

Abstract
The highest incidence of seizures during lifetime is found in the neonatal period and neonatal seizures lead to a propensity for epilepsy and long-term cognitive deficits. Here, we identify potential mechanisms that elucidate a critical role for AMPA receptors (AMPARs) in epileptogenesis during this critical period in the developing brain. In a rodent model of neonatal seizures, we have shown previously that administration of antagonists of the AMPARs during the 48 h after seizures prevents long-term increases in seizure susceptibility and seizure-induced neuronal injury. Hypoxia-induced seizures in postnatal day 10 rats induce rapid and reversible alterations in AMPAR signaling resembling changes implicated previously in models of synaptic potentiation in vitro. Hippocampal slices removed after hypoxic seizures exhibited potentiation of AMPAR-mediated synaptic currents, including an increase in the amplitude and frequency of spontaneous and miniature EPSCs as well as increased synaptic potency. This increased excitability was temporally associated with a rapid increase in phosphorylation at GluR1 S845/S831 and GluR2 S880 sites and increased activity of the protein kinases CaMKII (calcium/calmodulin-dependent protein kinase II), PKA, and PKC, which mediate the phosphorylation of these AMPAR subunits. Postseizure administration of AMPAR antagonists NBQX (2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline), topiramate, or GYKI-53773 [(1)-1-(4-aminophenyl)-3-acetyl-4-methyl-7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine] attenuated the AMPAR potentiation, phosphorylation, and kinase activation and prevented the concurrent increase in in vivo seizure susceptibility. Thus, the potentiation of AMPAR-containing synapses is a reversible, early step in epileptogenesis that offers a novel therapeutic target in the highly seizure-prone developing brain.
Introduction
The incidence of epilepsy is highest in the first year of life and peaks in the neonatal period (Volpe, 2001). Hypoxic encephalopathy in term infants is the most common cause of neonatal seizures, resulting in severe long-term consequences (Volpe, 2001; Jensen, 2006). The neonatal brain exhibits a predominance of excitatory neurotransmission over inhibition because of developmental regulation of expression of neurotransmitter receptors, ion channels, and transporters (Silverstein and Jensen, 2007). Early-life seizures cause permanent functional alterations in neuronal networks and render the brain susceptible to later epilepsy and cognitive deficits (Ben-Ari and Holmes, 2006; Silverstein and Jensen, 2007). Furthermore, current anticonvulsants are frequently ineffective in neonatal seizures (Sankar and Painter, 2005), emphasizing the need for better understanding of underlying mechanisms.
Animal model data strongly suggest that early-life seizures result in long-term hyperexcitability and long-lasting sequelae (Chen et al., 1999; Sogawa et al., 2001; Silverstein and Jensen, 2007), and even a single neonatal seizure may permanently alter glutamatergic synapses (Cornejo et al., 2007). However, the specific targets and mechanisms that mediate changes in synaptic strength in the acute period after seizures remain largely elusive.
We established a model of hypoxic seizures (hypoxia-induced seizures) in postnatal day 10 (P10) rats resulting in long-term increases in seizure susceptibility and decreased neurobehavioral performance (Jensen et al., 1992; Mikati et al., 2005). A role for AMPA receptors (AMPARs) in these changes was indicated when treatment with AMPAR antagonists, but not NMDA receptor antagonists or GABA agonists, preferentially suppressed seizures and their long-term consequences. Administration of AMPAR antagonists in the first 48 h after seizures attenuated long-term increases in seizure susceptibility and seizure-induced neuronal injury in hippocampus (Koh et al., 2004), suggesting that acute alterations in AMPARs in the 48 h after neonatal seizures may play a critical role in epileptogenesis. Previous studies with depth electrode recordings confirmed the presence of ictal activity in the hippocampus during hypoxia (Jensen et al., 1998). In addition, hypoxia-induced seizures at P10 have been reported to induce subacute and specific decreases in AMPAR GluR2 subunit as well as a calcineurin-mediated decrease in GABAergic transmission, leading to the potentiation of hippocampal network hyperexcitability in vitro (Sanchez et al., 2001, 2005).
The period of greatest susceptibility to early-life seizures is coincident with the critical period of brain development, characterized by maximal synaptic density (Rakic et al., 1986), as well as the highest neuronal expression of Ca2+-permeable, GluR2-subunit-deficient AMPARs (Kumar et al., 2002; Talos et al., 2006a). Activity-dependent phosphorylation/dephosphorylation of intracellular C-terminal sites on the AMPAR subunits GluR1 and GluR2 regulates their synaptic trafficking and functional properties, as described in long-term potentiation (LTP) and long-term depression (LTD) (Barria et al., 1997; Seidenman et al., 2003). For example, phosphorylation of GluR1 S831 and GluR1 S845 regulates both open channel conductance as well as synaptic incorporation of the receptor in the early phase of LTP (Lee et al., 2000; Malinow and Malenka, 2002). Similarly, phosphorylation of the GluR2 subunit S880 modulates its interaction with glutamate receptor-interacting protein (GRIP) and results in receptor endocytosis (Wyszynski et al., 1999). We hypothesized that similar dynamic mechanisms involving AMPARs may play an important role in epileptogenesis, given the efficacy of early postseizure AMPAR antagonist treatment in our seizure model.
Consistent with our hypothesis, we observed that hypoxia-induced seizures in the immature rat resulted in a rapid increase in AMPAR-mediated synaptic currents, an increase in GluR1 and GluR2 subunit phosphorylation, and a transient increase in the activity of calcium/calmodulin-dependent protein kinase II (CaMKII), PKA, and PKC. Furthermore, administration of AMPAR antagonists immediately after hypoxic seizures attenuated these alterations and the concurrent enhanced seizure susceptibility.
Materials and Methods
Animals.
Litters of male Long–Evans hooded rats (Charles River Laboratories; 10 pups per litter) were used in the study. Pups were subjected to hypoxia-induced seizure (HS), as described previously (Jensen et al., 1998) and allowed to survive for 1, 6, 12, 24, 48, and 168 h after hypoxia. Age- and weight-matched littermates not exposed to hypoxic conditions served as controls. Hypoxia-induced tonic-clonic seizures, automatisms followed by head and limb movements, and myoclonic jerks were observed. Only animals that had more than five tonic-clonic seizures were included in the study (93%). Littermate controls were kept at room air. All rats were returned to their dams within an hour after the experiment. All animals were housed in a temperature-controlled animal care facility with a 12 h light/dark cycle. All procedures were approved by and in accordance with the guidelines of the Animal Care and Use Committee at Children's Hospital (Boston, MA) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and the number of animals used.
NBQX, topiramate, and GYKI-53773 administration.
For drug-treatment studies, five groups of rat pups were used: HS plus 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), HS plus topiramate, HS plus (1)-1-(4-aminophenyl)-3-acetyl-4-methyl-7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine (GYKI-53773), vehicle-treated HS animals, and control littermates not exposed to hypoxia (normoxic controls). NBQX (20 mg/kg) and topiramate (30 mg/kg) were dissolved in PBS and injected intraperitoneally. GYKI-53773 was dissolved in DMSO at a concentration of 0.25 mg/ml for preparing a stock solution. The stock solution was diluted in PBS with 0.5% methyl cellulose and injected intraperitoneally (7.5 mg/kg). All drugs were administered within 30 min of the rats experiencing hypoxia-induced seizures, vehicle-treated animals were treated similarly.
Immunoblot analysis.
Long–Evans rat pups were killed at various time points between 1 and 168 h (7 d) after hypoxic seizures along with control littermates of the same age and weight. Brain tissue was dissected out immediately, and hippocampal samples were separated under a dissecting microscope. Tissue was rapidly frozen in ethanol and stored at −80°C until used for protein extraction. Membrane protein samples from whole hippocampal tissue were prepared as described previously (Wenthold et al., 1992; Talos et al., 2006a). Complete Mini Protease Inhibitor Cocktail Tablet (Roche) and phosphatase inhibitors PMSF (10 mm), Na-orthovanadate (10 mm), and okadaic acid (1 mm) were added to inhibit proteases and phosphatases. Total protein concentrations were measured using Bradford protein assay (Bio-Rad), and samples were diluted for equal amounts of protein in each sample. Samples were electrophoretically separated on 7.5% Tris-HCl gels and transferred to polyvinylidene difluoride membranes. Blots were blocked and incubated with primary and secondary antibodies. Phosphospecific antibodies raised against GluR1 S831 (1:1000 dilution), GluR1 S845 (1:1000 dilution) (Millipore), and GluR2 S880 (1:1000 dilution) (Upstate Biotechnology) were used for immunoblot studies. The membranes were stripped using Restore Stripping buffer (Pierce Biotechnology) as per the manufacturers' protocols and reprobed with antibodies raised against GluR1 (AB1504, 1:1000 dilution) and GluR2 (AB1768, 1:1000 dilution) subunits (Millipore). Appropriate anti-mouse or anti-rabbit IgG antibodies (Pierce; 1:5000 dilution) were used, and immunodetection was effected using Super-West Femto Maximum Sensitivity Substrate reagent (Pierce) as described previously (Talos et al., 2006a). Digital images were recorded using the Fuji Image 3000 chemiluminescence detection system. Densitometric analysis of the digital images was performed using Fuji Film MultiGauge image-analysis software to measure the optical signal density from each sample. The amount of phosphorylation observed was standardized to the amount of receptor subunit present in each sample.
Hippocampal slice preparation.
Hippocampal slices were prepared as described in detail previously (Jensen et al., 1998; Sanchez et al., 2001, 2005). Rat pups (male) were decapitated 1 h after hypoxia treatment with all procedures in accordance with guidelines set by the institutional animal care and use committee. Littermate rats that had not been exposed to hypoxia were used as normoxic controls. Rat brains were rapidly dissected from the skull and placed for sectioning in ice-cooled cutting solution bubbled with 95% O2/5% CO2 at 4°C. Coronal hippocampal slices (300 μm thickness) were sectioned from the middle third of hippocampus with a vibratome (WPI) in cutting solution containing (mm) 210 sucrose, 2.5 KCl, 1.02 NaH2PO4, 0.5 CaCl2, 10 MgSO4, 26.19 NaHCO3, and 10 d-glucose, pH 7.4. Slices were incubated in oxygenated artificial CSF (ACSF; composition as described previously) (Sanchez et al., 2005) and remained at 32°C for 30 min. Slices were kept at room temperature for at least 1 h before electrophysiological recordings.
Electrophysiology.
Whole-cell patch-clamp recordings were made from CA1 pyramidal neurons in hippocampal brain slices using infrared/differential interference contrast microscopy as described previously (Sanchez et al., 2001, 2005). All recordings were performed after a 1 h incubation period, allowing for washout of any systemically administered drugs (Kapus et al., 2000). The patch-pipette internal solution contained (in mm) 110 Cs-methanesulfonate, 10 tetraethylammonium-Cl, 4 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 4 ATP-Mg, and 0.3 GTP, pH 7.25, with QX-314 (N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium chloride) and creatine phosphokinase (17 unit/ml). Filled electrodes had resistances of 2–5 MΩ. Evoked EPSCs (eEPSCs) were elicited at 30 s intervals as described previously (Jensen et al., 1998). Minimally evoked unitary EPSCs were recorded as described previously (Raastad, 1995; Isaac et al., 1996). AMPAR-mediated spontaneous EPSCs (sEPSCs) and eEPSCs were pharmacologically isolated by blocking GABA and NMDA receptors with picrotoxin (30 μm) and dl-AP-5 (100 μm), respectively. TTX (1 μm) was added to the ACSF to record miniature EPSCs (mEPSCs). All recordings were performed at room temperature (22–24°C).
Data were collected using an Axopatch 200A amplifier (Molecular Devices) and Clampex 9.2 software (Molecular Devices) with compensation for series resistance (70%) and cell capacitance, filtered at 2 kHz, and digitized at 20 kHz using a Digidata 1320A analog to digital converter (Molecular Devices). EPSCs were detected automatically using Clampfit 9.2 (Molecular Devices), and frequency and amplitude histograms were constructed using this program essentially as described previously (Wyllie et al., 1994). Briefly, mEPSCs and sEPSCs were detected automatically using Clampfit 9.2 (Molecular Devices), and frequency and amplitude histograms were constructed using this program as described previously (Wyllie and Nicoll, 1994). The threshold for detection of sEPSC and mEPSC events was set at −5 pA (the baseline root mean square noise was 1.15 pA). This threshold remained constant throughout the analysis of whole experiments for all recordings in hippocampi of normoxic and post-hypoxic-seizure animals. All detected sEPSC and mEPSC were visually checked for a monotonic rising phase, and an approximately exponential decay time course. Total sEPSC event numbers were 368 and 3161 in the normoxic and post-hypoxic-seizure groups, respectively (n = 12 cells). Total number of mEPSC events was 46 and 468 in the normoxic and post-hypoxic-seizure groups, respectively (n = 5 cells). sEPSC and mEPSC events in each neuron were collected from consecutive 17.2 min recordings. The bin width for histogram and cumulative graphs was set to 1.2 pA. The total number of events in each group was set to 1 for the construction of the normalized cumulative histograms. Synaptic potency was defined as the mean amplitude of the eEPSCs from 10 consecutive trials divided by the success rate (1 minus the failure rate) (Isaac et al., 1996). The eEPSC rectification index and the synaptic potency were calculated as described previously (Isaac et al., 1996; Hayashi et al., 2000; McCormack et al., 2006). Statistical significance was assessed using Student's t test or one-way ANOVA test, when more than two groups were compared.
Kinase activity assay.
Animals subjected to graded global hypoxia were allowed to survive up to 1, 6, 12, 24, and 48 h along with littermate normoxic controls. The rat pups were killed at the specific time points and hippocampal tissue isolated as described previously. For drug-treatment studies, animals were administered either topiramate (30 mg/kg), NBQX (20 mg/kg), GYKI-53773 (7.5 mg/kg), or PBS immediately after hypoxia and were killed 3 or 6 h after hypoxia. Hippocampi were dissected out and immediately flash frozen in ethanol. Tissue was homogenized in lysis buffer with appropriate protease and phosphatase inhibitors added, to obtain a cytosolic protein fraction from the whole hippocampi as described previously (Talos et al., 2006a). BCA protein assay (Bio-Rad) was performed on the cytosolic fraction to ensure equal amounts of protein in each sample. Active PKC, PKA, and CaMKII were evaluated by ELISA using kits (Stressgen for PKC/PKA, Cyclex for CaMKII) per the manufacturers' protocols. Briefly, solid-phase ELISA assays that use specific synthetic peptide substrates for the protein kinases and use polyclonal antibodies directed against the phosphorylated form of the peptide substrate to measure the relative kinase activity were used. Results were expressed as concentration of active kinase determined by a standard curve and compared using a one-way ANOVA.
Recording behavioral and electrographic kainate-induced seizures.
P10 rat pups were subjected to graded global hypoxia and were administered NBQX (20 mg/kg) or PBS after hypoxia as described previously (Koh et al., 2004). The epileptogenic effect of neonatal seizures in the early period after hypoxic seizures was assessed by examining the susceptibility to kainate (KA)-induced seizures (2 mg/kg) at P13 (3 d after the hypoxia-induced seizures). Seizure activity was assessed based on the latency to onset of first behavioral seizure, latency to onset of forelimb clonus, and severity of maximal seizure stage attained. A seizure severity grade was assigned based on maximal response to KA on a scale of 0 to 5 as described previously in the modified Racine scale (Koh et al., 2004). Behavioral seizures were confirmed electrographically in a subset of animals. Electrographic seizures were also recorded using recording chambers with low-rotational-torque Slip-Ring Fluid Commutators (Dragonfly) and a LA Mont 32-channel amplifier with a head box (0.5–70 Hz signal filter with 4000 gain) integrated with a Stellate–Harmonie data-acquisition system (Staellate Systems). High-quality digital video integrated with EEG recording was acquired using a Panasonic WVCP 474 digital video camera with an ATI-WDM video card. Digital video–EEG recording files were archived using the Stellate–Harmonie database system (Stellate Systems).
Statistical analysis.
Group data were expressed as mean ± SE of mean, and n is the number of rats for a given data point. Data across multiple time points after induction of hypoxic seizures were compared with littermate normoxic control animals. Statistical significance was assessed for experiments having two sets of data involving animals experiencing hypoxic seizures and littermate controls using a matched-pair t test. One-way repeated-measures ANOVA with Tukey's post hoc analysis was used to assess the statistical significance of drug treatments and multiple comparisons across temporal profiles. p < 0.05 was considered statistically significant.
Results
Early enhancement of AMPAR-mediated spontaneous EPSCs after hypoxia-induced seizures
Neonatal seizures have been described to enhance the excitability of hippocampal networks as well as cause alterations in long-term network excitability (Jensen et al., 1991; Sanchez et al., 2001; Cornejo et al., 2007; Silverstein and Jensen, 2007), but their acute effects on AMPAR function have not been examined. We compared spontaneous and miniature AMPAR-mediated EPSCs in CA1 pyramidal neurons in P10 rat pup hippocampal slices removed 1 h after hypoxia-induced seizures and normoxic control rat pups. Slices from pups with hypoxic seizures showed significantly larger amplitude sEPSCs recorded in CA1 neurons (−24.4 ± 2.5 pA; n = 12; p < 0.05) compared with slices from normoxic control pups (−15.4 ± 1.0 pA; n = 12), and these events occurred at significantly higher frequency (0.3 ± 0.19 Hz; n = 12; p < 0.05) compared with normoxic controls (0.05 ± 0.01 Hz; n = 12) (Fig. 1). Consistent with the enhanced sensitivity of sEPSCs, postseizure AMPAR-mediated mEPSCs in CA1 neurons showed significantly increased amplitude and frequency compared with normoxic controls (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).

Enhanced AMPAR-mediated sEPSCs in CA1 pyramidal neurons immediately after hypoxic seizures. A , Representative recordings of AMPAR-mediated sEPSCs in CA1 neurons from normoxic control hippocampal slices (top) versus slices removed at 1 h after hypoxic seizures in vivo at P10 (bottom). Individual sEPSCs are shown in detail at expanded time scale below the continuous traces (whole-cell patch-clamp recordings at room temperature). B , Bath application of AMPAR antagonists GYKI-52466 (20 μm) or NBQX (10 μm) completely blocked the sEPSCs. C , D , Amplitude histograms ( C ) (normoxia sEPSC event, n = 368; post-hypoxic-seizure event, n = 3161) and cumulative distribution ( D ) show sEPSC events were increased with larger amplitudes in CA1 pyramidal neurons in slices removed after hypoxic seizures (−24.4 ± 2.5 pA; n = 12) compared with control slices (−15.4 ± 1.0 pA; n = 12; p < 0.05).
To elucidate whether modifications of postsynaptic AMPARs contributed to the increase in sEPSCs after hypoxic seizures, we compared minimally evoked AMPAR-mediated unitary EPSCs in single fibers from CA1 pyramidal neurons in slices removed 1 h after hypoxic seizures with CA1 neurons from normoxic control rats (Fig. 2 A,B). The distribution of successful eEPSC events was shifted to larger amplitudes in the slices removed after seizures (Fig. 2 C,D), and neurons in these slices exhibited a significant increase in synaptic potency (15.07 ± 0.87 pA; n = 5; p < 0.05) compared with slices from normoxic controls (10 ± 1.59 pA; n = 5) (Fig. 2 E). In addition, no significant change of paired-pulse facilitation was observed between the hippocampal slices from animals experiencing hypoxic seizures and normoxic animals at this age (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), similar to our observations with extracellular recordings in CA1 neurons (Jensen et al., 1998). These data suggest that hyperexcitability observed after neonatal seizures may result from changes in activation of postsynaptic neurotransmitter receptors, and this may contribute to the resultant epileptogenesis.

Increased synaptic potency in minimally evoked AMPAR-mediated EPSCs after hypoxic seizures. A , B , Left, Representative traces from single-fiber stimulation showing averaged unitary EPSCs at increasing stimulus intensities in CA1 pyramidal neurons in slices from normoxic control pups ( A ) and those removed at 1 h after hypoxic seizures ( B ). Middle, Stimulation profile of success rate of minimally evoked EPSCs as a function of stimulus intensity in slices from controls ( A ) and in those removed at 1 h after seizures ( B ). An abrupt threshold and a stable plateau for single axon fiber stimulation are observed in both conditions. Right panels in A (control) and B (after HS) show the corresponding individual EPSC event amplitude during the course of the experiment plotted against consecutive stimulus number. Averaged (□) and individual single response (●) amplitudes are plotted with a solid line, illustrating averaged EPSC amplitudes. C , D , Minimally evoked AMPAR-mediated EPSCs in slices from normoxic controls ( C ) and slices removed at 1 h after seizures ( D ). Top traces show 10 superimposed consecutive responses, and bottom traces show averaged successful EPSCs [synaptic potency, mean amplitude/(1 − failure rate)]. Bottom panels shows amplitude distribution histogram of EPSC events (n = 5 cells for each group) with a line showing the fitted Gaussian distribution of successful EPSC events. The amplitude distribution histogram shows a shift to the right in recordings from slices removed after hypoxic seizures. E , Neurons from slices removed after hypoxic seizures exhibited significant increase in synaptic potency (15.07 ± 0.87 pA; n = 5; *p < 0.05) compared with normoxic control (10 ± 1.59 pA; n = 5). Error bars indicate SEM.
Increased AMPAR GluR1 subunit phosphorylation after early-life seizures
Together, the electrophysiological data suggest that hypoxia-induced seizures in the immature rat resulted in increased AMPAR-mediated excitability of CA1 hippocampal neurons. The strength of synaptic transmission in intact neuronal networks, such as in LTP, can be regulated by altered AMPAR function mediated by phosphorylation of GluR1 S831 and GluR1 S845 (Barria et al., 1997; Lee et al., 2000, 2003). We observed an increase in the phosphorylation of GluR1 S831 as early as 1 h after hypoxic seizures (140%; n = 3; p < 0.05) compared with normoxic control littermates. The phosphorylation of GluR1 S831 was observed to be persistently increased at 6, 12, and 24 h after hypoxia before returning to baseline, with the maximal increase observed at 24 h after hypoxic seizures (183%; n = 3; p < 0.01) (Fig. 3 A). We similarly observed increased GluR1 S845 subunit phosphorylation as early as 1 h after hypoxic seizures (157%; n = 3; p < 0.05) (Fig. 3 B). Phosphorylation of GluR1 S845 remained persistently increased at 6, 12, and 24 h after hypoxia-induced seizures, with the maximal increase observed at 24 h after hypoxic seizures (190%; n = 3; p < 0.01). Thus, the seizure-induced increases in phosphorylation of GluR1 subunit sites were concurrent with the enhancement of AMPAR-mediated synaptic currents.

Rapid increase in phosphorylation of GluR1 S831/S845 is associated with early enhancement of excitability in hippocampus. A , B , Immunoblots showing significant increase in phosphorylation of GluR1 S831 ( A ) and GluR1 S845 ( B ) at 1, 6, 12, and 24 h after hypoxia. Densitometric analysis and relative quantitation of phosphorylation of GluR1 subunit revealed the maximal increase at 24 h after hypoxia for both GluR1 S831 (183%; n = 3; *p < 0.05) and GluR1 S845 (190%; n = 3, *p < 0.05) compared with normoxic controls. Error bars indicate SEM.
Rapid seizure-induced increase in inward rectification of AMPAR-mediated EPSCs along with increased GluR2 subunit phosphorylation
The age window of ∼P10–P12 is characterized by a relative preponderance of Ca2+-permeable AMPARs in hippocampal and neocortical neurons resulting from a developmentally regulated decrease in GluR2 subunit expression (Durand and Zukin, 1993; Sanchez et al., 2001; Kumar et al., 2002; Talos et al., 2006b). Additionally, hypoxic seizures have been associated previously with a decrease in the expression of GluR2 mRNA and protein 48 h after the induction of seizures (Sanchez et al., 2001). However, the acute alterations in the phosphorylation and function of GluR2-lacking AMPARs after hypoxic seizures have not been well characterized. We characterized the GluR2 deficiency of synaptic AMPARs by determining the rectification index of eEPSCs in slices removed at 1 h after in vivo seizures in the P10 pup and found that the rectification index was significantly increased in slices from animals after hypoxic seizures (2.98 ± 0.44; n = 9; p < 0.05) compared with normoxic controls (1.63 ± 0.31; n = 7) (Fig. 4 A,B). Furthermore, bath application of philanthotoxin-433 (10 μm), a specific antagonist for GluR2-lacking AMPARs, blocked the majority of eEPSCs in CA1 pyramidal neurons after hypoxia (65.35 ± 8.63%; n = 5; p = 0.02) compared with eEPSCs in CA1 neurons from normoxic controls (28.8 ± 10.1%; n = 5), confirming the presence of synaptic GluR2-deficient AMPARs after seizures. Expression of inwardly rectifying AMPAR-mediated currents can result from an increase in phosphorylation of GluR2 S880 and resultant trafficking of the GluR2 subunit out of the synaptic membrane (Chung et al., 2000; Kim et al., 2001; Seidenman et al., 2003). Indeed, GluR2 S880 phosphorylation was increased in the hippocampus of rats experiencing hypoxic seizures compared with normoxic litter mates, maximal at 12 h after hypoxia (178%; n = 5; p < 0.05) (Fig. 4 C,D). This seizure-induced phosphorylation of GluR2 S880 may contribute to the subacute changes in GluR2 expression reported previously (Sanchez et al., 2001), as well as increased inward rectification of EPSPs reported above.

Seizure-induced increase in inward rectification of AMPAR-mediated eEPSCs is associated with increased GluR2 S880 phosphorylation. A , Representative recordings of averaged traces (10–15 consecutive recordings at a 30 s interval) of AMPAR-mediated eEPSCs in CA1 hippocampus slices from normoxic controls (top) and those removed at 1 h after hypoxic seizures (bottom). The eEPSCs were recorded at holding potentials of −60 or +40 mV while the Schaffer collaterals were stimulated with a bipolar electrode in the stratum radiatum. B , Average of rectification index (the ratio of peak eEPSCs at −60/+40mv) of inward AMPAR-mediated eEPSCs from slices removed after hypoxic seizures were significantly larger (2.98 ± 0.44; n = 9; *p < 0.05) than those in control rats (1.63 ± 0.31; n = 7), demonstrating increased inward rectification. Increased inward rectification of AMPAR-mediated currents is mediated by internalization of GluR2-subunit containing AMPARs. C , Immunoblot analysis show increased phosphorylation of GluR2 S880 up to 12 h after hypoxic seizures. GluR2 S880 phosphorylation has been demonstrated to cause AMPAR internalization. D , Densitometric analysis and relative quantitation of GluR2 S880 phosphorylation showed maximal increase in phosphorylation at 12 h after hypoxic seizures (178%; n = 5; *p < 0.05) compared with normoxic littermate controls. Error bars indicate SEM.
Increased activity of protein kinase PKA, PKC, and CaMKII
PKA and CaMKII mediate the phosphorylation of GluR1 S845 and PKC is involved in the phosphorylation of both GluR1 S831 (Mammen et al., 1997; Kameyama et al., 1998) and GluR2 S880 (Seidenman et al., 2003). ELISA assays demonstrated a significant increase in PKA activity at 1 h after hypoxia-induced seizures (157%; n = 5; p < 0.01), returning to baseline levels observed in normoxic controls by 6 h (Fig. 5 A). Similarly, CaMKII activity increased at 1 h after seizures (223%; n = 4; p < 0.001) before returning to baseline levels at 6 h after hypoxia (Fig. 5 B). In contrast to PKA and CaMKII, the increases in PKC activity were more prolonged: PKC activity rapidly increased at 1 h after hypoxia (160%; n = 5; p < 0.01), was maximally increased at 6 h after hypoxia (193%; n = 5; p < 0.05), and returned to baseline levels by 12 h after hypoxic seizures (Fig. 5 C). The rapid seizure-induced activation of these protein kinases is occurring coincidentally with the alterations in phosphorylation state of their known substrates, GluR1 S831, GluR1 S845, and GluR2 S880.
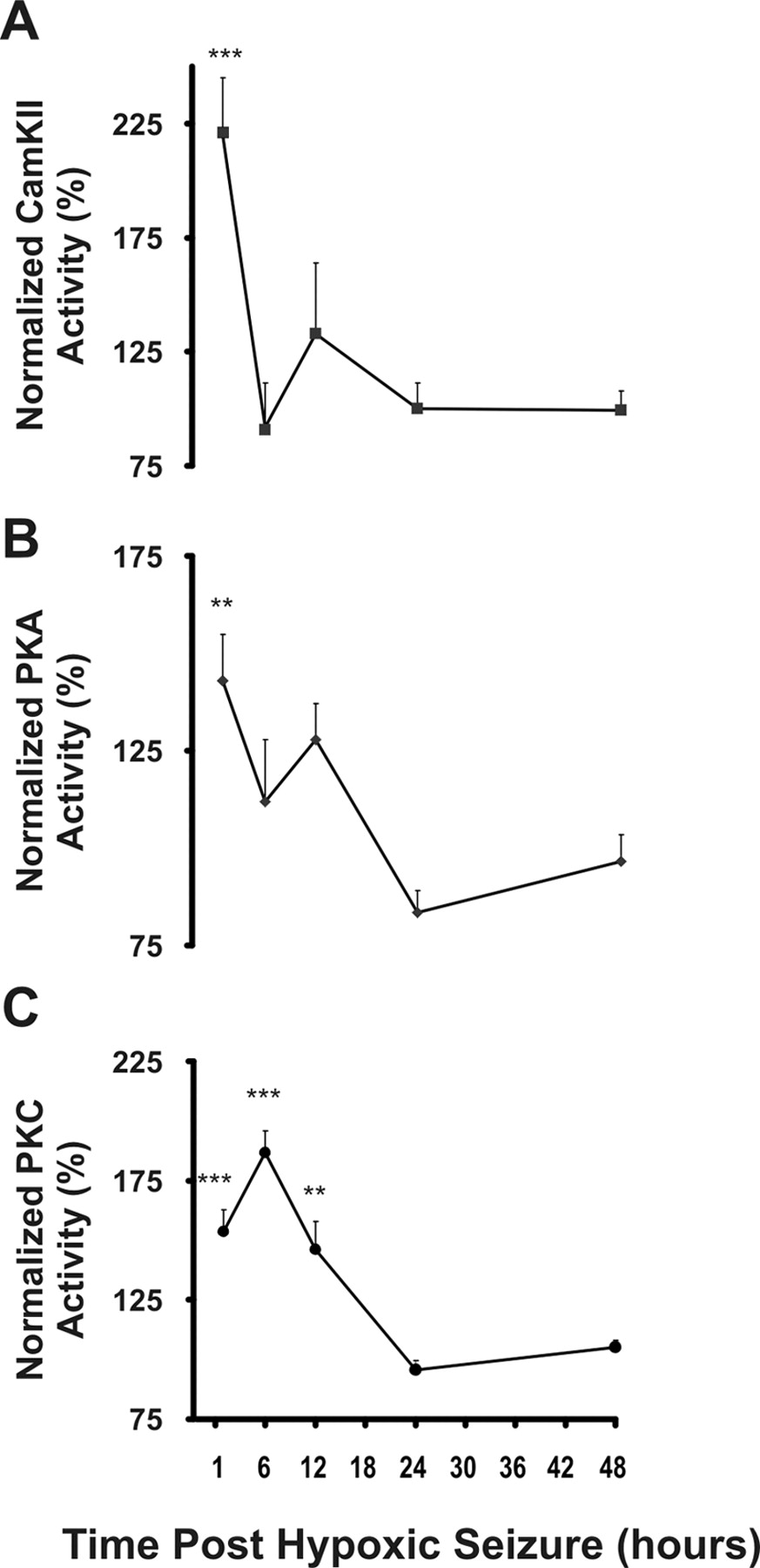
Seizures induce rapid and transient increases in kinase activity of PKA, PKC, and CaMKII. Whole-cell homogenates prepared from hippocampal tissue obtained 1, 6, 12, 24, and 48 h after hypoxic seizures from P10 rat pups were used to assay protein kinase activity. A , B , Significant increase was observed in the activity of CaMKII ( A ) (223%; n = 5; **p < 0.01), and PKA ( B ) (157%; n = 5, ***p < 0.001) 1 h after hypoxic seizures compared with littermate controls. C , Maximal increase in PKC activity (193%; n = 5; **p < 0.01) was observed at 6 h after hypoxic seizures. Error bars indicate SEM.
AMPAR antagonists attenuate seizure-induced phosphorylation and functional alterations in AMPARs and kinase activation
Thus far, our results demonstrate a rapid and transient increase in phosphorylation of AMPARs after seizures, coincident with altered receptor function, and these changes are occurring in the time window in which we have previously reported protective efficacy of postseizure treatment with AMPAR antagonists. To determine whether seizure-induced activation of AMPAR plays a critical role in the alterations in EPSCs and subunits reported here, we examined whether pharmacologic inhibition of AMPARs in vivo after hypoxic seizures leads to a decrease in the activity of Ca2+-dependent protein kinases and GluR1 subunit phosphorylation state. Notably, at P10, hippocampal and pyramidal neurons express Ca2+-permeable AMPARs (Sanchez et al., 2001; Kumar et al., 2002). Within 30 min after hypoxic seizures, rats were treated with a single dose of systemically administered AMPAR antagonists NBQX (20 mg/kg), topiramate (30 mg/kg), or GYKI-53773 (7.5 mg/kg), and compared with vehicle-treated (0.1 ml of PBS) hypoxic controls and normoxic littermates. These treatment doses are the same as the treatment paradigm in which we previously observed an attenuation of long-term epileptogenic changes (Koh et al., 2004). NBQX can block both AMPAR and KA receptors but preferentially blocks AMPARs, topiramate is a clinically available anticonvulsant demonstrated to have AMPAR antagonist properties but has actions at other voltage and ligand gated ion channels, and GYKI-53773 is currently in clinical trials and is a specific noncompetitive AMPAR antagonist. Importantly, blockade of AMPAR-mediated neurotransmission is the common mechanistic feature of these three structurally unrelated compounds and may represent the potential mode of mediating alterations. Although GluR1 S831 phosphorylation was increased at 12 h after vehicle treatment after hypoxic seizures (227 ± 20%; n = 5), consistent with the increased phosphorylation observed previously (Fig. 3 A), this increase was significantly reduced to baseline normoxia levels after post-treatment with NBQX (133 ± 20%; n = 5; p < 0.01), topiramate (87 ± 14%; n = 5; p < 0.01) or GYKI-53773 (110 ± 15%; n = 5; p < 0.001) (Fig. 6 A,B). Similarly, the increase in phosphorylation of GluR1 S845 observed at 12 h after hypoxia-induced seizures was significantly reduced to baseline normoxia levels after post-treatment with NBQX (110 ± 17%; n = 7; p < 0.05), topiramate (113 ± 11%; n = 7; p < 0.05) or GYKI-53773 (90 ± 12%; n = 7; p < 0.05) compared with vehicle treatment after hypoxic seizures (180 ± 15% increase compared with normoxia controls; n = 6) (Fig. 6 A,C).

Reversal of seizure-induced increases in GluR1 phosphorylation and protein kinase activity by in vivo administration of AMPAR antagonists after seizures. A , Immunoblots showing phosphorylation of GluR1 S831 and GluR1 S845 observed after the in vivo administration of the AMPAR antagonists NBQX (20 mg/kg, i.p.), topiramate (30 mg/kg, i.p.), or GYKI-53773 (7.5 mg/kg, i.p.) after HS. B , C , Densitometric analysis and quantification of the relative phosphorylation of GluR1 S831 ( B ) and GluR1 S845 ( C ) showed attenuation with administration of AMPAR antagonists compared with vehicle treatment at 12 h after hypoxic seizures. Similarly, the increased activity of protein kinases involved in AMPAR phosphorylation is attenuated after the in vivo administration of AMPAR antagonists after induction of HS. D–F , CaMKII ( D ) and PKA activity ( E ) measured in hippocampal tissue lysates collected 1 h and PKC activity ( F ) measured in hippocampal tissue lysates collected 6 h after in vivo administration of NBQX, topiramate and GYKI-53773 was significantly reduced compared with vehicle-treated animals subjected to hypoxic seizures (*p < 0.05). Error bars indicate SEM.
These treatment paradigms also resulted in a significant attenuation of the seizure-induced increases in activity of CaMKII, PKA, and PKC. The activity of CaMKII at 1 h after hypoxic seizures was reduced to near baseline levels observed in normoxic animals, after in vivo administration of NBQX (130%; n = 4; p < 0.05), topiramate (150%; n = 4; p < 0.05), and GYKI-53773 (140%; n = 4; p < 0.05) versus vehicle treatment after hypoxic seizures (310% compared with normoxic controls; n = 6; p < 0.05) (Fig. 6 D). Similarly, seizure-induced PKA activation at 1 h after hypoxic seizures was reduced significantly after administration of NBQX (160%; n = 4; p < 0.05) and topiramate (180%; n = 4; p < 0.05) versus vehicle treatment after hypoxic seizures (330% compared with normoxic controls; n = 4; p < 0.05) (Fig. 6 E). Unlike the assessment of activity at 1 h for PKA and CaMKII, we assayed PKC activity at 6 h after hypoxic seizures because this was when it was maximally increased (Fig. 5 C). The seizure-induced increase in PKC activity was significantly reduced after administration of NBQX (140%; n = 8; p < 0.05) and GYKI-53773 (160%; n = 8; p < 0.05) versus vehicle treatment after hypoxic seizures (260%; n = 8) compared with normoxic controls (Fig. 6 F). Similar results were observed on assaying PKC activity at 3 h after hypoxia after administration of AMPAR antagonists (data not shown) Thus, our results suggest that the attenuation in the activity of protein kinases PKA, CaMKII, and PKC at early time periods is associated with a temporally relevant decrease in the phosphorylation of their known substrate sites on the AMPARs, specifically GluR1 S831 and S845.
AMPAR antagonist treatment after seizures attenuates enhanced AMPAR-mediated sEPSCs
Given the observation that treatment with AMPAR antagonists immediately after seizures suppressed seizure-induced kinase activation and subunit phosphorylation, we next determined whether the treatment also attenuated the alterations in AMPAR sEPSCs. After hypoxic seizures, NBQX, topiramate, or GYKI-53773 were administered to the rat pups experiencing hypoxic seizures, and hippocampal slices were prepared 1 h after administration of AMPAR antagonists. The in vivo administration of AMPAR antagonists significantly decreased the sEPSC amplitude (for representative traces, see Fig. 7 A 1,A2 for topiramate; B 1,B 2 for NBQX; C 1,C 2 for GYKI-53773) observed in the ex vivo hippocampal slices as shown in the cumulative distribution histogram (Fig. 7 D). sEPSC amplitude was significantly attenuated in hippocampal slices from animals after hypoxic seizure administration of NBQX (106.4 ± 5.14%; n = 6), topiramate (116.29 ± 10.9%; n = 8), and GYKI-53773 (109.3 ± 19.2%; n = 5), compared with vehicle treatment (158 ± 9.2%; n = 6; p < 0.05) (Fig. 7 E). Similarly, we observed an attenuation of sEPSC frequency after administration of NBQX (98.7 ± 9.4%; n = 6), topiramate (48.02 ± 7.4%; n = 8), and GYKI-53773 (54.6 ± 8.5%; n = 5) compared with vehicle treatment after hypoxic seizures (261.57 ± 83.09%; n = 6; p < 0.05) (Fig. 7 F).

Postseizure in vivo administration of AMPAR antagonists attenuates the enhanced AMPAR-mediated sEPSCs in ex vivo slices. A 1–C 2 , Representative traces and histograms of pharmacologically isolated sEPSCs in CA1 pyramidal neurons from hippocampal slices removed from rat pups that experienced HSs and were post-treated in vivo with topiramate ( A 1 , A 2 ), NBQX ( B 1 , B 2 ), or GYKI-53773 ( C 1, C 2 ). D , Normalized cumulative distribution of sEPSCs showed a significant decrease in amplitude after in vivo administration of NBQX, topiramate, and GYKI-53773 compared with vehicle-treated animals experiencing HS. E , sEPSC amplitude was significantly decreased after in vivo administration of NBQX (106.4%; n = 6), topiramate (116.3%; n = 8), and GYKI-53773 (109.3%; n = 5) after hypoxic seizures compared with enhanced amplitudes observed in vehicle-treated animals experiencing HSs (158%; n = 6; **p < 0.05). F , Frequency of sEPSC was significantly decreased after in vivo administration of NBQX (98.75%; n = 6), topiramate (48.02%; n = 8), and GYKI-53773 (54.62%; n = 5) after hypoxic seizures compared with increases seen after HS in vehicle-treated controls (261.6%; n = 6; *p < 0.05). Error bars indicate SEM.
Increased seizure susceptibility observed after hypoxic seizures is reversed by AMPAR antagonist post-treatment
Given that here we showed that AMPAR antagonist postseizure treatment suppressed seizure-induced alterations in AMPAR subunits and function, we next evaluated whether in the present model there was an associated in vivo effect on later seizure susceptibility. We reported previously that enhanced hippocampal excitability observed in rat pups after hypoxia-induced seizures is reflected in the increased susceptibility to KA-induced seizures in these animals at both short (72–96 h) and long (40+ d) intervals after the initial seizures at P10 (Koh et al., 2004). KA (2 mg/kg, i.p.) was administered 3 d after hypoxic seizures at P10, and the latency to onset of both the first behavioral seizure and forelimb clonus was observed to be significantly shorter and seizure severity higher in animals with previous hypoxic seizures compared with normoxic littermate controls (Fig. 8 A,B). The administration of the AMPAR antagonist NBQX (20 mg/kg i.p.) after hypoxic seizures significantly attenuated the enhanced seizure susceptibility at P13. The latency to seizures in NBQX-treated animals was significantly longer (forelimb clonus, 54.3 ± 1.8 min; n = 9; p = 0.007) compared with vehicle-treated hypoxic controls (39.6 ± 2.1 min; n = 8) (Fig. 8 B). In addition, the maximal seizure severity was significantly lower in the NBQX-treated group (3.22 ± 0.45; n = 9; p = 0.001) compared with vehicle-treated pups that had previously experienced hypoxic seizures (4.37 ± 0.15; n = 8) (Fig. 8 C). Together, these data suggest that the attenuation of the enhanced AMPAR phosphorylation and function may be associated in mediating the decrease in seizure susceptibility and neuronal injury, as similar brief postseizure treatment paradigms have been shown previously to prevent long-term epileptogenic changes in this model (Koh et al., 2004).

Postseizure administration of NBQX attenuates enhanced seizure susceptibility observed after hypoxic seizures. A , B , Rat pups with previous exposure to hypoxia at P10 had ( A ) shorter latency to onset of first behavioral seizures (16.27 ± 0.97 min; n = 8) and ( B ) forelimb clonus (39.6 ± 2.13 min; n = 8) compared with normoxic littermate controls (24 ± 0.95 min and 72 ± 4.36 min respectively; n = 9; *p < 0.05) after administration of kainic acid (KA, 2 mg/kg, i.p.) at 3 d after HS, suggesting enhanced seizure susceptibility even in the early postseizure period. C , The maximal severity of KA-induced seizures at P13 was significantly higher in pups experiencing HS at P10 (4.37 ± 0.15; n = 8) compared with littermate controls (3.2 ± 0.15; n = 9; *p < 0.05). Administration of AMPAR antagonist NBQX (20 mg/kg, i.p.) in vivo after HS significantly attenuated the latency to onset of first behavioral seizure (20.95 ± 1.6 min; n = 9; *p = 0.03) and forelimb clonus (54.3 ± 1.8 min; n = 9; *p = 0.007) as well as the maximal seizure level (3.22 ± 0.45; n = 9; *p = 0.001) compared with vehicle-treated pups that had previously experienced HS. Error bars indicate SEM.
Discussion
Here, we observed that early-life seizures lead to a rapid enhancement of AMPAR-mediated synaptic currents and this was associated with rapid increases in AMPAR phosphorylation at GluR1 S831, GluR1 S845, and GluR2 S880 sites, in addition to activation of CaMKII, PKA, and PKC. Systemic administration of AMPAR antagonists immediately after seizures attenuated the early increase in AMPAR function and phosphorylation. Furthermore, AMPAR antagonist administration in P10 rats after early-life seizures also attenuated the increased seizure susceptibility observed in vehicle-treated pups. These results suggest that the potentiation of AMPAR-containing synapses is a reversible, early step in epileptogenesis that offers a novel therapeutic strategy in the highly seizure-prone developing brain.
Posttranslational modifications of AMPARs mediate hippocampal hyperexcitability after seizures in the immature brain
A central and novel finding of this study is that seizures cause rapid alterations in AMPAR subunit composition and function in the developing brain. Enhanced synaptic AMPAR strength may be an important component of the acute and long-term seizure-induced hyperexcitability that constitutes epileptogenesis. Our study showed that both sEPSCs and mEPSCs were increased in amplitude and frequency in CA1 neurons from hippocampal slices removed 1 h after hypoxic seizures; this enhanced excitability in AMPARs resembles the modifications in synaptic efficacy observed in hippocampal LTP (Nicoll and Malenka, 1999; Collingridge et al., 2004). In models of LTP, similar rapid changes in synaptic potentiation are mediated by trafficking and endocytosis of AMPARs (Ehlers, 2000; Derkach et al., 2007). Neonatal seizures also lead to an enhancement of AMPAR-mediated minimally evoked unitary EPSCs and synaptic potency; similar enhanced AMPAR function is proposed as direct evidence for postsynaptic modification of AMPARs in LTP (Isaac et al., 1996; Pratt et al., 2003). Here, paired-pulse facilitation was not changed after hypoxic seizures, suggesting the involvement of postsynaptic mechanisms. The increase in mEPSC frequency observed in hippocampal slices from animals experiencing hypoxic seizures also may reflect a reduction in the number of “silent synapses” and their conversion into functional AMPAR-containing synapses (Petralia et al., 1999). The early alterations resulting in hyperexcitability of hippocampal networks occur rapidly after seizure activity, and may represent the early and rapid transition from normal to epileptic networks. In addition to the alterations in hippocampal networks, there may also be enhanced excitability in the neocortex, as hypoxic seizures have been reported previously to induce epileptiform activity in the neocortex (Jensen et al., 1998).
Coincident with the rapid enhancement of synaptic efficacy observed in the hippocampus after hypoxic seizures, we observed posttranslational modifications of AMPAR subunits consistent with those reported in models of LTP (Lee et al., 2000). Phosphorylation of GluR1 S831 and GluR1 S845 was increased in hippocampi of rat pups as early as 1 h after experiencing hypoxic seizures in vivo. GluR1 subunit-containing heteromeric receptors have been shown to be delivered into synapses during LTP after phosphorylation of the GluR1 receptor subunit (Song and Huganir, 2002), and these modifications influence bidirectional changes in efficacy of synaptic transmission (Matsuda et al., 1999; Lee et al., 2000). Phosphorylation of GluR1 S831 has been shown to increase single-channel conductance in GluR1 subunit-containing AMPAR heteromers (Mammen et al., 1997; Derkach et al., 1999). Phosphorylation of GluR1 S845 has been reported to be critical for increased open-channel probability and activity-dependent insertion of GluR1-containing receptors into the membrane-associated pool of AMPARs (Esteban et al., 2003; Oh et al., 2006; Man et al., 2007), thereby modulating bidirectional synaptic plasticity. Furthermore, transgenic mice with mutations in these phosphorylation sites exhibit impaired LTP and spatial memory (Lee et al., 2003; Hu et al., 2007). Hence, phosphorylation of GluR1 S831 and S845 subunit would both be predicted to enhance AMPAR EPSCs as seen here.
We also observed a rapid seizure-induced increase in phosphorylation of GluR2 S880. Previous reports have shown that dynamic regulatory mechanisms modulate the GluR2 phosphorylation and its subsequent interaction with synaptic proteins like GRIP1/2, AMPA-binding protein, and PKC-interacting protein 1 that promote internalization from the synaptic surface (Chung et al., 2000; Seidenman et al., 2003). Neonatal seizures induce phosphorylation of GluR2 S880 that in turn may cause GluR2 subunit internalization, suggested by the rapid increase in the inward rectification ratio of AMPAR currents after hypoxic seizures. This seizure-induced decrease in GluR2 would accentuate the existing preponderance of Ca2+- permeable AMPARs in the immature brain (Sanchez et al., 2001; Kumar et al., 2002; Talos et al., 2006a,b). In the immature brain, the Ca2+- permeable heteromeric AMPARs may act as source of synaptically mediated Ca2+ influx (Sanchez et al., 2001) in addition to that mediated by NMDARs (Malenka and Nicoll, 1993; Malenka and Bear, 2004).
The present study has identified a novel and critical role for AMPAR modifications that begins within hours after neonatal seizures and may represent the early mediators of an epileptogenic cascade. Cellular and molecular mechanisms previously associated with physiological synaptogenesis and synaptic plasticity may be co-opted by seizures for creation of hyperexcitable epileptogenic circuits. Our results would suggest that hypoxic seizures induce Ca2+ influx and mediate the rapid increase in the activity of protein kinases CaMKII, PKA, and PKC in the hippocampal neurons, which in turn may lead to enhanced phosphorylation of the GluR1 and GluR2 receptor subunits. AMPAR receptor subunit phosphorylation may promote enhanced excitability and synaptic potentiation in hippocampal networks, thus initiating a persistent loop of enhanced AMPAR function and neuronal dysplasticity, which potentially may be a target for intervention, such as with AMPAR antagonists (Fig. 9). This unique dysplasticity may underlie the severe consequences of early-life seizures, including enhanced epileptogenesis observed in neonates as well as later impairment in cognitive function (Mikati et al., 2005; Silverstein and Jensen, 2007).

AMPARs as mediators and targets of seizure-induced epileptogenesis in the immature brain: AMPAR expression peaks between the first and second postnatal weeks in the rat. A , In the immature rat brain, the majority of AMPARs lack GluR2 and are Ca2+ permeable. B , Neonatal seizures result in intense synaptic activity and AMPA-mediated Ca2+ influx resulting in the rapid increase in activity of protein kinases (PKA, CaMKII, and PKC), which phosphorylate the GluR1 and GluR2 subunits. The enhanced phosphorylation of these AMPAR subunits contributes to enhanced receptor trafficking, increased excitability and synaptic potentiation, initiating a feedforward loop of enhanced AMPAR function and neuronal dysplasticity. Treatment with AMPAR antagonists after neonatal seizures attenuates the Ca2+ influx-mediated receptor phosphorylation and enhanced kinase activity, mitigating AMPAR-dependent epileptogenic effects (see Figs. 6 ⇑–8).
Postseizure attenuation of AMPAR potentiation and enhanced kinase activity
A second major and novel finding is that seizure-induced alterations of the AMPAR and kinases can be attenuated or possibly reversed even after the onset of the pathologic trigger. Post-treatment with the AMPAR antagonists NBQX, topiramate or the specific AMPAR antagonist GYKI-53773 blocked the acute and subacute enhancement in AMPAR EPSCs, GluR1 subunit phosphorylation, and PKA, CaMKII, and PKC activation observed after hypoxia-induced seizures, when administered during the early period after seizure induction. NBQX can block both AMPAR and KA receptors but preferentially blocks AMPARs. Topiramate is a noncompetitive AMPAR antagonist with actions at other ligand-gated channels, and GYKI-53773 is a specific noncompetitive AMPAR antagonist. Importantly, they share a common action of being able to block AMPAR-mediated neurotransmission. Here, we show that AMPAR antagonists reverse seizure-induced potentiation and it is possible that activation of AMPARs, which are predominantly Ca2+ permeable at this age (Sanchez et al., 2001; Kumar et al., 2002; Talos et al., 2006a,b; Silverstein and Jensen, 2007), may be responsible for activating PKA, PKC and CaMKII as observed in this study.
Postseizure treatment with NBQX or topiramate within the first 48 h after hypoxic seizures prevents the long-term increase in susceptibility to seizures and seizure-induced neuronal injury (Koh et al., 2004). In the present study, the same postseizure treatments that result in attenuation of the physiological alterations and subunit phosphorylation also resulted in prevention from enhanced seizure susceptibility at the level of the whole animal. We observed that rats post-treated with the AMPAR antagonist NBQX demonstrated a reversal of the increased seizure susceptibility in vivo at 72 h after hypoxic seizures, providing support for the notion that there is an in vivo correlate to the reversal of the cellular changes observed in vitro.
Implications of AMPAR potentiation for intervention in the epileptogenic process
The current study with pharmacological inhibition in vivo suggests that AMPARs may play an important role in the epileptogenic process, either directly or indirectly, by their effects on synaptic plasticity. Whereas early-life seizures promote epileptogenesis, repeated neonatal seizures have been shown to lead to long-term impairment of neurobehavior, cognition, and memory (Chen et al., 1999; Holmes et al., 1999; Sogawa et al., 2001; Silverstein and Jensen, 2007). The specific alterations in AMPAR function and GluR1 and GluR2 phosphorylation observed here may not only be associated with epileptogenesis, but also may produce impairment of normal network plasticity leading to deficits in neurobehavior and cognition. Similar activity-mediated alterations in AMPAR phosphorylation state and function have been reported to be involved in physiological phenomena such as LTP and LTD (Song and Huganir, 2002; Boehm and Malinow, 2005) and also in pathological settings including the stress response as well as addiction (Carlezon and Nestler, 2002; Saal et al., 2003; Dong et al., 2004; Self and Choi, 2004; Kauer and Malenka, 2007; Xiang and Tietz, 2007).
Given the importance of AMPAR-mediated neurotransmission in both health and disease, the clinical use of AMPAR antagonists may provide significant improvement in neurological dysfunction. However, because of the important role of AMPAR-mediated neurotransmission, antagonist treatment might also be associated with adverse effects on physiological synaptic plasticity. Interestingly, the threshold for memory impairment with topiramate in healthy volunteers and patients with epilepsy is lower than lamotrigine and valproate, which are currently clinically used as anticonvulsants (Meador et al., 2003, 2005). In light of our results, the memory impairments reported with topiramate may in part be caused by its action on AMPAR synapses in hippocampus. However, more preliminary human studies with the administration of specific AMPAR antagonist GYKI-53773, which has been used in this study, show less cognitive impairment (Chappell et al., 2002; Howes and Bell, 2007).
The process leading to the development of epilepsy after early-life seizures is likely to be multifactorial (Noebels, 2003; Silverstein and Jensen, 2007). In addition to AMPARs, expression and function of other neurotransmitter receptors and their regulators undergo major changes because of developmental regulation, including that of GABAA (Sanchez et al., 2005; Raol et al., 2006), NMDA (Sutula et al., 1996; McNamara et al., 2006), chloride cotransporters (Dzhala et al., 2005), and metabatropic glutamate receptors (Doherty and Dingledine, 2002). These developmental phenomena increase the susceptibility of the developing brain to seizures and may contribute to epileptogenesis in early life. In addition to these changes in neurotransmitters, acute and chronic changes in translation, transcription, and epigenetic regulation of molecular substrates may be playing a role mediating this process (McNamara et al., 2006). Our findings are a novel demonstration in that at least the changes in AMPARs can be reversed after the initial insult to modify the process of epileptogenesis.
The efficacy of AMPAR antagonists in attenuating epileptogenesis, especially when administered after neonatal seizures, may be particularly useful clinically given their relative safety profile and lack of apoptotic cell death induction at pharmacologically tolerated doses in the developing brain (Ikonomidou et al., 1999; Bittigau et al., 2002). Conventional agents like phenytoin and barbiturates, that currently constitute the first line of therapy, lack efficacy for acute seizure suppression in almost 50% of infants with neonatal seizures and have no effect on long-term outcome (Painter et al., 1999; Sankar and Painter, 2005). AMPAR antagonists, even when administered after seizure onset, may be an effective age-specific therapeutic strategy that may have a role in preventing epileptogenesis and epilepsy-related cognitive impairment.
Footnotes
-
This work was supported by National Institutes of Health Grants NS 31718 (F.E.J.) and DP1 OD003347 (F.E.J.) (from the Office of the Director), Mental Retardation Developmental Disorders Research Center Grant P30 HD18655 (National Institute of Child Health and Human Development), the Epilepsy Therapy Development Project (F.E.J.), and Parents Against Childhood Epilepsy (S.N.R., F.E.J.). We thank Erin Straw and Neil Marya for their help with animal handling and GluR1 S831 immunoblot experiments. GYKI-53773 was a generous gift from Mike Fetell at Teva Pharmaceutical. We thank Drs. Kevin Staley and Takao Hensch for valuable discussion.
- Correspondence should be addressed to Dr. Frances E. Jensen, 300 Longwood Avenue, Enders 348, Children's Hospital and Harvard Medical School, Boston, MA 02115. Frances.Jensen{at}childrens.harvard.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}