

Abstract
Dentate granule cell (DGC) neurogenesis persists throughout life in the mammalian hippocampal dentate gyrus and increases after epileptogenic insults. The DGC layer in human and experimental mesial temporal lobe epilepsy (mTLE) often shows abnormal dispersion and the appearance of hilar-ectopic DGCs. In the pilocarpine mTLE model, hilar-ectopic DGCs arise as a result of an aberrant chain migration of neural progenitors. Reelin is a secreted migration guidance cue that persists in the adult rodent and human hippocampus. We tested the hypothesis that loss of Reelin in the epileptic dentate gyrus leads to aberrant chain migration of DGC precursors. We found that interneuron subsets typically lost in human and experimental mTLE express Reelin, and DGC progenitors express the downstream Reelin signaling molecule Disabled 1 (Dab1). Prolonged seizures decreased Reelin immunoreactivity in the adult rat dentate gyrus and increased Dab1 expression in hilar-ectopic neuroblasts. Exogenous Reelin increased detachment of chain-migrating neuroblasts in dentate gyrus explants, and blockade of Reelin signaling increased chain migration. These findings suggest that Reelin modulates DGC progenitor migration to maintain normal DGC integration in the neonatal and adult mammalian dentate gyrus. Loss of Reelin expression in the epileptic adult hippocampus, moreover, likely contributes to ectopic chain migration and aberrant integration of newborn DGCs.
Introduction
Reelin is a key factor that regulates neuronal migration during cerebral development. Its expression in the embryonic marginal zone is required because reeler mice with spontaneous Reelin mutations develop an inverted cortex in which early born neurons migrate to the superficial cortical plate instead of stopping at deeper layers (D'Arcangelo et al., 1995) (for review, see Rice and Curran, 2001; Tissir and Goffinet, 2003). Reelin binds to the apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) expressed on neurons or radial glia (D'Arcangelo et al., 1999; Hiesberger et al., 1999; Trommsdorff et al., 1999; Luque et al., 2003). Binding and internalization leads to phosphorylation, downstream signaling, and subsequent degradation of the adaptor protein Disabled 1 (Dab1) (Howell et al., 1997; Sheldon et al., 1997; Ware et al., 1997). Inactivation of Dab1 by gene targeting or spontaneous mutation causes a reeler-like phenotype (Howell et al., 1997; Sheldon et al., 1997; Ware et al., 1997).
Reelin signaling also is necessary for dentate granule cell (DGC) layer lamination in the hippocampal dentate gyrus. Dab1, Reelin, and Reelin receptor mutant or knock-out mice have markedly abnormal DGC layers (Stanfield and Cowan, 1979; Förster et al., 2002; Weiss et al., 2003). Dentate lamination defects in reeler hippocampal cultures are rescued by coculture with Reelin-expressing tissue (Zhao et al., 2004, 2006). Reelin may indirectly influence DGC layer lamination by affecting formation of the radial glial scaffold on which dentate neuroblasts migrate (Förster et al., 2002; Weiss et al., 2003; Zhao et al., 2004, 2006); however, a direct action of Reelin on DGC progenitor migration has not been excluded. In the adult rodent olfactory bulb, for example, Reelin acts directly to detach chain-migrating neuroblasts so they may reach their appropriate laminar destinations (Hack et al., 2002). Reelin expression persists also in the adult rodent and human dentate gyrus (Impagnatiello et al., 1998; Pesold et al., 1998), but its role there is unknown.
Abnormal DGC layer lamination is a feature of human mesial temporal lobe epilepsy (mTLE) (Houser, 1990), a common and intractable epilepsy. The dentate gyrus often shows mossy fiber sprouting, DGC layer dispersion, loss of hilar interneurons, and ectopic granule-like neurons in the hilus and molecular layer (de Lanerolle et al., 1989; Houser, 1990; Houser et al., 1990; Parent et al., 2006). The role these abnormalities play in mTLE pathogenesis is unknown, but similar pathology is recapitulated in adult rodent models. Mossy fiber sprouting, DGC layer dispersion, and loss of hilar interneuron subsets occur in the pilocarpine mTLE model (Mello et al., 1993; Obenaus et al., 1993; Mello and Covolan, 1996; Kobayashi and Buckmaster, 2003). Pilocarpine- or kainic acid-induced status epilepticus (SE) also markedly stimulates DGC neurogenesis and leads to the appearance of hilar-ectopic granule cells (Bengzon et al., 1997; Parent et al., 1997, 2006; Scharfman et al., 2000; Dashtipour et al., 2001). Ectopic DGCs integrate abnormally, are hyperexcitable, and thus may contribute to seizures (Scharfman et al., 2000; Dashtipour et al., 2001).
Evidence suggests that loss of Reelin expression correlates with the degree of DGC layer dispersion in human mTLE (Haas et al., 2002). The same group has shown that dentate Reelin expression and neurogenesis both decrease after intrahippocampal kainate-induced SE in adult mice (Heinrich et al., 2006). These findings led to the conclusion that DGC layer dispersion after SE arises from abnormal movement of mature DGCs secondary to Reelin deficiency disrupting the radial glial scaffold. This mTLE model, however, is atypical in having severe dentate injury and suppressed neurogenesis. Studies of multiple different adult rodent models of mTLE or acute seizures, in contrast, show markedly upregulated subgranular zone (SGZ) cell proliferation or DGC neurogenesis (Bengzon et al., 1997; Parent et al., 1997, 1998; Gray and Sundstrom, 1998; Scott et al., 1998; Nakagawa et al., 2000; Hattiangady et al., 2004; Mohapel et al., 2004; Jessberger et al., 2005; Overstreet-Wadiche et al., 2006). We found that hilar- and molecular layer-ectopic DGCs arise after pilocarpine-induced SE from aberrant chain migration of DGC progenitors (Parent et al., 2006). Although not seen in the intact dentate, chain migration is typical of adult rodent subventricular zone (SVZ) neuroblasts that migrate to the olfactory bulb (Gates et al., 1995; Lois et al., 1996); as mentioned above, Reelin is necessary for neuroblasts to exit from the chains and disperse in the bulb (Hack et al., 2002).
Given our observation of abnormal dentate neuroblast chain migration after SE, we hypothesized that Reelin maintains the short-distance migration of neuroblasts into the DGC layer and that decreased Reelin expression after SE induces aberrant chain migration. We found in the pilocarpine mTLE model that SE decreases dentate Reelin expression and that ectopically located DGC progenitors show evidence of deficient Reelin signaling. Reelin also directly modulated neuroblast migration in dentate gyrus explant cultures. These findings suggest that Reelin is required to maintain the DGC layer and that loss of Reelin in the epileptic hippocampus contributes to mTLE pathology by altering DGC precursor migration.
Materials and Methods
Pilocarpine-induced SE.
Young adult, male Sprague Dawley rats (200–240 g) were pretreated with injections of atropine methylbromide (5 mg/kg, i.p.; Sigma, St. Louis, MO) and, 15 min later, were given pilocarpine hydrochloride (340 mg/kg, i.p.; Sigma) to induce SE. If seizure activity was not initiated within 1 h after the initial pilocarpine hydrochloride dose, an additional dose of 170 mg/kg was given. Seizures were monitored behaviorally and then terminated after 2 h of convulsive SE (head and forelimb clonus) with diazepam (10 mg/kg, i.p.; Elkins-Sinn, Cherry Hill, NJ); additional doses of 5 mg/kg diazepam were given at 45–60 min intervals if subtle convulsive seizure activity persisted. Control rats were treated identically except that 0.9% sodium chloride solution was substituted for pilocarpine. Between 7 and 21 d after SE, rats received an overdose of pentobarbital sodium (Abbott Laboratories, Irving, TX) and were transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA), pH 7.4 (360 ml). After postfixation in 4% PFA overnight, brains were cryoprotected, frozen, and sectioned coronally at 40 μm thickness through the entire rostrocaudal extent of the hippocampal formation. All procedures were approved by the University Committee for the Use and Care of Animals at the University of Michigan Medical Center and followed guidelines contained in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Matrigel/organotoypic slice cultures and Reelin/CR-50/receptor-associated protein treatment.
For three-dimensional cultures (referred to as “Matrigel” cultures), brains from postnatal day 7 (P7) CD-1 mice were removed from the skull and placed in ice-cold OPTI-MEM I (Invitrogen, Carlsbad, CA). The hippocampus was removed, and the dentate gyrus was dissected under a dissecting microscope and cut into pieces 50–200 μm in diameter. A 75% Matrigel/collagen gel was prepared by mixing Matrigel (Collaborative Research, Bedford, MA) with collagen [rat tail tendon, type I (Sigma); 2 mg/ml dissolved in 0.2% acetic acid] in a 3:1 ratio. Explants were mixed with the gel, and 200 μl of gel/explant suspension was placed onto the center of each well in 12-well plates with membrane inserts (Fisher Scientific, Houston, TX). Plates were placed in an incubator (37°C, 5% CO2) to allow gel formation. Once the gel had set, 1.5 ml of Neurobasal A medium supplemented with B27 and 0.5 mm l-glutamine (all from Invitrogen) was added to each well. Explants were returned to the incubator and cultured for 24 h before analysis. SVZ explants were prepared similarly, except that 100% Matrigel was used. To prepare Reelin-conditioned media, COS7 cells were transfected with 1 μg of myc-tagged full-length Reelin plasmid DNA (1.0 μg/μl; pCrlM; a gift from T. Curran, Children's Hospital of Philadelphia, Philadelphia, PA) per 100 μl of Fugene6/DMEM complex and incubated at 37°C/5% CO2 for 5 h. The media in each well then was replaced with 2 ml of serum-free DMEM, and cells were incubated for 48 h at 37°C/5% CO2. Medium from transfected COS7 cells was concentrated using centrifugal concentration tubes (Ultra Centrifugal Filter Devices, 50,000 molecular weight cutoff; Amicon, Beverly, MA) and stored at −70°C. Matrigel-cultured dentate gyrus explants were treated for 24 h with Reelin- or control-conditioned media (added 1:1 to culture media). Some cultures were treated with 2 μl of CR-50 antibody (100 μg/ml) in 1.5 ml of culture medium instead of conditioned media; additional antibody was added to the media after 14 h.
Organotypic hippocampal slice cultures were prepared from P15 mice. Mice were given a single intraperitoneal injection of bromodeoxyuridine (BrdU) (100 mg/kg; Roche Products, Welwyn Garden City, UK), and, 2 h later, brains were removed and placed in ice-cold OPTI-MEM I. Hippocampi were dissected out under a dissecting microscope and embedded in low-melting point agarose (Invitrogen). Hippocampal slices (300 μm thick) were prepared using a vibratome (VT-1000S; Leica, Nussloch, Germany) and cultured on polycarbonate membrane filters in media consisting of 50% MEM/25% normal horse serum/25% Ca2+/Mg2+-free HBSS/20 mm HEPES/1 mm glutamine/5 mg/ml glucose (w/v). Receptor-associated protein (RAP) (1 μm; a gift from D. Strickland, American Red Cross, Rockville, MD) or vehicle was added to the media every 12 h for 48 h, and cultures were fixed in 4% PFA for 1 h and processed for BrdU immunohistochemistry.
Immunohistochemistry and Western blotting.
Single-and double-labeled immunofluorescence histochemistry was performed on free-floating tissue sections, cells, or explants according to previously described methods (Wang et al., 2005; Parent et al., 2006). Primary antibody dilutions used were as follows: 1:1000 for CR-50 (mouse monoclonal; a gift from M. Ogawa, RIKEN Brain Science Institute, Wako, Japan); 1:50 for somatostatin (rat monoclonal; Accurate Chemicals, Westbury, NY); 1:2000 for neuropeptide Y (NPY) (rabbit polyclonal; Bachem, Bubendorf, Switzerland); 1:10,000 for calretinin (rabbit polyclonal; Chemicon, Temecula, CA); 1:500 for polysialylated neural cell adhesion molecule (PSA-NCAM) (mouse IgM 5A5 clone; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); 1:500 for Dab1 (rabbit polyclonal; a gift from B. Howell, National Institute of Neurological Disorders and Stroke, Bethesda, MD); 1:200 for BrdU (rat monoclonal; Accurate Chemicals); 1:400 for class III-β-tubulin (mouse TuJ1 clone; Covance Research Products, Berkeley, CA); and 1:500 for glial fibrillary acidic protein (rabbit polyclonal; Sigma). Secondary antibodies for immunofluorescence were goat anti-mouse or rabbit IgG conjugated to Alexa-594 or -488 (1:400 dilutions; Invitrogen) or goat anti-mouse IgM conjugated to FITC (1:200; Jackson ImmunoResearch, West Grove, PA). Tissue was incubated in secondary antibody overnight at 4°C. For Matrigel cultures, washes were extended to 6 h after each antibody incubation step.
Western blotting was performed on the concentrated media of Reelin-transfected COS7 cells. Protein (40 μg) was resolved on a SDS-PAGE gel (1.5 m Tris, pH 8.8, 30% acrylamide/bis-acrylamide, 10% SDS, 10% ammonium persulfate, and N,N,N',N'-tetramethyl-ethylenediamine) using an electrophoresis apparatus (Bio-Rad, Hercules, CA). Proteins were transferred from the gel onto a 0.45 μm nitrocellulose membrane using a Trans-Blot semidry system (Bio-Rad), and the membrane was washed twice in TBS/0.1% Tween 20 (TTBS) and then blocked in a solution of 5% nonfat dry milk (in TTBS) for 1 h. The membrane was probed with anti-Reelin antibody (G10, 1:1000; Abcam, Cambridge, MA), washed twice (in TTBS), and incubated for 1 h with a horseradish peroxidase-conjugated anti-mouse IgG secondary antibody (diluted 1:5000 in 5% milk). Immunoreactivity was detected by chemiluminescence (Immobilon Western Chemiluminescent HRP substrate; Millipore, Bedford, MA).
Microscopy and cell counts.
For double-labeled immunofluorescence, images were obtained using a Zeiss (Oberkochen, Germany) LSM 510Meta confocal laser microscope as z-series stacks (1.0 μm optical thickness) and transferred to Adobe Photoshop for color merging (Adobe Systems, San Jose, CA). Phase-contrast and single-labeled epifluorescence microscopic images were acquired using a Leica DMIRB inverted microscope and SPOT-RT digital camera (SPOT; Diagnostic Instruments, Sterling Heights, MI). Counts of hilar CR-50-immunoreactive cells were made from digitized epifluorescent images acquired under a 20× objective using three to four sections per animal spanning the rostrocaudal extent of the hippocampus with the observer blinded to experimental condition. Quantification of migrating cells and chains in Matrigel explants was performed on digitized images acquired with a 20× objective under phase-contrast optics from 6–14 explants per condition, with the observer blinded to experimental condition.
Statistical analyses.
Statistical comparisons between groups for counts of dentate hilus CR-50-positive cells were made using an unpaired Student's t test. For dispersed cell and chain numbers in explant cultures, ANOVA and Fisher's PLSD post hoc test was used. Analyses were performed with StatView software (Abacus Concepts, Calabasas, CA). Data are presented as means ± SEM, and the significance level set at p < 0.05.
Results
Prolonged seizures decrease Reelin expression in the adult rat dentate gyrus
To test the hypothesis that altered DGC neuroblast migration results from loss of dentate gyrus Reelin, we compared Reelin expression in saline- and pilocarpine-treated adult rats using the CR-50 antibody that recognizes Reelin (Ogawa et al., 1995). Saline-treated controls displayed Reelin immunoreactivity in putative inhibitory neurons in the dentate SGZ and hilus (Fig. 1 a), as described previously (Pesold et al., 1998). After SE, however, Reelin expression decreased substantially in these regions (Fig. 1 b). The numbers of CR-50-immunoreactive cells were quantified in the dentate hilus of adult rats 14 d after saline or pilocarpine treatment, the time point when aberrant neuroblast chain migration first appears after SE (Parent et al., 2006). Pilocarpine-induced SE significantly decreased the numbers of hilar Reelin-expressing cells compared with controls (mean ± SE of 3.9 ± 1.1 cells per hilar section in pilocarpine-treated rats vs 10.6 ± 0.9 in controls; p = 0.009).

Reelin-immunoreactive neurons in the dentate gyrus decrease after SE. Coronal sections from adult rats immunostained with the CR-50 antibody against Reelin 14 d after treatment with saline ( a , c ) or pilocarpine ( b , d ) are shown. Note the marked decrease in Reelin immunoreactivity in the hilus (h; a , b ) and molecular layer (ml; c , d ) of pilocarpine-treated rats compared with the controls. Scale bar, 50 μm. gcl, Granular cell layer.
A similar effect was seen in the dentate molecular layer of the superior blade. Two weeks after SE, CR-50 expression was greatly reduced compared with controls in some animals (Fig. 1 c,d). Similar to the variability in ectopic molecular layer neurogenesis of pilocarpine-treated rats (Parent et al., 2006), the loss of dentate molecular layer Reelin expression was less consistent than that found in the hilus. Our results do not distinguish between an isolated loss of Reelin protein expression versus death of Reelin-expressing cells in these regions; however, Reelin is expressed by GABAergic interneurons in the adult rat dentate gyrus (Pesold et al., 1998) (Fig. 2), and cell death is likely given the known loss of subsets of GABAergic interneurons after pilocarpine-induced SE (Obenaus et al., 1993; Mello and Covolan, 1996). The loss of hilar and molecular layer Reelin expression correlates with the appearance of aberrant neuroblast chain migration after SE (Parent et al., 2006) and suggests a potential mechanism for the seizure-induced alteration of migratory behavior in DGC progenitors.

Phenotypes of dentate gyrus Reelin- and Dab1-expressing cells. a–d , Many Reelin-immunoreactive cells (CR-50; red in all panels) in the dentate hilus (h) coexpress somatostatin (SS; green, arrows in a ), whereas few double labeled for NPY (green, arrows in b ) and no CR-50-positive cells express calretinin (CR; green in c ) or glutamate receptor 3/4 (GluR; green in d ), a marker of principal neurons that include hilar mossy cells. e–f″ , Dab1/PSA-NCAM double labeling in representative brain sections from an intact control ( e–e″ ) and a rat 14 d after status epilepticus (14 d Sz; f–f″ ). Dab1-expressing cells in the inner granule cell layer (gcl) of the control (arrows in e ) coexpress the neuroblast marker PSA-NCAM (arrows in e″ ) but not the larger Dab1-positive cell in the hilus (arrowhead in e″ ). The 14 d Sz animal, in contrast, shows many Dab1-positive ectopic neuroblasts in the hilus that coexpress PSA-NCAM (arrows in f , f″ ). Dab1 is red and PSA-NCAM is green in the merged images shown in e″ and f″ . Scale bar: a–d , 125 μm; e–f″ , 50 μm.
Reelin and Dab1 expression in the intact and epileptic adult rat dentate gyrus
Based on the above findings, we examined Reelin signaling components in the adult rat dentate gyrus. We first used immunofluorescence double labeling for Reelin and neuropeptides or calcium binding proteins known to be expressed by subsets of dentate hilar interneurons (for review, see Freund and Buzsaki, 1996). Confocal microscopic images showed that many Reelin-expressing hilar neurons coexpressed the neuropeptide somatostatin (Fig. 2 a). Of note, somatostatin interneurons are one of the dentate hilar populations that die in human and experimental mTLE (de Lanerolle et al., 1989; Kobayashi and Buckmaster, 2003). A subset of hilar CR-50-immunoreactive interneurons also coexpressed NPY (Fig. 2 b), and NPY-containing interneurons also are lost after pilocarpine-induced SE (Lurton and Cavalheiro, 1997). Unlike in the developing embryonic cortex, in which calretinin-positive Cajal–Retzius cells express Reelin, no calretinin-immunoreactive cells in the dentate hilus were immunoreactive for Reelin (Fig. 2 c). The principal cells of the hilus are the glutamatergic mossy cells, and they are known to express the AMPA-sensitive glutamate receptor 2 subunit (Leranth et al., 1996). These cells, which also are vulnerable to SE, are not immunoreactive for Reelin in the intact adult rat (Fig. 2 d). Thus, subsets of hilar interneurons that undergo cell death in human and experimental mTLE are the Reelin-expressing cells in the adult rat dentate gyrus.
To determine what cell types respond to Reelin signaling, we used double-labeled immunofluorescence and confocal microscopy to identify the downstream Reelin signaling molecule Dab1 and the neuroblast marker PSA-NCAM. Most DGC progenitors in the SGZ were colabeled, and some PSA-NCAM-negative cells with interneuron morphology and location also expressed Dab1 (Fig. 2 e–e″). One to 3 weeks after SE, more Dab1/PSA-NCAM double-labeled neuroblasts appeared, and many were found in the hilus 2–3 weeks after pilocarpine treatment in association with decreased CR-50 immunoreactivity compared with controls (Figs. 2 e–f″, 3). Because Dab1 protein expression identifies Reelin-responsive cells and it accumulates in the absence of Reelin signaling (Rice et al., 1998), these findings suggest that DGC progenitors in the adult rat respond to Reelin expressed by subsets of dentate interneurons. Moreover, pilocarpine-induced SE injures the interneuron subsets (Kobayashi and Buckmaster, 2003) that we have shown comprise many Reelin-expressing cells, and aberrantly migrating DGC precursors that accumulate in the hilus show evidence that they fail to receive the appropriate Reelin signal.
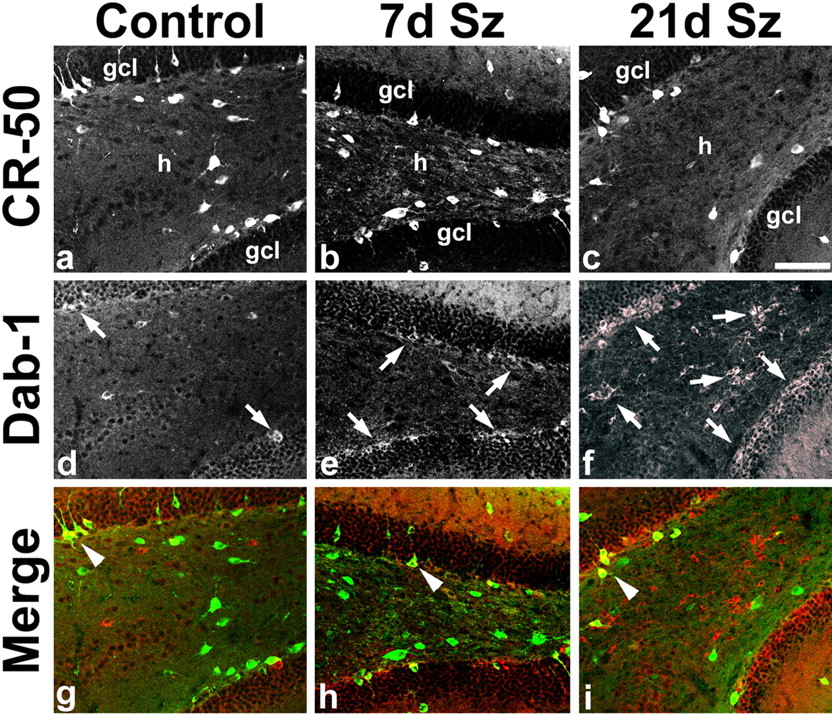
Time course of altered Reelin and Dab1 immunoreactivity after SE. a–c , Dentate gyrus CR-50 immunoreactivity is decreased in the hilus (h) at 21 d after SE (21 d Sz; c ) compared with a saline-treated control ( a ) and an animal 7 d after SE (7 d Sz; b ). d–f , Dab1 expression in the same sections is increased in the inner granule cell layer (gcl) in the 7 d Sz animal compared with control (arrows in d , e ). By 21 d after SE, Dab1 immunoreactivity has increased markedly in the hilus as well as inner gcl (arrows in f ). g–i , Merged images of a–e with Dab1 in red and CR-50 in green. Arrowheads point to double-labeled cells that are found in small numbers. Scale bar, 100 μm.
Reelin detaches chain-migrating dentate neuroblasts in vitro
To more directly test the influence of Reelin on dentate gyrus progenitor migration, we used two in vitro techniques. The first is a three-dimensional matrix Matrigel cell migration assay in which tissue explants are cultured in a Matrigel/collagen (3:1) gel for 24 h. We obtained Reelin protein by concentrating conditioned media from COS cells transfected with full-length Reelin and grown in serum-free media. The presence of Reelin protein and its proteolytic cleavage products in the media was confirmed by Western blot with the G10 monoclonal antibody that recognizes an N-terminal Reelin epitope (Fig. 4 a). No protein was present in serum-free media conditioned by control enhanced green fluorescent protein (eGFP)-transfected COS cells; as expected, low levels of Reelin were detected in media conditioned by control cultures grown with serum because of the presence of some Reelin protein in serum (Smalheiser et al., 2000).

Reelin increases detachment of chain-migrating dentate gyrus neuroblasts in explant cultures. a , Western blot showing bands corresponding to full-length (400 kDa) and cleaved (∼300 and 180 kDa) Reelin protein in concentrated media conditioned by Reelin-transfected COS7 cells (Reln). No bands are present in the control eGFP-transfected COS7 cell conditioned serum-free media (arrows), although the control cells cultured in 1% serum show slight bands attributable to the presence of Reelin in serum (eGFP at right). b , c , Dentate gyrus explants from P7 mouse cultured for 24 h in Matrigel with control ( b ) or Reelin-conditioned ( c ) media. Note the cells migrating in chains out of the control explant (arrows in b ) versus the more individually migrating cells in the explant treated with Reelin ( c ). d–g , Matrigel dentate gyrus explants cultured for 24 h and immunostained for neuron-specific β-tubulin. Neuroblasts migrate out of the control explant in chains ( d , e ), but those migrating out of the Reelin-treated explant are detached and separated ( f , g ). h , Quantification of the mean number of migrating cell chains per explant (left) or detached migrating cells per explant (right) after treatment with conditioned media from eGFP-transfected cells (Con), Reelin-transfected cells (Reln), or with Reelin function-blocking antibody (CR-50). n = 6–14 explants per condition, 3 separate experiments. *p = 0.046 control versus CR-50 for chains, p = 0.02 CR-50 versus Reelin for cells, and p = 0.001 control versus Reelin for cells; **p = 0.0009 Reelin versus CR-50 for chains; control versus Reelin for chains was not significant (p = 0.10). Scale bar: b , c , 25 μm; d , f , 50 μm; e , g , 15 μm.
To confirm the activity of Reelin protein in the supernatant, we cultured neonatal mouse SVZ explants in Matrigel and added Reelin- or eGFP-transfected control-conditioned media to the cultures. As described previously (Hack et al., 2002), controls showed extensive chain migration out of explants, whereas explants exposed to Reelin-conditioned media had markedly fewer chains and more isolated cells (supplemental figure, available at www.jneurosci.org as supplemental material). We then cultured P7 mouse dentate gyrus explants in Matrigel. Control media showed cell migration out of the explants into the matrix mainly in chains over the first 24 h (Fig. 4 b), similar to the SVZ explants although less extensive (Wichterle et al., 1997; Hack et al., 2002) (supplemental figure, available at www.jneurosci.org as supplemental material), and they expressed the immature neuronal marker β-III-tubulin (Fig. 4 d,e). Dentate gyrus explants incubated with Reelin-conditioned media for 24 h, in contrast, showed significantly more individual detached cells and a trend toward fewer chains outside the explants on average than controls (Fig. 4 c,f–h). To block Reelin signaling, we took advantage of the function-blocking activity of the CR-50 antibody (Ogawa et al., 1995). Dentate gyrus explants cultured in Matrigel/collagen with CR-50 antibody added to the media showed significantly more chains than control or Reelin-treated cultures and the fewest detached cells of the three groups (Fig. 4 h). These findings indicate that Reelin prevents chain migration of dentate gyrus progenitors in a manner similar to its effect on SVZ neuroblasts that migrate to the olfactory bulb.
We next examined the effects of inhibiting Reelin signaling on dentate gyrus progenitors in vitro using organotypic hippocampal slice cultures to preserve tissue architecture. Proliferating cells in the dentate gyrus of P15 mice were labeled in vivo by BrdU injection, and hippocampal slices were prepared 2 h later and cultured on polycarbonate membrane filters. This regimen of BrdU administration efficiently labeled proliferating cells in the dentate SGZ (Fig. 5 a). We then treated cultures with the 39 kDa protein RAP, a molecular chaperone used extensively as a tool to block lipoprotein receptors (for review, see Bu and Schwartz, 1998) such as ApoER2 and VLDLR for the study of Reelin signaling (Niu et al., 2004). Slice cultures were exposed to 1 μm RAP or vehicle for 2 d and then assessed for the pattern of dentate gyrus BrdU labeling (for a schematic of the experimental design, see Fig. 5 h). In control slices cultured for 2 d, the BrdU labeling pattern was very organized and localized mainly to the SGZ (Fig. 5 b–d). BrdU-labeled cells in slices exposed to RAP in the culture media for 2 d, however, showed a much more disorganized pattern, with BrdU-immunoreactive cells scattered throughout the hilus and molecular layer (Fig. 5 e–g). Together with data from the Matrigel cultures, our results suggest that Reelin acts directly on dentate gyrus neuroblasts to influence their migration.

Reelin inhibition alters the location of SGZ proliferating cells in hippocampal slices. a , BrdU-labeled cells (arrows) are located mainly at the border of the granule cell layer (gcl) and hilus (h) in an organotypic hippocampal (HC) slice culture derived from a P14 mouse and cultured for 48 h. The mouse was given a single BrdU injection 2 h before slice preparation. The granule cell layer is made visible by a light bizbenzimide counterstain. b–g , BrdU labeling in different hippocampal slices from a single mouse cultured for 48 h with vehicle (Veh; b–d ) or RAP ( e–g ) to block Reelin receptors. BrdU labeling is confined mainly to the hilar/granule cell layer border (dashed arrows) of controls ( b–d ), but BrdU-immunoreactive nuclei are scattered throughout the hilus and molecular layer (ml) in RAP-treated cultures ( e–g ). h , Schematic diagram of the experimental design. Scale bar, 50 μm.
Discussion
We identified persistent hippocampal Reelin expression preferentially in adult rat hilar inhibitory interneuron subsets known to be vulnerable to epileptogenic insults. Many of the Reelin-responsive cells identified by Dab1 expression, moreover, are PSA-NCAM-expressing SGZ neuroblasts; a smaller subset appears to be more immature progenitors or interneurons, some of which coexpress Reelin, consistent with autocrine signaling (Deguchi et al., 2003) (Fig. 3, arrowheads). Reelin immunoreactivity declines dramatically after pilocarpine-induced SE, likely attributable to cell death of Reelin-expressing inhibitory interneuron subsets. The loss of Reelin protein in the dentate hilus and molecular layer after prolonged seizures occurs in parallel with changes in migratory behavior of SGZ neuronal progenitors and the appearance of ectopic DGCs (Parent et al., 2006). Moreover, our in vitro data using dentate gyrus explants support a direct effect of Reelin on dentate gyrus progenitor migration. These findings, therefore, provide evidence that Reelin functions to maintain the normal integration of newborn neurons in the neonatal and adult mammalian hippocampal formation and that an acquired loss of Reelin contributes to aberrant plasticity in mTLE.
Several additional lines of experimental evidence support a role for Reelin in regulating the migration of adult DGC progenitors. Reelin is expressed by cells within and adjacent to the region of persistent DGC neurogenesis (Pesold et al., 1998) (Figs. 1 ⇑–3), and DGCs express mRNA for the Reelin receptors VLDLR and ApoER2 (Kim et al., 1996; Haas et al., 2002). Studies of mouse mutants with defects in Reelin signaling also implicate Reelin in directing DGC precursor migration. These mutants include reeler mice (Drakew et al., 2002; Förster et al., 2002; Weiss et al., 2003), which fail to express Reelin protein because of loss-of-function mutations (D'Arcangelo et al., 1995) (for review, see Lambert de Rouvroit and Goffinet, 1998), the scrambler mouse, which involves defects in Reelin signaling downstream of receptor-ligand interactions attributable to Dab1 mutation (Förster et al., 2002; Weiss et al., 2003), and mice containing single or double knock-outs of the ApoER2- and VLDLR-type Reelin receptors (Trommsdorff et al., 1999; Drakew et al., 2002; Weiss et al., 2003). In all of these mice, the DGC layer is abnormally dispersed or more severely disorganized, and ectopic DGCs are present in the dentate hilus. Moreover, the amount of Reelin mRNA expression in the dentate gyrus of humans with mTLE correlates inversely with the degree of abnormal DGC layer dispersion (Haas et al., 2002) (see below).
The loss of Reelin after SE may interfere with DGC progenitor migration through a number of mechanisms. The absence of Reelin signaling during development interferes with the radial glial scaffold in the dentate gyrus, and this alteration has been proposed to underlie the associated defects in DGC layer organization (Förster et al., 2002; Weiss et al., 2003). Because the radial glia-like scaffold persists in the adult rodent dentate gyrus after chemoconvulsant-induced SE (Huttmann et al., 2003) (J. M. Parent, unpublished data), however, this mechanism is less likely than the loss of a direct Reelin effect on DGC progenitors. The notion that decreased Reelin expression results in abnormal chain migration is supported by the findings of Hack et al. (2002), who elegantly demonstrated that Reelin serves as a detachment signal for chain-migrating SVZ neuroblasts. When the neuroblasts reach the olfactory bulb, they come into contact with Reelin expressed by mitral cells in the bulb, detach from the chains, and migrate radially into the olfactory bulb granule and periglomerular layers. Our previous finding of neuroblast chains in the dentate gyrus exclusively in pilocarpine-treated rats and not controls (Parent et al., 2006) fits with this function of Reelin and implicates an alteration of neuroblast migratory behavior as the cause of aberrant DGC progenitor migration. This conclusion is strengthened further by our in vitro data showing that Reelin leads to detachment of chain-migrating dentate gyrus neuroblasts and that blockade of Reelin signaling increases neuroblast chain migration and alters the location of proliferating SGZ progenitors. These findings are unlikely to result from an indirect effect of Reelin acting through radial glia given the short duration of the in vitro experiments and the absence of astrocytes migrating or extending processes out of the Matrigel explants (Fig. 4 and data not shown).
The present data suggest a model of how Reelin signaling regulates DGC neuroblast migration in the adult rat (Fig. 6). We propose that Reelin expressed in the dentate hilus and molecular layer prevents chain migration of DGC precursors and signals the neuroblasts to move into the DGC layer in which they subsequently differentiate. Abnormal Reelin signaling in the dentate gyrus, either in mutant mice with defective Reelin signaling or attributable to an acquired loss of Reelin caused by prolonged seizures, allows the neuroblasts to form chains and migrate aberrantly into the hilus and molecular layer. Although seizure-induced chain migration is less robust in the molecular layer than the hilus after pilocarpine-induced SE, molecular layer neuroblast chains are observed, and ectopic DGC-like neurons are seen in both the hilus and molecular layer in human mTLE (Parent et al., 2006). The abnormal migration leads to lamination defects, dispersion of the DGC layer, and the presence of hilar and molecular layer ectopic granule neurons. Abnormal integration of the hilar ectopic DGCs, moreover, contributes to hyperexcitability (Scharfman et al., 2000; Dashtipour et al., 2001) that may induce more seizure activity and result in additional Reelin loss and aberrant neuroblast migration. Because adult-generated DGCs are thought to be required for certain forms of hippocampal learning and memory (Shors et al., 2001), abnormally integrated DGCs also may play a role in the disabling memory dysfunction commonly observed in human mTLE (Helmstaedter et al., 2003). Other migration cues in addition to Reelin likely are involved in DGC layer development (Bagri et al., 2002), and Reelin therefore is probably not the sole factor influencing aberrant cell migration after injury. Nonetheless, we have shown that it plays an important role during both development and persistent neurogenesis in the adult.

Model of altered DGC precursor migration and ectopic neurogenesis after seizure-induced injury. Top, In the intact adult rodent dentate gyrus, a DGC progenitor in the subgranular zone (asterisk) gives rise to cells in the granule cell layer (GCL). Reelin expressed by inhibitory interneurons in the hilus and molecular layer (ML) prevents chain migration of the differentiating DGCs, thereby restricting them to the GCL. Bottom, Several weeks after an episode of status epilepticus, loss of Reelin-expressing interneurons (1) allows DGC progenitors to form chains (2; white arrows) and migrate aberrantly into the hilus and ML (3), in which they differentiate into ectopic DGCs.
We cannot exclude the possibility that DGC layer dispersion in human or experimental mTLE results from the abnormal movement of mature DGCs or arises from a developmental insult during initial DGC layer formation. Evidence from the mouse intrahippocampal kainate mTLE model supports the idea that dispersion in some instances involves movement of mature cells, because it can occur in the absence of neurogenesis (Kralic et al., 2005; Heinrich et al., 2006). This model, however, is the only epilepsy model in which neurogenesis decreases acutely, probably as a result of more severe dentate gyrus injury. In support of this idea, intrahippocampal injection of a 30% lower kainate dose stimulates, rather than suppresses, dentate gyrus BrdU labeling in mice after prolonged seizures (Lee et al., 2003). In addition, DGC neurogenesis increases in all other mTLE models examined to date (for review, see Parent, 2003). In the pilocarpine model, moreover, the appearance of mature, Prox-1 (prospero-related homeobox 1)-immunoreactive ectopic DGCs in the hilus and molecular layer occurs only after a delay and in a temporal sequence that follows the ectopic appearance of proliferating cells and neuroblasts (Parent et al., 2006). These findings argue against the movement of mature DGCs accounting for the ectopic neurons in this model. However, an important consideration is that dispersion, particularly in experimental mTLE, and the generation of ectopic DGCs may be unrelated or mechanistically distinct phenomena. Alternatively, multiple mechanisms may underlie the development of an abnormally dispersed DGC layer. The relationship between abnormalities of DGC layer structure and early developmental or persistent neurogenesis remains to be determined.
The finding of deficient Reelin signaling in experimental epilepsy has important implications for the pathophysiology of human mTLE. DGC neurogenesis persists in humans even as late as the eighth decade of life (Eriksson et al., 1998). Pathological findings in the syndrome of mTLE, including dispersion of the DGC layer and the appearance of hilar and molecular layer ectopic granule-like neurons (Houser, 1990; Parent et al., 2006), implicate defects in persistent DGC neurogenesis as a cause of dentate gyrus structural abnormalities. Importantly, recent evidence suggests that the loss of Reelin expression plays a role in DGC layer dispersion in the epileptic human hippocampal formation. Haas et al. (2002) studied dentate gyrus morphology and Reelin expression in human autopsy cases and surgical specimens from patients with pharmacoresistent mTLE. They found that the degree of DGC layer dispersion correlated inversely with the amount of dentate gyrus Reelin mRNA expression. These data, together with the current findings and other recent studies using mTLE models (Heinrich et al., 2006; Parent et al., 2006), provide evidence that an acquired loss of Reelin underlies DGC structural abnormalities in the epileptic hippocampal formation. Replacement of Reelin or other cues that direct neural development after injury therefore is a potential therapeutic approach to prevent additional structural abnormalities that may continue to develop well after the initial insult. The acquired loss of Reelin, moreover, underscores the potential need to overcome changes in the local environment after injury that may be hostile to proper neural repair via progenitor cells.
Footnotes
-
This work was supported by a grant from the National Institute of Neurological Disorders and Stroke (J.M.P). We thank T. Curran, B. Howell, M. Ogawa, and D. Strickland for providing reagents, E. Gula for technical assistance, and F. Silverstein for helpful comments.
- Correspondence should be addressed to Dr. Jack M. Parent, University of Michigan Medical Center, 109 Zina Pitcher Place, 5021 Biomedical Science Research Building, Ann Arbor, MI 48109-2200. parent{at}umich.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}