorin expression and initial synapse formation
Varicosities with transmitter release sites form as growth cones at the tips of regenerating axons of the sensory neuron pause transiently while extending along the major processes of L7 (Hatada et al., 1999). After 24 h in culture, sensorin release into the bath is detected when sensory neurons are cultured with L7 but not with L11 (Hu et al., 2004b), and the release of sensorin at that time is required for the continued increase in synaptic efficacy, axon growth, and the formation of varicosities by sensory neurons contacting L7 (Hu et al., 2004b). Actions of sensorin contribute little to axon growth of sensory neurons cultured alone or with L11. To determine how initial target interaction affects the secretion of sensorin, we examined with immunocytochemistry whether the initial interaction with the target regulates the expression of the neuropeptide sensorin.
Expression of sensorin throughout the sensory neuron is significantly greater when sensory neurons interact with L7 compared with L11 (Fig. 1). Dishes containing both types of cocultures were fixed 16–18 h after plating and processed for sensorin immunoreactivity. Sensorin staining was restricted to the sensory neurons, and the target neurons were unstained (see also Hu et al., 2004a,b). Staining for sensorin was significantly greater in sensory neurons contacting L7 (Fig. 1A,C) compared with L11 (n = 5 dishes with both types of cocultures): 267 ± 21% in the cell body, 662 ± 83% in the sensory neuron axon, and 259 ± 18% in the regenerated neurites and varicosities. The staining in the axon and distal processes of sensory neurons contacting L11 (Fig. 1B,D) was diffuse, and large granules were poorly stained. In contrast, staining in these compartments in sensory neurons contacting L7 was primarily in granules, suggesting that the neuropeptide is stored in vesicular structures within distal processes and varicosities (Fig. 1, see insets). This increase in sensorin expression may contribute significantly to the target-dependent increase in sensorin secretion critical for synapse maturation and maintenance (Hu et al., 2004b) (and see below).

Specific target induces an increase in sensorin expression. A, B, Phase-contrast micrographs of sensory neuron-target cultures (SN-L7 and SN-L11) 16 h after plating in the same culture dish. After fixation, the culture was processed for sensorin immunoreactivity. C, D, Sensorin immunoreactivity is greater in sensory neurons cultured with L7 compared with L11. Staining is confined to the sensory neurons. An ANOVA indicated a significant effect of target on sensorin immunostaining (df = 2, 16; F = 18.907; p < 0.001). A comparison for each compartment indicated that staining intensity was significantly greater when sensory neurons contact L7 (p < 0.01 for each compartment). The insets (a–c for SN-L7 and d–f for SN-L11) reveal the differences in staining in the sensory neuron axons and distal processes. Note that the sensory neuron axon that contacts L7 has intense staining within large granules. The axon stump (contacting the major processes of each target) accumulates sensorin, but the regenerated neurites contacting L7 are both more numerous and heavily stained. Scale bars, 25 μm.
At mature sensory neuron connections with L7, sensorin synthesis is regulated either by PKC or PI3K activity after stimuli that produce different forms of long-term facilitation (Hu et al., 2006, 2007). We therefore examined whether these signaling pathways mediate the target-induced increase in sensorin expression. Incubation with the PKC inhibitor chelerythrine, but not the PI3K inhibitor LY294002, blocked the increase in sensorin expression (Fig. 2). Compared with sensorin staining in control cultures (Cont; n = 6 normalized to 100% for each compartment), blocking PKC activity (n = 7) reduced sensorin expression in the cell body to 47 ± 3%, and in the axon plus stump to 59 ± 2%. Staining in varicosities was indeterminate, because no stained varicosities were detected in the presence of chelerythrine. Incubation with another PKC inhibitor bisindolylmaleimide I also blocked the increase in sensorin expression induced by interaction with L7. Compared with sensorin staining in control cultures (n = 8, normalized to 100% for each compartment), blocking PKC activity with bisindolylmaleimide I (n = 10) significantly reduced sensorin expression in the cell body to 50 ± 3%, and in the axon plus stump to 62 ± 3%. Because there were no detectable varicosities, staining in that compartment was indeterminate. Although incubation with the PI3K inhibitor (n = 7) reduced the overall axon growth of sensory neurons contacting L7 (see below), sensorin expression in the cell body (97 ± 4%), axon (97 ± 5%), and varicosities (87 ± 7%) was not significantly altered. Thus PKC activity, a signaling pathway that regulates sensorin expression after stimuli that produce an associative form of LTF (Hu et al., 2007), regulates the target-induced increase in sensorin expression.

PKC activity mediates the target-induced increase in sensorin expression. Phase-contrast and corresponding epifluorescent images of sensorin immunoreactivity 16 h after plating cultures in Cont, the PKCI chelerythrine (2 μm), or the PI3K inhibitor LY294002 (2 μm; PI3KI). Staining in the sensory neuron cell body, axon, and axon stump are markedly reduced with PKCI. Note the absence of growth from the axon stump with treatment with PKCI. In contrast, staining for sensorin in the cell body and axon with control or PI3KI treatments are strong. Intense staining can be seen in numerous granules distributed throughout the axon in both the control and PI3KI-treated cells. Although overall growth in the presence of PI3KI was reduced (see below), staining in the varicosities remained strong. An ANOVA indicated a significant effect of treatment on sensorin staining (df = 4, 34; F = 82.767; p < 0.001). Individual comparisons indicated that staining in the cell bodies and axons of controls were significantly greater than staining in the cell bodies and axons of sensory neurons treated with PKCI (F = 45.522, p < 0.01 and F = 22.642, p < 0.01), but not significantly different from sensorin staining observed in cell bodies and axons of cultures treated with PI3KI. Staining for sensorin in the varicosities of sensory neurons treated with PI3KI was not significantly different from the staining in the varicosities of sensory neurons treated with control. Scale bars, 80 μm.
The lack of growth by the sensory neurons after treatment with the PKC inhibitors (Fig. 2) suggested that PKC activity might be important for initiating synapse formation. We therefore examined axon growth of sensory neurons incubated with chelerythrine when cocultured with L7 and compared it to the growth of sensory neurons incubated with chelerythrine when cocultured with L11. After 16–18 h, the presence of synaptic connections was monitored with electrophysiology, and overall growth of each sensory neuron in contact with either L7 or L11 was measured after dye injection. Most culture dishes contained both types of cell pairs.
Inhibition of PKC blocked initial synapse formation and neuritic growth of sensory neurons contacting L7, but did not affect neuritic growth of L7 and L11 or growth of sensory neurons contacting L11 (Fig. 3). Incubation with the PKC inhibitor chelerythrine abolished the presence of EPSPs in 90% of cultures (0.1 ± 0.1 mV; n = 30 with a range of 0–1 mV) compared with the average EPSP amplitude in control cultures of 9.8 ± 1.0 mV (n = 23 with range of 3–21 mV). Another PKC inhibitor, bisindolylmaleimide I, also abolished synapse formation in 80% of cultures (0.3 ± 0.2 mV; n = 10 with range of 0–2 mV) compared with 13.4 ± 1.5 mV for controls (n = 8 with range of 8–21 mV). The absence of a detectable physiological response when PKC activity was inhibited was accompanied by the absence of significant growth from the axon stump of the sensory neuron contacting L7 (Figs. 3A and 3C). In control cultures (n = 12), the extent of sensory neuron growth and the number of varicosities in contact with the major processes of L7 was 381 ± 13 μm and 31 ± 3 varicosities. In contrast, in the presence of the PKC inhibitor chelerythrine (n = 15), axon growth from the stump was restricted to either a single varicose-like expansion from the stump (Fig. 3A, PKCI) or a few short filopodial-like processes emerging from the axon stump or the varicose-like expansion. Axon “growth” by sensory neurons along L7 was only 23 ± 3 μm with 0.4 ± 0.2 varicosities. Thus PKC activity is required for the initiation of synapse formation and for the outgrowth and varicosity formation associated with synapse formation.

PKC activity is required to initiate synapse formation and synapse-associated growth. A, B, Nomarski-contrast (top) and corresponding epifluorescent (bottom) images of sensory neurites (within outlined area in each micrograph) after injection of dye into sensory neurons cultured with either L7 (SN-L7; A) or L11 (SN-L11; B), plated in the same dish, and treated with control or the PKC inhibitor chelerythrine (2 μm) for 16 h. In the Cont, contact with L7 induces significantly more growth and varicosities compared with contact with L11. Incubation with chelerythrine blocked all growth from the sensory neurons contacting L7 with the exception of a varicose-like protrusion from the axon stump. In contrast, growth from the sensory neuron contacting L11 in the same culture dish was unaffected by the chelerythrine. In the presence of chelerythrine, growth from the sensory neuron contacting L11 was greater than the growth of the sensory neuron contacting L7. Scale bars, 50 μm. C, The PKC inhibitor significantly reduced sensory neuron growth and varicosity formation only when sensory neurons contact L7 but not when they contact L11. The histogram summarizes the effect of the PKC inhibitor on sensory neuron growth and varicosity formation when contacting different targets. An ANOVA indicated a significant effect of target and PKC inhibitor on axon growth and number of varicosities formed (df = 3, 35; F = 93.722; p < 0.001). Individual comparisons indicated that chelerythrine compared with Cont significantly reduced growth and number of varicosities when sensory neurons contacted L7 (F = 94.355, p < 0.001 and F = 45.78, p < 0.001), whereas there was no significant difference between Cont and chelerythrine on sensory neuron growth and varicosities formed when sensory neurons contact L11. As expected for cultures treated with Cont, growth and varicosity formation were greater when sensory neurons contacted L7 compared with L11 (F = 24.044, p < 0.01 and F = 8.848, p < 0.01, respectively). In contrast, when incubated with chelerythrine, growth of sensory neurons contacting L11 was now significantly greater than the growth in contact with L7 (F = 36.29, p < 0.01 for axon growth and F = 6.098, p < 0.01 for varicosities).
Although growth of sensory neurons in contact with L7 was severely curtailed by inhibitors of PKC, overall growth from the stumps of L7 and L11 did not appear to be significantly affected (Figs. 2, 3, Nomarski-contrast images). Because of the complexity of the growth from L7 and L11, it was not possible to quantify any differences between drug treatment and control. We quantified the effects of chelerythrine on sensory neuron growth after dye injection into the sensory neurons. The PKC inhibitor chelerythrine did not affect axon growth from sensory neurons when their stumps contacted L11 (Fig. 3B,C). Axon growth and varicosity number in contact with L11 was 219 ± 19 μm and 14 ± 2 varicosities in control cultures (n = 6) and 216 ± 22 μm and 13 ± 2 varicosities in cultures treated with the PKC inhibitor (n = 6). No EPSP was detected when sensory neurons contacted L11 either in the presence or absence of drug. Incubation with the PKC inhibitor reversed the behavior of sensory neuron growth contacting the different targets; in controls, sensory neuron growth contacting L7 is significantly greater than the growth contacting L11, whereas with PKC inhibition, sensory neuron growth contacting L11 is significantly greater than the growth from sensory neurons contacting L7. Thus PKC activity is required for target selection (initiating synapse formation with specific targets and the growth associated with synapse formation) but is not essential for regulating growth per se or growth of sensory neurons when they contact postsynaptic targets that fail to induce synapse formation.
To rule out the possibility that the PKC inhibitor chelerythrine was irreversibly damaging sensory neurons specifically when they contact L7, we examined how the cultures responded after chelerythrine was washed out. After exposure to the inhibitor for the first 18 h after plating, cultures were rinsed of drug or control medium and maintained in control medium for another 24 h. Synapse formation, growth, and sensorin expression all resumed after washout of the PKC inhibitor (Fig. 4). After the first 18 h, average EPSP amplitude in control cultures (n = 9) was 5.9 ± 0.7 mV compared with 0.1 ± 0.1 mV in cultures treated with the PKCI (n = 10). After washout of PKCI, average EPSP amplitude at 42 h was now 8.8 ± 1.2 mV, comparable with EPSP amplitudes recorded in controls after 18 h (Fig. 4A,B). After washout, axon growth along the major processes of L7 was now 370 ± 21 μm with 35 ± 4 varicosities (Fig. 4C) (n = 10). In addition, sensorin expression throughout the sensory neuron returned to high levels and was comparable with the sensorin levels observed in control cultures (Fig. 4D,E). Thus, the effects of the PKC inhibitor on synapse formation, growth, and sensorin expression are reversible. The results support the finding that PKC activity is required for the initiation of synapse formation, the growth associated with synapse formation, and the target-induced regulation of sensorin expression.

Synapse formation, growth, and sensorin expression resumes after washout of the PKC inhibitor. A, B, After washout of the PKCI (chelerythrine), sensory neurons form functional connections with L7. EPSPs were recorded once again in the same cultures after washout of the control medium or PKCI. Incubation with the PKCI resulted in no detectable EPSP. After washout, EPSP amplitudes increased in control cultures, and EPSPs were now recorded in cultures treated initially with the PKCI. Calibration: 10 mV, 25 ms. The histogram in B indicates the reversal of PKC inhibition on synapse formation. ANOVA indicated a significant effect of treatment on EPSP amplitude (df = 1, 17; F = 29.59; p < 0.001). Individual comparisons indicated that the presence of the PKCI (18 h) blocked synapse formation compared with control (F = 5.618; p < 0.01), but EPSP amplitude after washout of the inhibitor (PKCI-Washout; 42 h) was significantly increased (F = 13.923; p < 0.01) but was not significantly different from the EPSP amplitude for controls after 18 h. C, Growth and varicosity formation resume after washout of the PKCI. Nomarski-contrast and epifluorescent images (within outlined area in each micrograph) after dye injections reveal that growth and varicosities are considerable after PKCI washout and is consistent with a reversal of the blockade by the inhibitor. Because of the additional day of growth, control cultures have more growth and have formed more varicosities (F = 6.709, p < 0.01 and F = 7.459, p < 0.01). Scale bars, 50 μm. D, E, Sensorin expression returns to control levels after washout of the PKCI. Epifluorescent images of sensorin immunostaining in a 42 h control culture and in a 42 h culture 24 h after washout of the PKCI (D). Staining intensities in all compartments are comparable. Scale bar, 50 μm. The histogram in E indicates that staining in each compartment after PKCI washout was not significantly different from the staining in controls (normalized to 100%).
PKC mediates target-dependent release of sensorin required for synapse-associated growth
During the first 4 d in culture, the amplitude of the EPSPs evoked in L7 increase as sensory neurons form more synaptic contacts with L7 (Bank and Schacher, 1992; Zhu et al., 1994; Schacher and Wu, 2002). Bath application of anti-sensorin Ab between day 1 and day 3 affects the maturation of sensory neuron synapses. Blocking sensorin's actions reduces significantly both the change in synaptic strength and the growth and formation of new sensory neuron varicosities (Hu et al., 2004b). In contrast, incubations with exogenous sensorin further enhanced changes in synapse efficacy and sensory neuron growth. We therefore tested whether anti-sensorin Ab applied during the first 20 h of culture affected the formation of synapses in terms of synapse efficacy and growth and whether the antibody affected the growth of sensory neurons contacting L11. At plating, the cultures were bathed in culture medium containing either 400 ng/ml rabbit anti-sensorin Ab (SEN Ab; n = 12) or 400 ng/ml rabbit IgG (Cont Ab; n = 10). After 20 h, the cultures were examined for the presence of synaptic connections, and the sensory neurons were injected with dye to image growth of sensory neuron axons along the major processes of the target neurons (L7 or L11). We also tested EPSPs in L7 evoked by stimulation of sensory neurons after an 8 h incubation with anti-sensorin Ab and compared the amplitudes to those recorded in cultures 8 h after plating in control medium.
Blocking the actions of released sensorin with anti-sensorin Ab significantly reduced synaptic efficacy and growth at 20 h after plating (Fig. 5), but failed to block the initiation of synapse formation at 8 h after plating. In control cultures (n = 10) the average EPSP amplitude after 20 h was 10.5 ± 1.1 mV compared with an average EPSP amplitude of only 3.2 ± 0.8 mV in cultures incubated with anti-sensorin Ab (n = 12) (Fig. 5A). No EPSP was detected when sensory neurons contacted L11. However, at 8 h after plating, incubation with anti-sensorin Ab did not have a significant effect on EPSP amplitude (3.3 ± 0.5 mV; n = 7) compared with controls (3.5 ± 0.8 mV; n = 6). Although anti-sensorin Ab did not appear to interfere with the initial formation of synapses, the reduction in EPSP amplitude with anti-sensorin Ab at 20 h was correlated with a significant reduction in axon growth and varicosity number contacting the major processes of L7 (Fig. 5B,C). Sensory neuron growth extended 375 ± 13 μm and formed 29 ± 3 varicosities along the major axons of L7 when incubated with Cont Ab (n = 10) compared with growth of only 122 ± 8 μm with 5 ± 0.8 varicosities when cultures were incubated with anti-sensorin Ab (n = 12). The effect of anti-sensorin Ab on growth was restricted to sensory neurons contacting L7. Growth of sensory neurons contacting L11 plated in the same culture dishes (n = 6 for each treatment) was not significantly affected by anti-sensorin Ab (growth of 259 ± 23 μm with control Ab compared with 253 ± 22 μm with anti-sensorin Ab) (Figs. 5B,C). With anti-sensorin Ab, growth of sensory neurons contacting L11 was now greater than the growth of sensory neurons contacting L7 plated in the same dishes. The number of varicosities formed by sensory neuron axons contacting L11 was also unaffected by anti-sensorin Ab (Fig. 5B,C). Thus target-dependent release of sensorin affects synapse-associated growth by sensory neurons during the maturation of the synapse after synapse formation was initiated with L7, but not growth of sensory neuron axons contacting another target (L11) that fails to induce synapse formation.
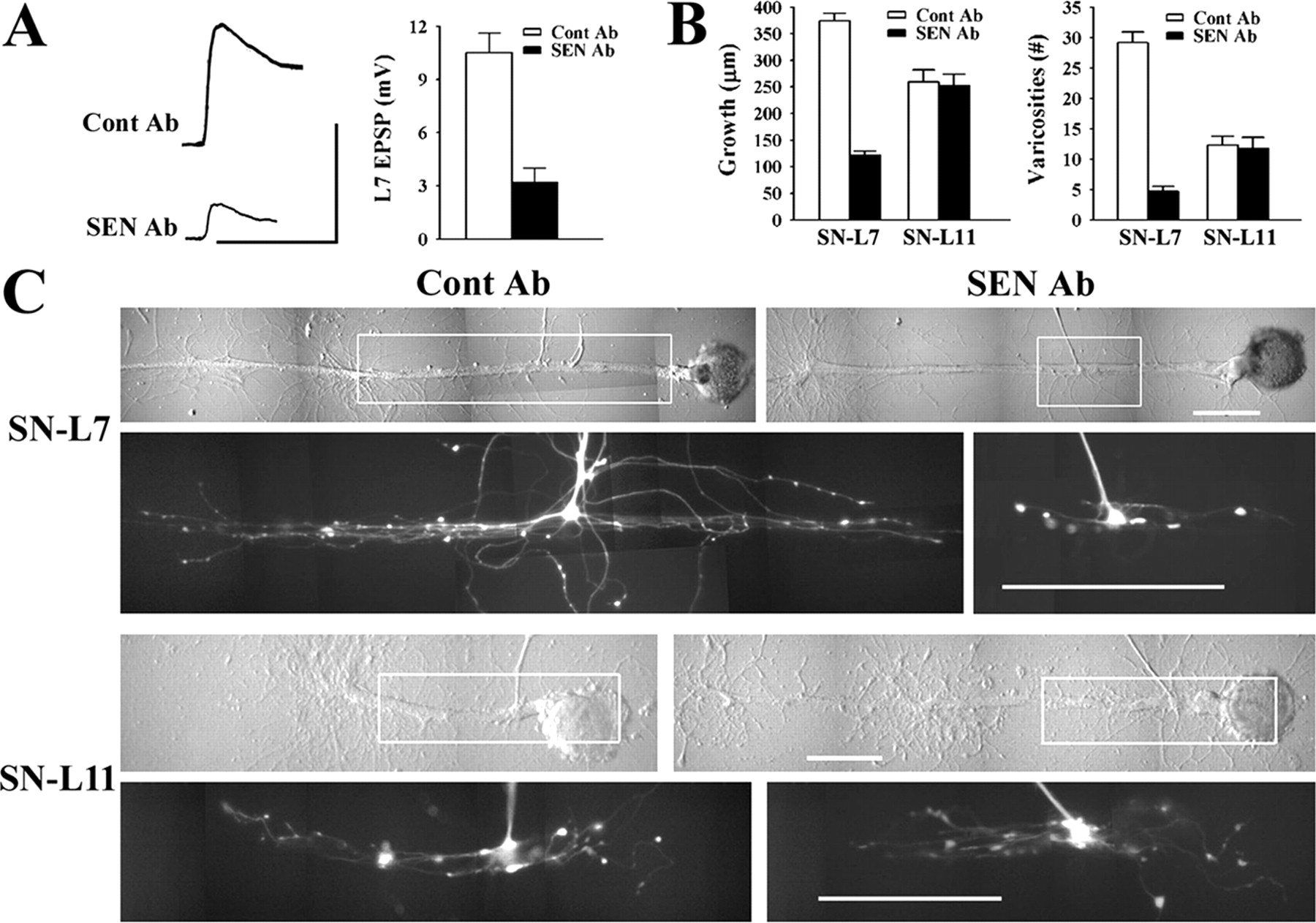
Exogenous anti-sensorin Ab blocked synapse maturation and synapse-associated growth. A, Synaptic efficacy is attenuated significantly with incubation with SEN Ab. Representative EPSPs were recorded in L7 after 20 h in the presence of Cont Ab or SEN Ab. Calibration: 5 mV, 25 ms. Incubation with SEN Ab significantly reduced EPSP amplitude (F = 14.509; p < 0.01). B, SEN Ab reduces significantly growth and varicosity formation when sensory neurons contact L7, but not L11. The histograms summarize axon growth and the number of varicosities formed by sensory neurons in contact with the major processes of the targets. An ANOVA indicated a significant effect of target and SEN Ab on synaptic efficacy, neuritic growth and number of varicosities formed (df = 6, 60; F = 58.125; p < 0.001). Individual comparisons indicated that SEN Ab compared with Cont Ab significantly reduced growth and number of varicosities when sensory neurons contacted L7 (F = 65.559, p < 0.001 and F = 64.318, p < 0.001, respectively), whereas there was no significant difference between Cont Ab and SEN Ab on growth and varicosities formed when sensory neurons contact L11. As expected for cultures treated with Cont Ab, growth and varicosity formation was greater when sensory neurons contacted L7 compared with L11 (F = 9.369, p < 0.01 and F = 20.9, p < 0.01, respectively). In contrast, when incubated with SEN Ab, growth of sensory neurons contacting L11 was now significantly greater than the growth in contact with L7 (F = 12.853; p < 0.01). C, Target and exogenous SEN Ab affect sensory neuron growth. Nomarski-contrast and corresponding epifluorescent image of sensory neurites (within outlined area in each micrograph) after dye injection are displayed for each condition. Each column contains the pair of cocultures (SN-L7 and SN-L11) in the same dish treated with either Cont Ab or SEN Ab. Note that contact with L7 induced significantly more growth and more varicosities compared with contact with L11 when cultures are incubated with Cont Ab. Incubation with SEN Ab significantly reduced growth of the sensory neuron contacting L7 to a level that was now less than the growth detected for the other sensory neuron in the same dish contacting L11. Scale bars, 100 μm.
At mature synapses, sensorin secretion is once again upregulated by stimuli that produce LTF. After the stimuli, the secretion of newly synthesized sensorin is regulated by both PKC and PKA activities (Hu et al., 2006, 2007). We therefore examined whether the activities of these kinases regulated sensorin secretion during the early phases of synapse formation. We tested whether the addition of exogenous sensorin during the application of the PKC inhibitor would reverse the actions of the kinase inhibition. We applied sensorin plus PKC inhibitor either at the initial plating of the sensory neuron with L7 or 6 h after plating the coculture. After a 16 h incubation, the EPSP amplitudes were measured and compared with companion cultures treated with control or the PKC inhibitor alone.
The addition of sensorin at the time of plating failed to reverse the blockade of synapse formation by the PKC inhibitor (Fig. 6A). Whereas EPSP amplitudes in control cultures at 16 h after plating was 9.8 ± 1.8 mV (n = 23), PKC inhibitor blocked synapse formation both in the absence of sensorin (0.1 ± 0.1; n = 20) and with sensorin (0.5 ± 0.2 mV; n = 20). The failure of exogenous sensorin to initiate synapse formation when PKC activity is blocked suggests that target-induced initiation of synapse formation mediated by PKC is independent of the target-induced increase in sensorin secretion.

PKC activity is required both for initiating synapse formation and later for the secretion of sensorin. A, Addition of sensorin at plating does not rescue the blockade of synapse formation by the PKCI. Representative EPSPs recorded in L7 after stimulation of each sensory neuron (stimulation artifact) 16 h after plating cocultures in control, PKCI chelerythrine, or chelerythrine plus sensorin neuropeptide (80 ng/ml; PKCI + SEN) added to the cultures at time of plating. Very weak EPSPs were evoked in cultures treated with PKCI with or without sensorin. Calibration: 10 mV, 25 ms. The histogram summarizes the average EPSP amplitudes recorded after treatments. Incubation with the PKC inhibitor (PKCI or PKCI + SEN) blocked synapse formation. An ANOVA indicated a significant effect of treatment (df = 2, 60; F = 79.727; p < 0.001). Individual comparisons indicated that EPSP amplitude with control treatment was significantly greater than treatment with PKCI (F = 67.136; p < 0.01) or PKCI + SEN (47.034; p < 0.01). There was no significant difference in the EPSP amplitude in cultures treated with PKCI compared with cultures treated with PKCI + SEN. B, Addition of sensorin at 6 h after plating rescues the block of synapse formation by the PKC inhibitor. Representative EPSPs recorded in L7 after stimulation of each sensory neuron (simulation artifact) 22 h after plating cocultures in control, PKCI chelerythrine added 6 h after plating or PKCI + SEN (80 ng/ml) added to the cultures 6 h after time of plating. Note that PKCI added at 6 h reduced EPSP amplitude compared with control, whereas the addition of sensorin reversed the block produced by PKCI. Calibration: 10 mV, 25 ms. The histogram summarizes the average EPSP amplitudes recorded after treatments. Incubation with the PKC inhibitor 6 h after plating allowed the initiation of synapse formation but attenuated subsequent synapse maturation. The addition of sensorin reversed most of the attenuation produced by PKCI. An ANOVA indicated a significant effect of treatment (df = 2, 29; F = 12.195; p < 0.001). Individual comparisons indicated that EPSP amplitude with control treatment was significantly greater than treatment with PKCI at 6 h (F = 11.738; p < 0.01), but not significantly different from treatment with and PKCI + SEN at 6 h (p > 0.3). The addition of sensorin together with the PKCI at 6 h significantly increased EPSP amplitude (F = 4.64; p < 0.03) compared with incubation with PKCI alone at 6 h.
In contrast, exogenous sensorin succeeded in reversing the action of PKC inhibition when it was added with the PKC inhibitor at 6 h after plating (Fig. 6B). Incubation with the PKC inhibitor for 16 h starting at 6 h after plating (n = 12) significantly reduced EPSP amplitude to 4.8 ± 1.2 mV compared with an average EPSP amplitude in control cultures (n = 11) of 14.5 ± 2.2 mV at 22 h after plating. Thus PKC activity is required also after the initiation of synapse formation. In contrast to the situation when sensorin was added at the initial plating, sensorin added at 6 h together with the PKC inhibitor (n = 9) reversed the actions of the PKC inhibition, and the average EPSP amplitude was now 10.1 ± 1.9 mV, which was not significantly different from the average EPSP amplitude recorded in the control cultures. These results suggest that PKC activity after synapse initiation regulates the secretion of sensorin required for the maturation of sensory neuron synapses.
Does PKA activity regulate sensorin synthesis, secretion, or synapse-associated growth? We incubated cocultures with an inhibitor of PKA (5 μm KT5720) at plating for 18 h. We then measured EPSP amplitudes with electrophysiology, axon growth by visualizing sensory neurons after fluorescent dye injection, and sensorin expression with immunocytochemistry. Inhibition of PKA activity did not affect synapse formation, growth, or sensorin synthesis (Fig. 7). Average EPSP amplitude in the presence of the PKA inhibitor (n = 14) was 8.6 ± 0.8 mV and was not significantly different from the average EPSP amplitude of 8.8 ± 0.8 mV in controls (n = 13). In the presence of the PKA inhibitor, axon growth and varicosity number in contact with L7 was 385 ± 19 μm and 31 ± 2 varicosities (n = 10). This growth was not significantly different from axon growth of 390 ± 23 μm and 34 ± 3 varicosities contacting L7 in controls (n = 8; Figs. 7A). Sensorin expression in cell body, axons, and distal processes were also unaffected by PKA inhibition (Fig. 7B,C). Compared with controls (n = 6, normalized to 100% in each compartment), staining in cell bodies (102 ± 10%), axons (98 ± 9%), and varicosities (97 ± 9%) was not significantly affected by the PKA inhibitor (n = 6). Thus unlike the critical role of PKA activity after stimuli that produce long-term synaptic facilitation at sensory neuron synapses, PKA activity does not appear to play a critical role in regulating the initial formation and maturation of sensory neuron synapses.
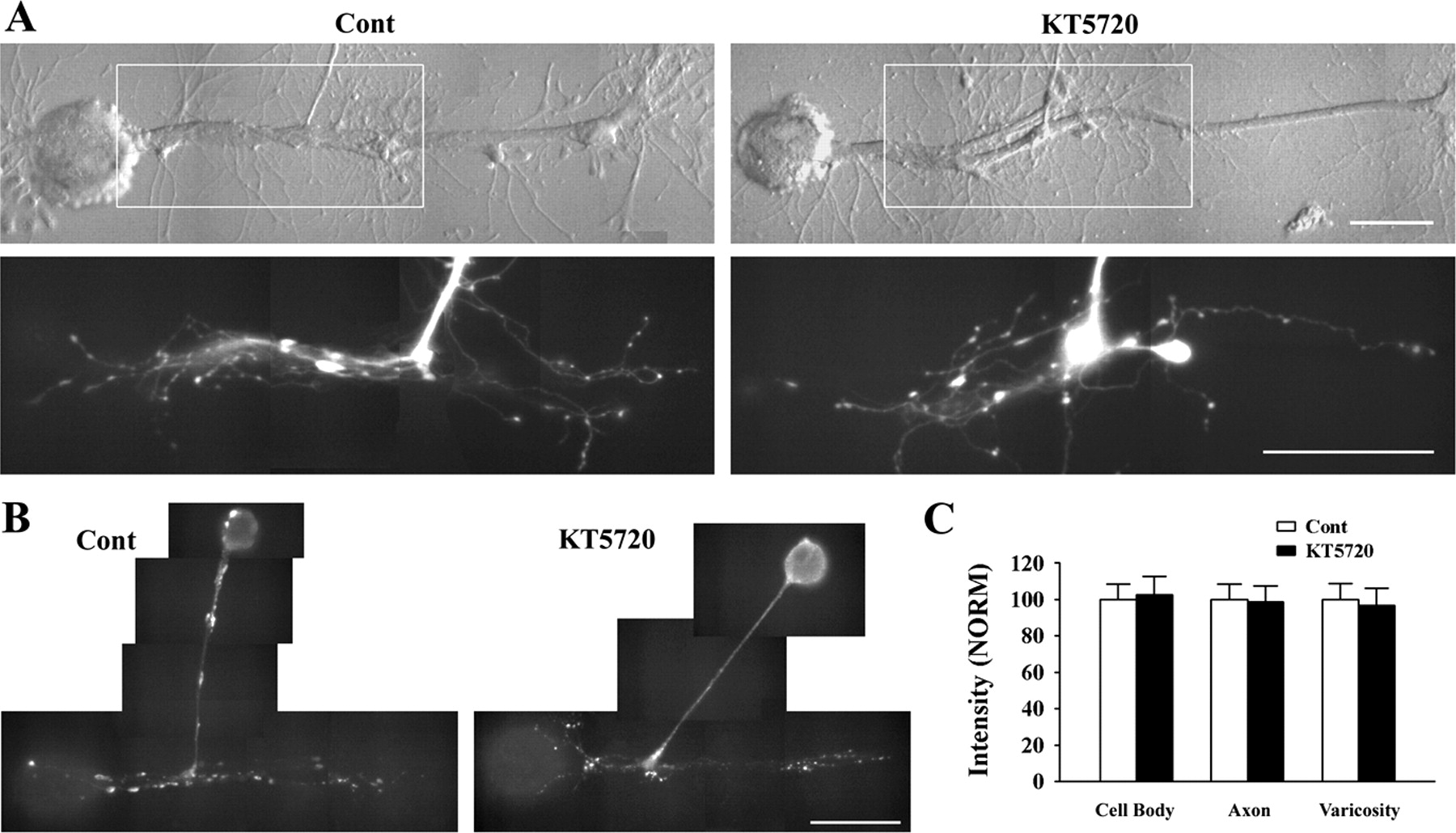
PKA inhibitor failed to affect synapse formation, growth, and varicosity formation by sensory neurons and sensorin expression. EPSP amplitude was not affected by incubation with the PKA inhibitor (5 μm KT5720) for 18 h after plating. A, Growth and varicosity formation was not affected by incubation with the PKA inhibitor for 18 h. Nomarski-contrast and corresponding epifluorescent image of sensory neurites (within outlined area in each micrograph) after dye injection are displayed for each condition. Growth from the axon stump of both sensory neurons is extensive and has numerous varicosities. ANOVA indicated no significant effect of treatment, and individual comparisons indicated no significant difference in growth and varicosity formation by the sensory neurons. Scale bars, 100 μm. B, C, Sensorin expression in sensory neurons is not affected by incubation with the PKA inhibitor KT5720. Epifluorescent images of sensorin immunostaining in a control culture and in a culture incubated with the PKA inhibitor KT5720 (B). Staining intensities in all compartments are comparable. Scale bar, 100 μm. The histogram in C indicates that staining in each compartment of the sensory neurons exposed to KT5720 was not significantly different from the staining in controls (normalized to 100%).
Secreted sensorin activates and translocates MAPK in sensory neurons and is required for synapse-associated growth
Long-term facilitation of mature sensory neuron synapses with 5-HT involves a rapid increase in the secretion of sensorin, which leads to the activation and translocation of p42/44 MAPK into nuclei of sensory neurons that is required for long-term synaptic facilitation (Martin et al., 1997; Hu et al., 2004a). We therefore tested whether the target-dependent secretion of sensorin leads to activation and translocation of MAPK in sensory neurons contacting L7 but not in sensory neurons contacting L11. Immunocytochemistry was used to examine the staining intensity and distribution of both phosphorylated p42/44 MAPK and total p42/44 MAPK in sensory neurons contacting L7 or L11 for 20 h in the presence of control Ab or anti-sensorin Ab. All dishes contained both types of coculture.
MAPK activation and translocation was pronounced in sensory neurons contacting L7 but not L11 (Fig. 8). This activation and translocation in sensory neurons contacting L7 was blocked when cultures were incubated with anti-sensorin Ab (Fig. 8A,B). The increase in phosphorylated MAPK staining was not a result of an increase in overall MAPK expression. Overall staining for phosphorylated MAPK in the cell bodies of sensory neurons contacting L7 (n = 6) was 1.5-fold greater than overall staining in sensory neurons contacting L11 plated in the same cultures and 1.4-fold greater than the staining in sensory neurons contacting L7 and incubated with anti-sensorin Ab (n = 6). The staining in nuclei for sensory neurons contacting L7 was even greater: 1.9-fold greater than staining in nuclei of sensory neurons contacting L11 and 2.1-fold greater than staining in nuclei of sensory neurons contacting L7 in the presence of anti-sensorin Ab. Staining for all forms of p42/44 MAPK in the cell bodies of sensory neurons contacting L7 in the presence of control Ab (n = 6) or in the presence of anti-sensorin Ab (n = 6) did not differ significantly from the staining in cell bodies of sensory neurons contacting L11 (n = 6) plated in the same culture dishes. However, the distribution of the staining differed and indicated that existing MAPK was translocated into the nuclei of sensory neurons contacting L7 as a result of sensorin secretion. Staining intensity (arbitrary units) of all forms of p42/44 MAPK for sensory neurons contacting L7 in the presence of control Ab was 64 ± 4 units in the entire cell body compared with 62 ± 3 units for sensory neurons contacting L7 and incubated with anti-sensorin Ab or 61 ± 4 units for sensory neurons contacting L11. However, the distribution of total MAPK staining differed significantly (df = 4, 30; F = 32.613; p < 0.001). Staining intensity in the nucleus for sensory neurons contacting L7 (72 ± 3 units) was significantly greater than staining intensity in the nucleus of sensory neurons contacting L7 in the presence of anti-sensorin Ab (50 ± 3; F = 14.347; p < 0.01) or the staining intensity in the nucleus of sensory neurons contacting L11 (51 ± 3 units; F = 12.975; p < 0.01). Staining in the cytoplasm of sensory neurons contacting L7 was reduced significantly compared with the other treatments in compensation for the increase in the nucleus: 55 ± 3 units compared with 71 ± 4 for sensory neurons contacting L7 in the presence of anti-sensorin Ab (F = 5.011; p < 0.03) and 69 ± 4 units for sensory neurons contacting L11 (F = 4.518; p < 0.03). Thus p42/44 MAPK phosphorylation and translocation into nuclei of sensory neurons at 20 h is mediated by target-dependent secretion of sensorin. Because blocking MAPK activated by sensorin with U0126 interferes with long-term facilitation produced by 5-HT (Hu et al., 2004a), we tested whether blocking MAPK with the inhibitor U0126 during the first 20 h interfered with synapse maturation and synapse-associated growth.

Contact with L7, which increased the secretion of sensorin, resulted in the activation and translocation of p42/44 MAPK in sensory neurons. A, Epifluorescent images of sensory neurons immunostained for phospho-MAPK. The sensory neurons contacting L7 (SN-L7) and the sensory neuron contacting L11 (SN-L11) were plated in the same dish and exposed to control Ab for 20 h before fixation. The other dishes contained SN-L7 cocultures incubated with SEN Ab. Note that staining for phospho-MAPK in the nuclei of the SNs is less than staining in the cytoplasm when SNs contact L11 or when SN-L7 cocultures are incubated with SEN Ab. Scale bar, 25 μm. B, Summary of the effect of target and incubation with SEN Ab on staining intensity (arbitrary units) for phospho-MAPK in the entire cell body, nucleus, and cytoplasm of sensory neurons. An ANOVA indicated a significant effect of treatment and target (df = 4, 30; F = 41.493; p < 0.001). Individual comparisons indicated that overall cell body and nuclear staining were significantly greater in sensory neurons contacting L7 incubated with Cont Ab compared with sensory neurons contacting L7 and incubated with SEN Ab (F = 9.605, p < 0.01 and F = 46.78, p < 0.01, respectively) or compared with sensory neurons contacting L11 and incubated with Cont Ab (F = 11.272, p < 0.01 and F = 40.259, p < 0.01, respectively). Staining and its distribution in sensory neurons contacting L7 and incubated with SEN Ab were not significantly different from the staining in sensory neurons contacting L11.
Incubation with MAPK inhibitor U0126 (2 μm) produced the same changes as incubations with anti-sensorin Ab (Fig. 9). Average EPSP amplitude at 18 h was 3.6 ± 0.6 mV when cultures were incubated with U0126 (n = 8) compared with 11.7 ± 1.4 mV when incubated with control U0124 (2 μm; n = 7) (Fig. 9A). The effects of the MAPK inhibitor and control compound on growth and the number of varicosities formed in contact with L7 correlated with the effects produced on synaptic strength (Fig. 9B,C). For cultures incubated with U0126 (n = 8), axon growth and varicosities formed by sensory neurons contacting L7 were reduced significantly to 111 ± 11 μm and 4 ± 1 varicosities compared with 377 ± 14 μm and 27 ± 2 varicosities when incubated with the control compound (n = 7). The axon growth by sensory neurons contacting L7 was reduced significantly by U0126 compared with the axon growth by sensory neurons contacting L11 in the same dishes (263 ± 17 μm). In contrast, axon growth of sensory neurons contacting L11 in the presence of control compound (281 ± 11 μm) did not differ significantly from the growth in the presence of the inhibitor (Fig. 9B,C). The number of varicosities formed by sensory neuron axons extending in contact with L11 was also unaffected by the MAPK inhibitor compared with the control compound. Thus activation of MAPK by target-dependent secretion of sensorin is required for synapse-associated growth and not for axon extension by sensory neurons contacting nonsynaptic targets.

MAPK inhibitor U0126 blocked synapse maturation and synapse-associated growth. A, Synaptic efficacy was attenuated significantly with incubation with inhibitor U0126 compared with the control compound U0124. EPSPs were recorded in L7 with a single action potential in each sensory neuron plated with L7 for 18 h in the presence of inhibitor U0126 (2 μm) or control U0124 (2 μm). Incubation U0126 significantly reduced but did not abolish synaptic efficacy (F = 20.495; p < 0.01). Calibration: 5 mV, 25 ms. B, Incubation with MAPK inhibitor U0126 reduced significantly both growth and varicosity formation when sensory neurons contacted L7, but not L11. The histograms summarize axon growth and the number of varicosities formed by sensory neurons in contact with the major processes of the targets. An ANOVA indicated a significant effect of the target and inhibitor U0126 on synaptic efficacy, axon growth, and number of varicosities formed (df = 6, 52; F = 58.591; p < 0.001). Individual comparisons indicated that MAPK inhibitor U0126 compared with control U0124 significantly reduced growth and number of varicosities when sensory neurons contacted L7 (F = 60.601, p < 0.001 and F = 38.351, p < 0.01, respectively), whereas there was no significant difference between incubation with U0126 and U0124 on growth and number of varicosities formed by sensory neurons contacting L11. As expected for cultures treated with the control U0124, growth and varicosity formation were greater when sensory neurons contacted L7 compared with L11 (F = 7.408, p < 0.01 and F = 16.932, p < 0.01, respectively). In contrast, when incubated with the inhibitor U0126, growth by sensory neurons contacting L11 was now significantly greater than sensory neuron growth in contact with L7 in the same culture dishes (F = 21.322; p < 0.01). C, Target and inhibitor U0126 affect sensory neuron growth. Nomarski-contrast and corresponding epifluorescent image of sensory neurites (within outlined area in each micrograph) after dye injection are displayed for each condition. Each column contains the pair of cocultures (SN-L7 and SN-L11) in the same dish treated with either control U0124 or MAPK inhibitor U0126. Contact with L7 induces significantly more growth and more varicosities compared with L11 when cultures are incubated with U0124. Incubation with U0126 significantly reduced growth of sensory neurons contacting L7 to a level that was now less than the growth detected for the sensory neuron in the same dish contacting L11. Scale bars, 100 μm.
PI3K activity is required for synapse-associated growth
Incubation with an inhibitor of PI3K did not affect target-induced regulation of sensorin expression, but appeared to alter growth of sensory neurons in contact with L7 (Fig. 2). We therefore tested whether PI3K activity contributed to synapse-associated growth after the initiation of synapse formation.
Incubation with PI3K inhibitor LY294002 (2 μm) produced the same changes as incubations with MAPK inhibitor; the inhibitor reduced the amplitude of the EPSP compared with controls (Fig. 10A). Average EPSP amplitude at 18 h was 3.1 ± 0.4 mV when cultures were incubated with the PI3K inhibitor (n = 15) compared with 9.0 ± 0.9 mV for control (n = 13). The effects of the inhibitor on growth and the number of varicosities formed in contact with L7 correlated with the effects produced on synaptic strength (Fig. 10B,C). For cultures incubated with the PI3K inhibitor (n = 7), axon growth and varicosity number formed by sensory neurons contacting L7 was reduced significantly to 113 ± 12 μm and 4 ± 1 varicosities compared with 362 ± 30 μm and 27 ± 3 varicosities in control cultures (n = 6). Axon growth by sensory neurons contacting L7 was reduced significantly by PI3K inhibitor compared with the axon growth by sensory neurons contacting L11 in the same dishes (231 ± 19 μm; n = 6). In contrast, axon growth of sensory neurons contacting L11 in control cultures (239 ± 22 μm; n = 5) did not differ significantly from growth in the presence of the inhibitor (Fig. 10B,C). The number of varicosities formed by sensory neuron axons extending in contact with L11 was also unaffected by the PI3K inhibitor compared with the controls. Thus, as was the case for MAPK, activation of PI3K is required for synapse-associated growth and not for axon extension by sensory neurons contacting a nonsynaptic target.

PI3K inhibitor LY294002 blocked synapse maturation and synapse-associated growth. A, Synaptic efficacy was attenuated significantly with incubation with the PI3K inhibitor compared with the control. EPSPs were recorded in L7 with a single action potential in each sensory neuron plated with L7 for 18 h in the presence of inhibitor LY294002 (2 μm) or control. Incubation with the inhibitor significantly reduced synaptic efficacy (F = 50.964; p < 0.01). Calibration: 5 mV, 25 ms. B, Incubation with PI3K inhibitor reduced significantly both growth and varicosity formation when sensory neurons contacted L7, but not L11. The histograms summarize axon growth and the number of varicosities formed by sensory neurons in contact with the major processes of the targets. An ANOVA indicated a significant effect of the target and PI3K inhibitor on synaptic efficacy, axon growth, and number of varicosities formed (df = 3, 19; F = 24.937; p < 0.001). Individual comparisons indicated that the PI3K inhibitor compared with control significantly reduced growth and number of varicosities when sensory neurons contacted L7 (F = 26.167, p < 0.01 and F = 23.262, p < 0.01, respectively), whereas there was no significant difference between incubation with LY294002 and control on growth and number of varicosities formed by sensory neurons contacting L11. As expected for cultures treated with the control, growth and varicosity formation were greater when sensory neurons contacted L7 compared with L11 (F = 5.509, p < 0.02 and F = 7.955, p < 0.01, respectively). In contrast, when incubated with the PI3K inhibitor, growth by sensory neurons contacting L11 was now significantly greater than sensory neuron growth in contact with L7 in the same culture dishes (F = 6.491, p < 0.01 for growth and F = 3.995, p < 0.05 for varicosities). C, Target and inhibitor LY294002 affect sensory neuron growth. Nomarski-contrast and corresponding epifluorescent image of sensory neurites (within outlined area in each micrograph) after dye injection are displayed for each condition. Each column contains the pair of cocultures (SN-L7 and SN-L11) in the same dish treated with either control or PI3K inhibitor LY294002. Contact with L7 induces significantly more growth and more varicosities compared with L11 when cultures are incubated with control. Incubation with LY294002 significantly reduced growth of sensory neurons contacting L7 to a level that was now less than the growth detected for the sensory neuron in the same dish contacting L11. Scale bars, 100 μm.
Discussion
Our results suggest that sequential PKC activities linked to the activation of additional signal pathways by a secreted neuropeptide first induce synapse formation with specific targets and then the extensive branching and synapse formation with those appropriate partners (Fig. 11). PKC is necessary for initiating synapse formation and the axon growth from sensory neurons associated with the formation of synapses with an appropriate target. PKC is also required for synapse maturation through its regulation of sensorin synthesis and secretion. The secreted sensorin activates and translocates p42/44 MAPK into the sensory neuron nucleus to produce synapse-associated growth. Consistent with sensorin acting as a neurotrophin-like molecule affecting several signaling pathways, PI3K activity also contributes to synapse-associated growth.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of the events and signaling pathways mediating the formation and maturation of sensory neuron synapses with a specific postsynaptic target. Interaction between appropriate cells leads to PKC-dependent initiation of synapse formation and growth followed by a PKC-dependent increase in the synthesis and secretion of the neuropeptide sensorin [for the role of PKC activity in regulating sensorin synthesis and secretion in activity-dependent LTF, see Hu et al., (2007)]. We do not know whether enhanced or constitutive PKC activity is required to initiate synapse formation (dashed arrow a). Because sensorin application in the presence of PKC inhibition failed to initiate synapse formation, sensorin's capacity to modulate synapse maturation may require a prerequisite step that is completed during the initial formation of the synapse (dashed arrow b). PKA activity is required neither for initial synapse formation nor for the increase in sensorin synthesis and secretion. Secreted sensorin is required for synapse maturation and growth with the appropriate target by activating and translocating MAPK into the nuclei of the sensory neurons. PI3K activity is also required for synapse maturation, but we do not have direct evidence that PI3K is activated by sensorin during this phase of synapse formation (dashed arrow c). Activation of PI3K by sensorin is required for sensorin-dependent LTF at mature sensory neuron synapses (J. Y. Hu and S. Schacher, unpublished observations).
PKC regulates the initial formation of synapses with specific targets
Several synaptic cell adhesion molecules have been identified and characterized that initiate the assembly of presynaptic and postsynaptic structures [for review, see Akins and Biederer (2006), Craig and Kang (2007), Dalva et al. (2007), and Takeichi (2007)]. Downstream signaling molecules may include small GTPases (Elia et al., 2006; Paradis et al., 2007) and specific kinases or phosphatases (Kaufmann et al., 2002; Patel et al., 2006; Sytnyk et al., 2006) that affect the organization of the cytoskeleton required for the formation of synapses. One kinase that might regulate synaptic adhesion and assembly is PKC (Loeb et al., 1998; Xia et al., 2003; Schmidt et al., 2004; Kolkova et al., 2005). PKC inhibition selectively blocked all growth and varicosity formation from sensory neurons when their axon stumps contacted L7, but not when they contacted L11. Thus growth associated with the initial formation of synapses has different requirements than growth associated with axon extension along a target that fails to induce synapse formation.
We do not know the nature of the signal from L7 associated with the PKC-dependent growth and synapse formation and the increase in cell-wide expression of sensorin in sensory neurons (see below). The signal(s) must work at close range because synapse specificity and branch-specific cellular changes are observed even when a single sensory neuron interacts with L7 and L11 simultaneously (Santarelli et al., 1996; Schacher et al., 1999; Hu et al., 2002). Local cell–cell communication through ligand-receptor binding of a secreted product or through binding of homologous or heterologous surface receptors may trigger this cell-specific response. We also do not know whether the PKC activities regulating the initial formation of the synapse and growth are increased by target interaction; whether they are presynaptic, postsynaptic, or both; or the types of PKC isoforms involved (Sossin, 2007). PKC-dependent changes in the local expression and distribution of cell surface molecules on L7 or the secretion of a retrograde signal from L7 might regulate the presynaptic response to initiate synapse formation and growth. Whatever these initial events might be, they do not appear to be mediated by the actions of PKC on sensorin synthesis and secretion (see below). High levels of exogenous anti-sensorin Ab did not block the initial formation of synapses, and exogenous sensorin applied at the time of plating did not induce synapse formation during PKC inhibition. Timely PKC activities are required for multiple functions in the formation and maturation of a specific synapse.
PKC-dependent changes in sensorin expression and secretion affect synapse-associated growth
Release of local factors plays important roles in synapse maturation. Maintenance of transmitter receptor clusters at the neuromuscular junction requires activation of neurotrophin receptors (Gonzalez et al., 1999), whereas increase in the number of presynaptic structures at the Drosophila neuromuscular junction requires release of BMP-like molecules and downstream signaling (McCabe et al., 2003, 2004). Synapse number in developing hippocampus and other CNS areas are regulated by levels of neurotrophins such as BDNF (Martinez et al., 1998; Vicario-Abejon et al., 1998; Alsina et al., 2001; Shen et al., 2006). PKC and neuropeptide signaling are critical for the formation of cholinergic synapses in C. elegans (Sieburth et al., 2005). It is unclear, however, whether these secreted neuropeptides are instructive for the formation and maturation of synapses with specific target neurons or are only permissive for synapse maturation in the target region.
Our results suggest that interaction with a synaptic target induced rapid PKC-dependent increases in sensorin expression and secretion. Because sensorin mRNA transport to and accumulation at distal sites are affected by target interaction (Schacher et al., 1999; Hu et al., 2002; Lyles et al., 2006), the rapid increase in sensorin in sensory neurons contacting L7 may be mediated by a PKC-dependent change in sensorin expression (mRNA and protein) throughout the sensory neuron. Protein synthesis, including synthesis at distal sites of sensory neurons, is required for synapse maturation and growth (Schacher and Wu, 2002). Protein synthesis inhibitors blocked synapse-associated growth of sensory neurons (Schacher and Wu, 2002; Hu et al., 2004a) and interfered with the local synthesis of sensorin (Hu et al., 2002, 2004a, 2006, 2007; Liu et al., 2003). Inhibition of PKC also blocked the rapid increase in local synthesis of sensorin after paired stimuli that produce an associative form of LTF (Hu et al., 2007). Because paired stimuli activate the calcium-dependent PKC in the sensory neurons (Zhao et al., 2006), it is possible that interaction with the correct target leads to the activation of this calcium-dependent PKC in the sensory neurons to regulate sensorin expression. In contrast, PI3K, which is required for local synthesis of sensorin after stimuli that produces nonassociative LTF (Hu et al., 2006), does not play a role in regulating the target-induced increase in sensorin expression.
The secretion of sensorin apparently requires PKC. In contrast, PKA activity does not appear to be involved with sensorin release or synapse maturation. This was surprising because at mature synapses after stimuli that produce LTF, PKA and PKC are both required for the rapid release of newly synthesized sensorin, and PKA activity is required for sensorin-induced activation and translocation of MAPK (Hu et al., 2004a, 2006). During the initial phases of synapse formation, PKC may specifically regulate sensorin release from varicosities, structures containing both high levels of sensorin and transmitter release sites (Glanzman et al., 1989; Hatada et al., 1999; Liu et al., 2003; Hu et al., 2004b). Because varicosities contacting L11 contain low levels of sensorin and no active zones (Glanzman et al., 1989), these results might explain the low levels of sensorin release when sensory neurons contact L11 (Hu et al., 2004b). Sensorin's downstream signaling pathways do not require “priming” by PKA during synapse maturation because constitutive release of sensorin alone appears to influence synapse maturation (Hu et al., 2004b). The priming of these downstream events may occur during the initial phase of synapse formation because synapse initiation is a prerequisite for sensorin's role in synapse maturation (Figs. 6, 11).
The secretion of sensorin has important consequences on sensory neuron growth along a specific target (L7). Branch growth, varicosity formation, and synaptic efficacy are all attenuated when the actions of sensorin are blocked by exogenous anti-sensorin antibody. The presence of anti-sensorin antibody had no detectable impact on growth by sensory neurons contacting L11 or on sensory neurons plated alone (Hu et al., 2004b). In the presence of anti-sensorin antibody, growth of sensory neurons contacting L11 was greater than the growth of sensory neurons contacting L7 plated in the same dish. Sensorin appears to be critical for presynaptic axon growth associated specifically with the formation of additional synapses (synapse maturation). Sensorin may also act on L7 to regulate postsynaptic properties that compliment the formation of presynaptic varicosities (Conrad et al., 1999). Some targets of sensory neurons respond with membrane polarizations to exogenous sensorin (Brunet et al., 1991), but L7 does not (Hu et al., 2004a). In addition, applications of sensorin failed to activate or translocate MAPK in L7 (Hu et al., 2004b). By analogy to neurotrophins such as BDNF, sensorin may activate other signaling pathways in L7 (PI3K and PLC, for example) that could facilitate both presynaptic growth and the formation of apposed postsynaptic structures required for functioning synapses (Zhu et al., 1994; Gonzalez et al., 1999; Wells et al., 1999; Bamji et al., 2006).
Activation and translocation of MAPK in sensory neurons and activation of PI3K are required for synapse-associated growth
Our results indicate that sensorin-mediated activation and translocation into nuclei of p42/44 MAPK in sensory neurons and the activation of PI3K are required for synapse-associated growth with L7, but not axon growth along targets (L11) that fail to initiate synapse formation by sensory neurons. Sensorin-mediated activation and translocation of p42/44 MAPK into sensory neuron nuclei are also critical for synaptic growth associated with long-term facilitation of mature sensory neuron synapses (Hu et al., 2004a, 2006, 2007). Thus the same secreted peptide and signaling pathway required for activity-dependent long-term plasticity at mature sensory neuron synapses [a cellular correlate of long-term memory (Kandel, 2001)] is critical for the maturation of specific synapses in the neural circuit mediating the behavior. The expression of numerous neuropeptides in the mature nervous system may reflect the persistent expression of molecules first required for the maturation of specific synapses.
The decision to form and maintain specific synapses requires both local and cell-wide changes. Target-induced regulations of gene expression, translation, and posttranslational modification of specific substrates are all required for synapse formation and maintenance (Greenough et al., 2001; Meems et al., 2003; Javaherian and Cline, 2005). Signaling pathways such as p42/44 MAPK and PI3K could regulate both local and cell-wide changes. Timely activations of these signaling pathways could influence the distribution of cell adhesion or other molecules or local protein synthesis critical for establishing synaptic contacts (Zhu et al., 1994; Bailey et al., 1997; Ginty and Segal, 2002; Koh et al., 2002; Kelleher et al., 2004; Bamji et al., 2006). Translocation of MAPK into sensory nuclei may serve to phosphorylate transcription factors that regulate expression of gene products required for the formation of new synaptic contacts (Martin et al., 1997; Davis et al., 2000; Sweatt, 2004). The potential activation of a receptor tyrosine kinase by sensorin (Hu et al., 2004a; Ormond et al., 2004) may set in motion the activation of a number of signal transduction pathways, as when Trk-like receptors bind neurotrophins (Huang and Reichardt, 2001; Segal, 2003) to regulate the maturation and maintenance of synapses contacting specific targets.
Footnotes
- Received July 20, 2007.
- Revision received August 27, 2007.
- Accepted September 13, 2007.
-
This work was supported by National Institutes of Health (NIH) Grants MH 60387 and NS 42159. Animals were provided by the National Center for Research Resources for Aplysia at the University of Florida in Miami, which is supported by NIH Grant RR-10294. We thank Drs. John Koester and Alejandro Hernandez for comments on earlier drafts of this manuscript.
- Correspondence should be addressed to Samuel Schacher, Center for Neurobiology and Behavior, Columbia University College of Physicians and Surgeons, New York State Psychiatric Institute, 1051 Riverside Drive, New York, NY 10032. sms2{at}columbia.edu
- Copyright © 2007 Society for Neuroscience 0270-6474/07/2711712-13$15.00/0