

Abstract
The development of the olfactory system in vertebrates is a multistep process, in which several regulatory molecules are required at different stages. The development of the olfactory sensory epithelium and its projection to the olfactory bulb are both known to require the LIM-homeodomain transcription factor Lhx2. We examined whether Lhx2 plays a role in the development of the OB itself, as well as its projection to the olfactory cortex.
Although there is no morphological OB protuberance in the Lhx2 mutant, mitral cells are normally specified and cluster in a displaced olfactory bulb-like structure (OBLS). The OBLS is not able to pioneer the lateral olfactory tract (LOT) projection in vivo or when provided control (host) telencephalic territory in an in vitro assay. Strikingly, the mutant OBLS is capable of projecting along the LOT if provided with an existing normal LOT in the host explant. This is the first report of a role for a transcription factor expressed in the OB that selectively affects the axon guidance but not the specification of mitral cells.
Furthermore, the Lhx2 mutant lateral telencephalon does not support growth of an LOT projection from control OB explants. The defect correlates with the disruption of a cellular mechanism that is thought to be critical for LOT pathfinding: a specialized cell population, the “lot cells,” is mislocalized in the Lhx2 mutant. In addition, the expression of Sema6A is aberrantly upregulated. Together, these findings reveal a dual role for Lhx2, in the OB as well as in the lateral telencephalon, for establishing the LOT projection.
Introduction
The olfactory system is conserved across all vertebrates and is distinct from other sensory processing modalities in that primary sensory information enters the brain via the olfactory bulb (OB), a specialized protrusion at the rostral end of the brain. The principal projection neurons in the OB, the mitral cells, receive inputs from olfactory sensory neurons and in turn project via the lateral olfactory tract (LOT) to the olfactory cortex. This projection generates a complex odor-specific map in the olfactory cortex (Zou et al., 2005). The specificity of innervation of LOT axons is therefore crucial to olfactory function, but little is known about the mechanisms that regulate the formation of the LOT.
Several transcription factors and signaling molecules regulate distinct aspects of OB development. Analysis of mice mutant in each of these genes has provided important insights into specific developmental events in OB formation. First, OB morphogenesis is independent of mitral cell specification. In the Tbr1 mutant mouse, mitral cells are absent although an OB protrusion is present (Bulfone et al., 1998). The opposite scenario is seen in the FGFR1 (fibroblast growth factor receptor 1) mutant, in which OB morphogenesis is severely impaired, but mitral cells accumulate at the anterior end of the telencephalon (Hebert et al., 2003). Similarly, in mice lacking functional Pax6, the OB protrusion is absent, but a mislocated olfactory bulb-like structure (OBLS) is seen, which contains mitral cells that have migrated aberrantly because of a cell-nonautonomous role of Pax6 (Jimenez et al., 2000; Nomura and Osumi, 2004). Second, the specification of different OB cell types is controlled by independent cell-autonomous mechanisms: Tbr1 is required by mitral cells (Bulfone et al., 1998), whereas Arx, Sp8, and Dlx family members are required for aspects of olfactory interneuron production but not for mitral cell specification (Anderson et al., 1997; Bulfone et al., 1998; Levi et al., 2003; Long et al., 2003; Yoshihara et al., 2005; Waclaw et al., 2006). Finally, the LOT projection is guided by repulsive cues from the septum, such as Slit1 and Slit2 (Li et al., 1999; Nguyen-Ba-Charvet et al., 2002), and attractive or repulsive signals by distinct members of the Semaphorin family (de Castro et al., 1999). The LOT trajectory is also guided by a unique cellular mechanism, the “lot cells,” which form a precise “channel” within which the LOT fibers grow (Tomioka et al., 2000). However, the guidance of the LOT projection remains poorly understood, because the only mutant reported to lack an LOT projection, the Tbr1 mutant, also lacks the mitral cells themselves (Bulfone et al., 1998).
Here, we examine the role of LIM-homeodomain transcription factor Lhx2 in the development of the LOT. In the absence of Lhx2, there is no detectable OB protrusion, but a mislocated OBLS is present, and mitral cells are specified. Axons from these cells do not form an LOT, however. We uncover a dual basis for the LOT phenotype in the Lhx2 mutant. Lhx2 is required in the OB, selectively for“pioneering” the LOT projection, but not for projecting along an existing LOT tract. Lhx2 is also critical in the lateral telencephalon for the proper migration of lot cells that guide the LOT and for the normal expression of guidance molecules.
Materials and Methods
Timed pregnant Swiss mice were obtained from the Tata Institute of Fundamental Research mouse breeding facility. Lhx2 mutant embryos were obtained by crossing heterozygous mice (Porter et al., 1997). All procedures followed the Institutional Animal Ethics Committee guidelines.
In vitro explant culture and immunostaining.
Green fluorescent protein (GFP)-expressing mice (FVB.Cg-Tg(GFPU)5Nagy; The Jackson Laboratory, Bar Harbor, ME) were crossed into the Lhx2+/− background. The OB/OBLS was dissected from GFP-expressing embryonic day 12.5 (E12.5) control and Lhx2 mutant embryos, respectively. In the mutant, a portion of the rostrolateral telencephalon that encompassed the entire OBLS primordium was dissected. The accuracy of the dissection was confirmed by testing a few preparations for the absence of AP2ε expression in the remaining portion of the telencephalon (see Fig. 3). Nonfluorescing wild-type and mutant lateral telencephalic explants with the OB/OBLS removed served as “recipient” explants. Mutant OBLS explants were juxtaposed to wild-type lateral telencephalic preparations, and wild-type OB explants were juxtaposed to mutant lateral telencephalic recipient explants, in the position that had been occupied by the OBLS. These were maintained on Millipore (Bedford, MA) filters for 3 d in serum-free medium consisting of DMEM with B27 supplement (Invitrogen, Carlsbad, CA) and l-glutamine. Explants were fixed in 4% paraformaldehyde and were processed for immunohistochemistry using rabbit anti-GFP at 1:1000 dilution (Invitrogen, Eugene, OR) and Alexa-568-labeled goat anti-rabbit at 1:500 dilution (Invitrogen). In each experiment, littermate controls were used.
mAb Lot1 immunohistochemistry.
This procedure was performed as described by Sato et al. (1998).
In situ hybridization.
In situ hybridization using digoxigenin-labeled probes was performed as reported previously (Vyas et al., 2003). Lhx2 mutant brains were used from the original C57BL/6 background (Porter et al., 1997), as well as from the mixed GFP-expressing background (above). Similar results were seen in either background. Between two and six different mutant brains were examined for each marker, and littermate controls were used in each case.
DiI placement.
Control (n = 5) and Lhx2 mutant (n = 5) E14.5 brains were fixed overnight in 4% PFA. Single crystals of Carbocyanin DiI (Molecular Probes) were placed on the OB/OBLS and incubated at 37°C in 4% PFA for 2 d, after which the brains were imaged.
Results
Morphogenesis of the OB is disrupted in the Lhx2 mutant, but an OBLS forms at a displaced location and contains mitral cells
In mice, OB morphogenesis starts at E12.5, and the distinct OB protruberance is seen shortly thereafter. In Lhx2−/− mice, however, no such morphological protuberance is detected 2 d later, at E14.5 (Fig. 1a,b). To test whether OB specification had occurred despite the lack of morphogenetic development, we examined specific molecular markers. AP2ε expression labels the OB primordium at the rostral tip of the E12.5 telencephalon in normal embryos and is expressed in mitral cells at this early stage (Fig. 1c) (Feng and Williams, 2003). In the Lhx2 mutant, AP2ε expression identifies a small patch in the frontolateral telencephalon suggestive of the presence of mislocated OB tissue containing mitral cells (Fig. 1d). Mitral cell marker Neuropilin1 (NP1) (Kawakami et al., 1996) and a new marker, LIM-only gene Lmo4, are both expressed in the control OB at E14.5 (Fig. 1e,g). These genes identify a cluster of cells in the rostrolateral Lhx2 mutant telencephalon, indicating that mitral cells are specified in the absence of Lhx2 (Fig. 1f,h). In control brains, Lmo4 expression encompasses both the main olfactory bulb (MOB) as well as the accessory olfactory bulb (AOB), whereas NP1 expression is limited to the MOB. Comparison of NP1 and Lhx2 expression in adjacent sections indicates that Lhx2 is present in the MOB mitral cells as well as in a portion of the AOB. Lhx2 expression is in a thicker band than NP1 staining, suggesting that Lhx2 is also expressed in cells outside the mitral cell layer at E14.5 (Fig. 1i–n).

Mitral cells are specified in the Lhx2 mutant. a, b, At E14.5, the morphological protuberance of the olfactory bulb is prominent in control brain (a) but is not seen in the Lhx2−/− mutant (b). c, AP2ε marks the olfactory bulb primordium at the rostral end of control brains at E12.5 (arrow). d, In Lhx2−/− mutant, AP2ε identifies an olfactory bulb-like structure that is laterally displaced (arrow). e–h, In parasagittal sections of E14.5 brains, NP1 and Lmo4 expression labels mitral cells in control brains (e, g, arrow), and also identifies these cells in sections through the laterally displaced OBLS of the Lhx2 mutant (f, h, arrow). i, NP1 expression defines mitral cells of the MOB (arrow) and excludes the AOB (arrowhead). j, Lhx2 is expressed in the MOB (arrow) as well as a portion of the AOB (arrowhead). k, A false-color overlay of NP1 and Lhx2 expression shows overlapping staining of the MOB mitral cell layer (arrow). Higher-magnification views of i–k are shown in l–n, respectively. p, Lhx2 is expressed in most of the cell layers of the postnatal OB, as seen in sections of P5 brains. The granule cell layer (gcl) and mitral cell layer (arrow) express Lhx2. In the glomerular layer, the arrowhead points to a glomerulus surrounded by Lhx2 expression; in the external plexiform layer, open arrowheads point to scattered Lhx2-expressing cells. o, An adjacent section shows NP1 expression in the mitral cell layer (arrow).
In fact, Lhx2 is in a position to play a role in most of the cell types in the OB, because it is expressed in the entire telencephalic ventricular zone (Bulchand et al., 2001). At postnatal stages, when the layers have formed and assignment of expression to specific layers is possible, it is clear that Lhx2 is expressed not only in the mitral cell layer but also in the granule and periglomerular layers that are populated by interneurons, as well as the external plexiform layer, which contains tufted cells (Fig. 1o,p). Tufted cells are also considered to be part of the projection neurons of the OB, because they receive sensory input and project along the LOT (Bayer, 1983). The projection neurons are the first populations to be born in the OB (Bayer, 1983; Blanchart et al., 2006). Because definitive markers for tufted cells have not been reported, we could not ascertain specifically whether tufted cells are present in the E14.5 Lhx2 mutant OBLS. In accordance with current literature, however, mitral and tufted cells (“M/T”cells) are collectively referred to as “projection neurons” of the OB (Yamatani et al., 2004; Nagai et al., 2005; Blanchard et al., 2006; Walz et al., 2006).
The Lhx2 mutant OBLS does not form an LOT
To determine whether the mutant projection neurons are capable of forming an LOT, we placed tiny DiI crystals in the OB/OBLS of E14.5 control and mutant brains, respectively. In control hemispheres (n = 5), DiI traces a bundle of LOT fibers that courses in a stereotypical path in the lateral telencephalon (Fig. 2a,d). No such LOT bundle is seen in the mutants, however (n = 5). Instead, a few axons are found to project in aberrant dorsal and ventral directions (Fig. 2b,e,f). Thus, Lhx2 function appears to be critical for the formation of a proper LOT.
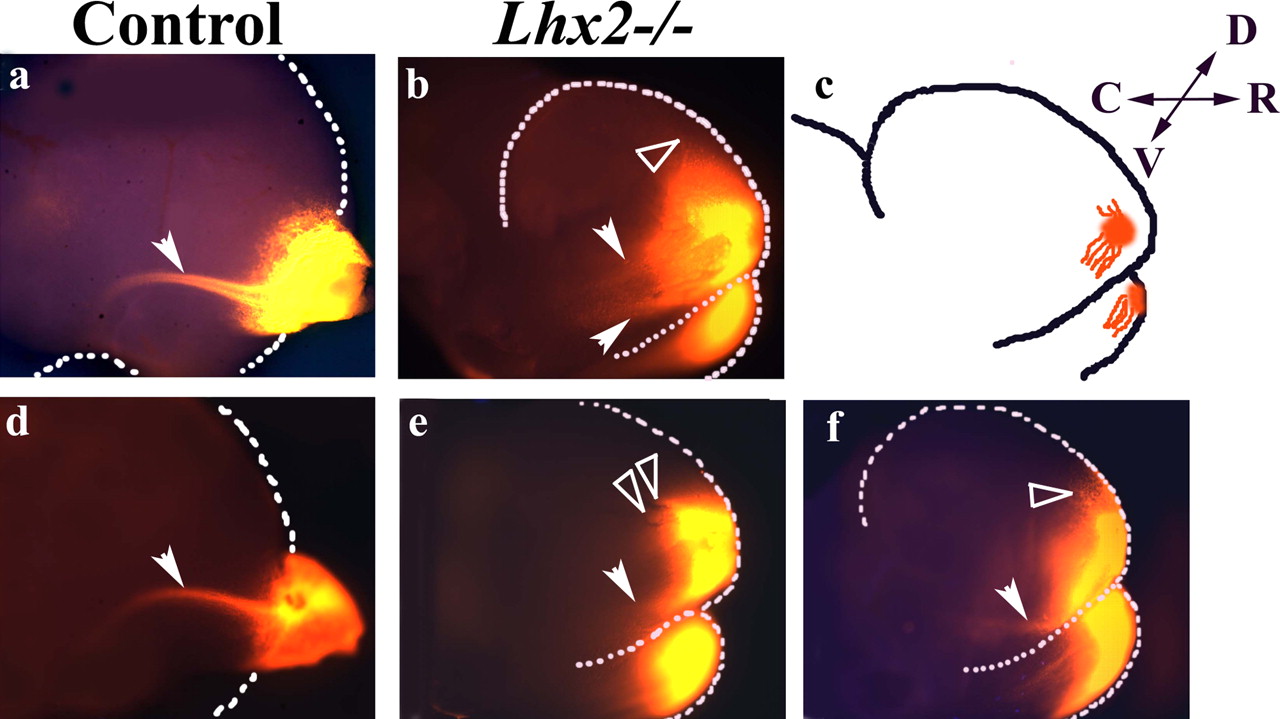
The Lhx2 mutant fails to generate a LOT. a, d, The LOT is labeled by a DiI placement in the control olfactory bulb at E14.5 (arrowhead). b, e, f, When DiI is placed in the site of the OBLS in Lhx2−/− brains, multiple short and aberrantly directed projections are seen dorsally (open arrowheads) and ventrally (filled arrowheads). These aberrant projections are schematized in c. D, Dorsal; R, rostral; V, ventral; C, caudal.
Because Lhx2 is expressed in the entire ventricular zone of the developing forebrain (Bulchand et al., 2003), it is in a position to play a role both within the OB as well as in the lateral telencephalic territory through which the LOT navigates. To clarify whether the defect in LOT formation is localized to the OBLS or whether it is attributable to a disrupted lateral telencephalic environment, we used an in vitro assay at E12.5 to examine LOT outgrowth. At this early stage, the OB/OBLS is expected to contain primarily projection neurons, because mitral cells are born first, “peaking” at E11.5 in mouse, and tufted cells are born shortly thereafter (Bayer, 1983; Blanchard et al., 2006). GFP-expressing OB/OBLS explants from donor brains were juxtaposed to GFP-negative lateral telencephalic recipient explants from which the OB/OBLS had been removed (Fig. 3a, schematic). Control cultures, in which both donor and recipient were from wild-type embryos, generated the normal curved trajectory of the LOT (Fig. 3b) (n = 11/11). However, when a control OB explant was cocultured with Lhx2 mutant telencephalic explants, axons failed to enter the recipient tissue (Fig. 3c) (n = 10/10). In the reverse experiment, when the mutant OBLS was juxtaposed to control telencephalic explants, although a few small fibers entered the telencephalon, they failed to show any directionality (Fig. 3d) (n = 11/11). It therefore appears that the LOT defect in Lhx2 mutant has a dual basis: a defect localized to the OBLS, as well as a defect in the territory through which the LOT navigates, possibly caused by disrupted guidance cues in this tissue.

LOT outgrowth in organotypic culture. GFP-positive control OB or Lhx2 mutant OBLS (donor) explants were juxtaposed to GFP-negative lateral telencephalic (recipient) tissue at E12.5. a, The recipient OB was removed (schematic). D, Dorsal; R, rostral; V, ventral; C, caudal. b, A normal LOT forms when both donor and recipient are from control brains (arrow). Cont, control; tel, telencephalic. c, d, However, few axons enter the lateral telencephalic explants when either recipient (c) or donor tissue (d, arrow) is from Lhx2 mutant (Mut) brains. e–h, In contrast, when the recipient OB is retained (e, schematic), both control OB (f) as well as Lhx2 mutant OBLS donor explants (g, h) are able to form an LOT (f–h, arrow). Only OB/OBLS tissue (j) can generate a projection when placed over the OB of the recipient: when portions of GFP-positive cortex (Ctx; k), hem (l), or subpallium (data not shown) are used as the donor tissue (as per schematic; i), no projection is seen. The accuracy of the OBLS dissection is illustrated by AP2ε staining. m, n, In control (m) and mutant (n) brains, AP2ε marks the prospective OB/OBLS region. A mutant hemisphere with the prospective OBLS removed (OBLS cut; o) and then processed for AP2ε expression shows a missing patch (dotted oval), confirming the identity of the dissected OBLS portion.
Presence of wild-type OB tissue can rescue the pathfinding defect of the mutant OBLS
To better understand the phenotype in the Lhx2 mutant OBLS, we tested whether the presence of wild-type OB tissue could rescue the LOT phenotype of the mutant. We created a “chimeric” OB by modifying the coculture assay, such that the wild-type E12.5 lateral telencephalic recipients now retained their OB (Fig. 3e, schematic). OBLS tissue from GFP-expressing Lhx2 mutants was placed over the normal OB of the recipient, creating a chimeric OB-containing mutant(GFP) plus wild-type tissue. In control cultures, wild-type GFP-expressing OB was placed over the recipient OB, creating a wild-type(GFP) plus wild-type control OB. After 3 d of culture in vitro, anti-GFP antibody staining reveals that mutant OBLS is indeed capable of extending axons along the LOT (Fig. 3g,h) (n = 8/11), similar to control OB tissue (Fig. 3f) (n = 9/14).
Might any neuronal tissue be able to “follow” an existing LOT projection? To test whether this ability is specific to OB tissue, we tested rostral cortex, cortical hem, and subpallial tissue in a similar chimeric assay (Fig. 3i, schematic). The recipient explant retained its OB and therefore provided an LOT, over which wild-type(GFP) OB explants projected as before, serving as controls (Fig. 3j). Neither GFP-expressing portions of cortex (Fig. 3k) (n = 8/8), nor cortical hem (Fig. 3l) (n = 7/7), nor subpallium (data not shown; n = 8/8) were able to project into the recipient, however. This finding illustrates that the ability to follow an existing LOT is specific to OB tissue and is not a general property of forebrain neurons. Furthermore, it confirms the accuracy of our dissection of the mutant: if tissue adjacent to the mutant OBLS were inadvertently used, it would consist of either residual rostral cortex or cortical hem, because the mutant telencephalon has a vast expansion of the hem (Bulchand et al., 2001; Monuki et al., 2001). Neither of these tissues is capable of sending projections along the LOT in the chimeric assay. The accuracy of the OBLS dissection is also illustrated by testing for absence of AP2ε staining on a mutant hemisphere after the OBLS is removed (Fig. 3n,o).
The finding that mutant OBLS tissue is capable of “following” an existing LOT in the chimeric coculture assay is consistent with a requirement for a “pioneer” population of neurons, the projections of which are able to guide mutant OBLS axons. The explant tissue used in the coculture experiments used the entire OB/OBLS primordium. The earliest projections from the OB are not from the MOB mitral cells but rather those of the AOB (Inaki et al., 2004). Although MOB tissue from normal mice has been shown to be capable of creating an LOT in the absence of AOB tissue (Sugisaki et al., 1996), it is worth establishing whether or not the AOB is specified in the Lhx2 mutant. We therefore examined the expression of markers specific to AOB mitral cells at E14.5. In control embryos, NP2 selectively and robustly labels the AOB mitral cells in the dorsal region of the OB (Inaki et al., 2004). T-bet and NetrinG1 are also AOB mitral cell-specific markers at this early stage (Nakashiba et al., 2000; Faedo et al., 2002). All of these markers show expression in the Lhx2 mutant, ventral to the MOB, instead of dorsal to it. The similar location of all three markers, however, indicates the presence of AOB mitral cells in the mutant, perhaps showing a greater degree of mislocalization than the MOB (Fig. 4).

The AOB is specified in the absence of Lhx2. a–f, AOB markers NP2 (a, d), T-bet (b, e), and Netrin G1 (c, f) reveal the presence of AOB mitral cells in control (a–c) and Lhx2 mutant brains (d–f) at E14.5.
In summary, both AOB and MOB mitral cells are specified in the Lhx2 mutant, but neither is able to pioneer an LOT projection in the explant culture assay, emphasizing the requirement of Lhx2 in this step of LOT formation.
Abnormal localization of guidance cues in the Lhx2 mutant telencephalon
The coculture assay experiments in which control OB tissue failed to create an LOT projection into Lhx2 mutant telencephalic explants suggested defective guidance cues in the territory through which the LOT axons navigate (Fig. 3c). Of the major players in axon pathfinding in other systems, secreted signaling molecules Slit1, Slit2, and Sema3F (SemaIV) have been reported to control the overall positioning of the LOT trajectory by providing repulsive cues (de Castro et al., 1999; Li et al., 1999; Nguyen-Ba-Charvet et al., 2002). Slit1 is expressed at the midline and in the ventricular zone of the subpallium in control and mutant brains, with some expansion of the mutant subpallial expression (Fig. 5a–d shows two different levels of sectioning). Slit2 shows a more restricted expression limited to the ventral midline. This expression is only detected at midlevels of sectioning (Fig. 5e–h), whereas expression at very rostral levels is not seen in either control or mutant brains (data not shown). Sema3F is robustly expressed deep in the subpallium, trailing off toward the lateral telencephalic margin, clearly excluding the “LOT region” in control brains. In the mutant, the subpallial expression is similar to that in control brains, and there is somewhat weaker expression at the lateral telencephalic margin, making the picture similar to that in the control (Fig. 5i–l). We examined another member of the Semaphorin family, Sema6A, which is implicated in guidance of the thalamocortical tract (Leighton et al., 2001). The expression of Sema6A in control brains is suggestive of a role in LOT guidance, because in control brains it appears to precisely delineate the location of the LOT (Fig. 5m,o). This guidance molecule displayed a striking overexpression in the rostrolateral region of the Lhx2 mutant telencephalon (Fig. 5n,p), raising the possibility that this region of the mutant telencephalon may be rendered inhibitory to the LOT projection.

Expression of guidance molecules in the Lhx2 mutant telencephalon. a–d, At E14.5, Slit1 (a–d) is expressed at the ventral midline (open arrowheads) and in the subpallial ventricular zone (filled arrowheads) of both control and Lhx2 mutant brains. Two levels of sectioning are represented in a and b and c and d. e–h, Control and Lhx2 mutant brains display Slit2 expression in a restricted manner in the ventricular zone of the ventral midline (arrowheads). i, k, In control brains, Sema3F is intensely expressed in the subpallial mantle (open arrowhead) but is undetectable in the region of the LOT in control brains (filled arrowhead). j, l, In Lhx2 mutant brains, Sema3F shows a similar strong labeling of the subpallial mantle (j, open arrowhead), but there is no discernable exclusion this region in the Lhx2 mutant (j, l, filled arrowhead). m, o, In control brains, Sema6A expression delineates the boundary of LOT region and is excluded from the LOT itself (arrowhead). o, p, In the Lhx2 mutant, there is a strong, aberrant upregulation of Sema6A in the lateral telencephalon (arrowhead). g, h, k, l, o, and p are higher-magnification views of the boxed regions in e, f, i, j, m, and n, respectively.
In addition to secreted signals, a unique population of cells, called lot cells, provides critical cellular guidance cues to the LOT. These cells are generated in the dorsal cortex at an early developmental age and migrate ventrally to form a cellular array presaging the LOT trajectory (Tomioka et al., 2000). We hypothesized that the lot cells may be missing or deficient in the Lhx2 mutant brain, because much of the dorsal cortex, in which these cells originate, is missing (Bulchand et al., 2001; Monuki et al., 2001). In normal brains, mAblot1 labels these cells, revealing a double-stripe pattern that extends rostrocaudally, creating a “tunnel” within which the LOT projects (Fig. 6a) (Tomioka et al., 2000). When DiI is placed in the same brains after mAblot1 staining, the LOT is seen to align perfectly within the lot cell tunnel (Fig. 6d,g). In Lhx2−/− brains, however, no such tunnel is seen when stained with mAblot1. Instead, diffuse labeling in the rostrolateral telencephalon is seen (Fig. 6b,c, arrows). Furthermore, this staining does not align with most of the short aberrant projections seen when DiI is placed in the same brains (Fig. 6e,f,h,i, arrowheads). Clearly, in the mutant, these cells are not in a position to guide the LOT, and they provide an additional basis for the LOT pathfinding defect observed in cocultures of normal OB with mutant telencephalon.

lot cells are mislocalized in the Lhx2 mutant. a, mAblot1 staining identifies a rostrocaudal array of lot cells in an E14.5 control brain hemisphere (arrows). d, DiI labeling of the same brain delineates the LOT (arrowhead), which navigates within the lot cell tunnel (g, merge of a and d). b, c, Two different Lhx2−/− whole-mount preparations reveal only diffuse surface staining with mAblot1 (arrows). e, f, DiI labeling of the same brains reveals the aberrant projections generated by the mutant OBLS (arrowheads), but no alignment with mAblot1 staining is seen (h and i represent merged images of b, e and c, f, respectively).
Discussion
Few mutants are known to have defective LOT outgrowth. In the absence of Tbr-1, a T-box family member, the mitral cells are missing, as is the LOT (Bulfone et al., 1998). Pdn/Pdn mice, which carry a mutation in Gli3, display apoptosis of precursor mitral cells in the anlagen of the olfactory bulb. Residual surviving mitral cells, however, create a slender LOT (Naruse and Keino, 1995). In both cases, the LOT defect is attributable to absence or severe reduction in the mitral cells. The Lhx2 mutation is the first report of a defect in which mitral cells are specified but cannot create an LOT projection in vivo or when juxtaposed to control telencephalic tissue in organotypic culture. This defect can be “rescued” by the presence of a normal LOT projection in the recipient explant, which reveals new insights into the mechanisms regulating LOT formation. The displaced location of the Lhx2 mutant OBLS is similar to that reported in the Pax6 mutant, but the significant difference is that the LOT projection forms in the absence of Pax6 (Jimenez et al., 2000). Thus, the mislocalization of the OBLS does not by itself explain the LOT defect in the Lhx2 mutant.
At E12.5, the age at which the in vitro assays were done, the projection neurons are the dominant population in the control OB, because all other cell types are born at later stages, and the overwhelming majority of interneurons are born at late embryonic or postnatal stages (Bayer et al., 1983). The earliest born interneurons originate at a distance, in the lateral ganglionic eminence, and begin their migration toward the OB at E12.5 (Tucker et al., 2006). This stream of interneurons is distinctly segregated from the control OB at this stage. In the mutant OBLS, because there is no out-pouching of the OB, the distance the interneurons have to travel is considerably shorter, which may explain why a thin stream of GAD67-expressing cells has just reached one side of the OBLS at E12.5 (supplemental figure, available at www.jneurosci.org as supplemental material). In the following discussion, although we have described the role of Lhx2 as “autonomous to the OB” as a structure, it is likely that the results we report are caused by a direct loss of function of Lhx2 in the projection neurons themselves, because they are the predominant cell types present in the OB/OBLS explants at E12.5.
Defective morphogenesis and disruption of the LOT in the absence of Lhx2
In Lhx2−/− mice, olfactory sensory neurons show increased apoptosis and fail to differentiate completely, and the olfactory sensory nerve does not reach the rostral telencephalon (Hirota and Mombaerts, 2004; Kolterud et al., 2004). Gong and Shipley (1995) proposed that the arrival of sensory innervation induces OB morphogenesis and differentiation. However, mutations in the transcription factors Mash1 or Dlx5, which eliminate nearly all of the sensory innervation, do not disrupt the OB formation (Guillemot et al., 1993; Levi et al., 2003). Thus, the arrival of sensory innervation may not be required for OB morphogenesis. Furthermore, the LOT projection from the OB starts well before synapse formation in the OB glomeruli, indicating that mitral cells do not need activity-based signals from the sensory innervation to send LOT projections (Lopez-Mascaraque et al., 1996). Consistent with this, mitral-like cells present in the Pax6−/− OBLS can extend axons to form a Lateral Tract, in the absence of olfactory sensory innervation (Jimenez et al., 2000). Therefore, the absence of a proper LOT in the Lhx2 mutant cannot be ascribed to the previously reported defective olfactory sensory development (Hirota and Mombaerts, 2004; Kolterud et al., 2004). Two possibilities remained: either the projection defect is intrinsic to the Lhx2−/− OBLS, or the mutant lateral telencephalon is defective in crucial guidance mechanisms on which LOT formation depends. We found evidence for both scenarios.
This deficiency of the Lhx2 mutant OBLS phenotype is apparently rescued by the presence of normal OB in the “chimeric OB” coculture assay. This reveals insights into the properties of both control and mutant OB/OBLS tissue: that the former can provide sufficient guidance, adhesion, or scaffolding for the mutant projection neurons to enable them to project normally along the LOT; and that the mutant OBLS cannot begin the LOT projection, but can follow an existing projection normally. Although both AOB and MOB mitral cells are present in the Lhx2 mutant, neither can initiate the LOT projection.
In summary, these data reveal a highly localized role for Lhx2 in the OB, specific to a pathfinding mechanism that is apparently critical in the first few projections along the LOT but redundant for later projections. In a series of elegant experiments, Sugisaki et al. (1996) demonstrated that much of the lateral telencephalic territory is not permissive for OB axon outgrowth and that this effect is specific to OB axons. Furthermore, they also found that only a very restricted portion of the telencephalon permits ingrowth from the OB, and this location becomes point of entry of the LOT. Lhx2 mutant OBLS tissue, when juxtaposed to control telencephalic explants, behaves as if it is unable to recognize this permissive region and fails to send projections into it (Fig. 3d). This raises the possibility that Lhx2 function enables projection neurons to recognize critical permissive cues in the lateral telencephalon and generate the LOT projection. These permissive cues may in fact be those presented by the lot cell array, in the absence of which projection neuron axons do not penetrate the telencephalon (Sato et al., 1998).
That Lhx2 mutant OBLS expresses normal markers and is capable of following existing LOT projections indicates that the projection neurons have been specified normally. Our results reveal a clear mechanistic separation between the specification of projection neurons and their ability to pioneer the LOT pathway, indicating that these are independent processes and that Lhx2 is responsible only for the latter. This requirement is intriguing, because the LOT pathway is not known to need specialized pioneering neurons. Indeed, Sugisaki et al. (1996) showed that the later-born MOB is capable of generating the LOT projection in the absence of the earlier-born AOB. Therefore, normally, any portion of the OB appears to be capable of pioneering (i.e., beginning to project along) the LOT. The subsequent “followers,” which are also capable of initiating this projection, do not normally need any “pioneers.” In the Lhx2 mutant, however, the ability to begin the LOT projection appears to be compromised. This may be caused by a failure of axon guidance mechanisms, because the ability to initiate axon extension per se is intact. However, the mutant OBLS retains the specific ability to follow an existing LOT, revealing a secondary mechanism of LOT guidance. This follower mechanism would normally be redundant, but it is revealed in the Lhx2 mutant, in which the pioneering mechanism is compromised.
Guidance molecules and guidepost cells in the Lhx2 mutant lateral telencephalon
The apparently normal ventral midline expression of both Slit genes is at odds with the aberrant ventral projection seen in the Lhx2 mutant (Fig. 2), because these molecules are known to repel LOT axons (Nguyen-Ba-Charvet et al., 2002). These aberrant projections, however, are not able to constitute themselves into an LOT when offered a wild-type lateral telencephalon (Fig. 3d).
Sema3F, known to repel mitral cell axons (de Castro et al., 1999), displays an apparently normal expression in the ventral telencephalon, and even in the vicinity of the LOT. However, another member of the Semaphorin family, Sema6A, is intensely and aberrantly expressed in the mutant lateral telencephalon and provides a possible basis for the failure of this tissue to support LOT outgrowth, when tested with control OB explants in the in vitro assay (Fig. 3c).
In addition to guidance molecules, interaction between growing axons and guidepost cells serves as a unique guidance mechanism that helps axons to grow along defined trajectories in other systems (Bentley and Caudy, 1983). The lot cells are generated in the dorsal telencephalon, and migrate ventrolaterally to the presumptive LOT position, where they provide a physical cellular array through which the LOT projects (Tomioka et al., 2000). Ablation of the lot cells in organotypic culture inhibits the growth of the LOT projection (Sato et al., 1998). In the Extra-toes mutant, which carries a deletion in the Gli3 gene, the lot cells are widely distributed over the entire dorsal telencephalon in small clusters (Tomioka et al., 2000). Our finding of disrupted lot cell localization in the Lhx2 mutant indicates that LIM-HD-dependent mechanisms are also involved in regulating this process. This raises questions as to whether the requirement for Lhx2 is cell-autonomous to the lot cells or in the territory through which they migrate. Future studies will explore these issues.
In conclusion, we find multiple roles for Lhx2 in multiple aspects of OB development. Lhx2 is required for OB morphogenesis, but not for the specification of mitral cells. There is a critical role in the OB to initiate the LOT projection. In the lateral telencephalon, Lhx2 is required for the proper localization of lot cells that guide LOT axons and for the appropriate expression of guidance molecules. Together, this reveals a complex function for a single transcription factor in distinct aspects of OB development.
Footnotes
-
This work was supported by a Wellcome Trust Senior Fellowship (056684/Z/99/Z), a Swarnajayanti Fellowship, a grant from the Department of Science and Technology (Government of India; to S.T.), and a Kanwal Rekhi Career Development Award from the Tata Institute of Fundamental Research (TIFR) Endowment Fund (B.S. and P.H.). We thank T. Hirata for the kind gift of mAb lot-1; F. Porter for Lhx2 breeding pairs; B. Anderson (Lmo4), A. Faedo (T-bet), J. Lin (NetrinGI), F. Porter (Lhx2), N. Tamamaki (NP1 and NP2), and T. Williams (AP2ε) for DNA reagents; V. Vaidya for critical comments on this manuscript; and the TIFR animal house staff for excellent support.
- Correspondence should be addressed to Shubha Tole, Department of Biological Sciences, Tata Institute of Fundamental Research, Homi Bhabha Road, Colaba, Mumbai 400 005, India. stole{at}tifr.res.in





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}