

Abstract
Within the Ras superfamily of GTPases, Rap1 and Rap2 are the closest homologs to Ras. In non-neural cells, Rap signaling can antagonize Ras signaling. In neurons, Rap also seems to oppose Ras in terms of synaptic function. Whereas Ras is critical for long-term potentiation (LTP), Rap1 has been shown to be required for long-term depression (LTD), and Rap2 has been implicated in depotentiation. Moreover, active Rap1 and Rap2 cause loss of surface AMPA receptors and reduced miniature EPSC amplitude and frequency in cultured neurons. The role of Rap signaling in vivo, however, remains poorly understood. To study the function of Rap2 in the brain and in behavior, we created transgenic mice expressing either constitutively active (Rap2V12) or dominant-negative (Rap2N17) mutants of Rap2 in postnatal forebrain. Multiple lines of Rap2N17 mice showed only weak expression of the transgenic protein, and no phenotype was observed. Rap2V12 mice displayed fewer and shorter dendritic spines in CA1 hippocampal neurons, and enhanced LTD at CA3–CA1 synapses. Behaviorally, Rap2V12 mice showed impaired spatial learning and defective extinction of contextual fear, which correlated with reduced basal phosphorylation of extracellular signal-regulated kinase (ERK) and blunted activation of ERK during fear extinction training. Our data support the idea that Rap2 opposes Ras–ERK signaling in the brain, thereby inhibiting dendritic spine development/maintenance, promoting synaptic depression rather than LTP, and impairing learning. The findings also implicate Rap2 signaling in fear extinction mechanisms, which are thought to be aberrant in anxiety disorders and posttraumatic stress disorder.
Introduction
Multiple lines of evidence support a prominent role for Ras–mitogen-activated protein kinase (MAPK) signaling in regulation of synaptic structure and function. Ras and its associated GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) are present in the postsynaptic density (PSD) of excitatory synapses (Husi et al., 2000; Peng et al., 2004; Sheng and Hoogenraad, 2007). Extracellular signal-regulated kinase (ERK) activation is necessary for formation of dendritic spines after depolarization (Wu et al., 2001). Both Ras and ERK are necessary for long-term potentiation (LTP), and activated Ras is sufficient to induce synaptic strengthening (English and Sweatt, 1996, 1997; Zhu et al., 2002). ERK function is also necessary for several kinds of learning, including spatial learning, fear conditioning, and fear extinction (Atkins et al., 1998; Blum et al., 1999; Fischer et al., 2007).
Other members of the Ras family (and their GAPs and GEFs) have also been identified at synapses, but their synaptic functions are less well understood (Husi et al., 2000; Jordan et al., 2004; Peng et al., 2004; Kim et al., 2005). Among these proteins are the Rap GTPases, Rap1 and Rap2. Rap1 and Rap2 share 50% identity with Ras, making them the closest relatives to Ras (Pizon et al., 1988). Moreover, Rap1 and Ras possess identical effector domains, whereas the effector domain of Rap2 differs by only one amino acid. Thus, Rap GTPases might stimulate the same effectors as Ras, or they might antagonize Ras by competing for common effectors. Consistent with the latter idea, Rap1 was first identified in a screen for suppressors of Ras transforming activity, and Rap2 inhibits Ras-dependent activation of the transcription factor Elk-1 (Kitayama et al., 1989; Ohba et al., 2000).
In neurons, recent data suggest that Rap1 and Rap2 may negatively regulate synaptic strength, thereby opposing Ras action. Constitutively active Rap1 and Rap2 induce loss of surface AMPA receptors, and Rap2 additionally causes shortening of dendrites and axons, and loss of synapses (Fu et al., 2007). SPAR (spine-associated RapGAP) inhibits Rap and enhances spine growth and synaptic strength (Pak et al., 2001; Pak and Sheng, 2003; Seeburg et al., 2008). In hippocampal slices, dominant-negative Rap1 blocks long-term depression (LTD), whereas dominant-negative Rap2 inhibits depotentiation, the weakening of potentiated synapses (Zhu et al., 2002, 2005). Therefore, multiple layers of evidence indicate that Rap1 and Rap2 are involved in mechanisms that suppress synaptic strength and synapse growth. Less consistent with this model is a report that transgenic mice expressing dominant-negative Rap1 exhibit reduced phosphorylated ERK (pERK) levels, suggesting that Rap1 can activate neuronal ERK in certain contexts (Morozov et al., 2003).
To date, there has been no study of the in vivo function of Rap2 in the nervous system. To address whether Rap2 functions to inhibit synaptic development or transmission in vivo, and to determine the behavioral correlates of Rap2 dysfunction, we constructed transgenic mice expressing dominant-negative (Rap2N17) or constitutively active (Rap2V12) mutants of Rap2 in postnatal forebrain. We present evidence of fewer spines and reduced ERK signaling in Rap2V12 mice, consistent with an inhibitory role for Rap2 at synapses. These biochemical and morphological abnormalities were associated with spatial learning deficits and impairment of fear extinction, a process that is thought to underlie psychiatric disorders such as posttraumatic stress disorder (PTSD) and anxiety disorders (Fischer et al., 2007; Myers and Davis, 2007).
Materials and Methods
Generation of Rap2V12 and Rap2N17 transgenic mice.
SV40 intron/polyadenylation [poly(A)] sequence from pMSG (GE Healthcare) was cloned to the 3′ end of pGW1-HA-Rap2V12 or pGW1-HA-Rap2N17 (gifts from D. Pak, Georgetown University, Washington, DC). HA-Rap2V12-SV40/poly(A) or HA-Rap2N17-SV40/poly(A) was then cloned into pNN19 vector containing the α-calcium/calmodulin-dependent protein kinase II (αCaMKII) promoter (gift from S. Tonegawa, Massachusetts Institute of Technology, Cambridge, MA). This final construct was cut with SalI, releasing a fragment containing αCaMKII promoter, hemagglutinin (HA)-tagged Rap2V12 or Rap2N17, and SV40/poly(A). This fragment was purified and microinjected into pronuclei of C57BL/6 zygotes to generate transgenic founders. For genotyping, PCR was performed on tail samples using a primer pair unique to the transgenic construct. All wild-type (WT) and transgenic mice in this study are from C57BL/6 background and were bred and maintained under Institutional Animal Care and Use Committee-approved conditions.
Biochemistry and antibodies.
For expression analysis, cortex, hippocampus, cerebellum, and brainstem were dissected from brains of 2-month-old littermates and homogenized in buffered sucrose (0.32 m sucrose, 6 mm Tris, pH 8, 1 mm MgCl2, 0.5 mm CaCl2, and protease inhibitors). Homogenates were spun 10 min at 800 × g, and the resulting supernatant was spun 10 min at 10,200 × g. The pellet (P2) was resuspended and 20 μg of protein/lane was loaded for SDS-PAGE. HEK293 cells were transfected with either Rap2V12 or Rap2N17 transgene constructs using Lipofectamine (Invitrogen), and extracts were harvested 48 h later. For subfractionation analyses, individual forebrains from 2-month-old mice were homogenized in buffered sucrose and subjected to differential centrifugation, as described previously (Huttner et al., 1983). Twenty micrograms of protein per lane were loaded for SDS-PAGE.
Levels of GTP-bound Rap2 and Rap1 were measured using EZ-Detect Rap Activation kit (Pierce). Individual forebrains from 2-month-old mice were homogenized in lysis buffer (25 mm Tris, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 0.5 mm CaCl2, 1% NP-40, 1 mm DTT, 5% glycerol, and protease inhibitors). The pulldowns were performed according to manufacturer instructions.
For pERK immunoblot analysis during fear extinction, mice were decapitated 1 h after the indicated training session, and brains were snap-frozen in liquid nitrogen. One-millimeter-thick coronal sections were cut from frozen brain, and amygdala and hippocampus were dissected from the appropriate section and homogenized in buffered sucrose (0.32 m sucrose, 6 mm Tris, pH 8, 1 mm MgCl2, 0.5 mm CaCl2, 1 mm Na3VO4, and protease inhibitors). Twenty micrograms of protein per lane were loaded for SDS-PAGE analysis.
Antibodies used in this study include those against Rap1 (Santa Cruz), Rap2 (Santa Cruz), HA (Santa Cruz), tubulin (Sigma-Aldrich), pERK (Cell Signaling), ERK (Cell Signaling), phosphorylated c-Jun N-terminal kinase (pJNK) (Cell Signaling), c-Jun N-terminal kinase (JNK) (Cell Signaling), and phosphorylated MAPK substrates (gift from D. Edbauer, Massachusetts Institute of Technology, Cambridge, MA).
Immunohistochemistry.
Two-month-old mice were transcardially perfused with 4% paraformaldehyde (PFA), and brains were removed and fixed in 4% PFA overnight at 4°C. Fifty micrometer sagittal or coronal sections were cut with a vibratome, blocked for 1 h at room temperature (RT) (5% normal horse serum, 0.2% Triton X-100 in 1× PBS), and incubated with either anti-HA or anti-Rap2 primary antibody (diluted in blocking buffer) overnight at 4°C. Sections were then washed in 1× PBS and incubated in Alexa 488-conjugated secondary antibody (Invitrogen) for 1 h at RT. Stained sections were visualized using epifluorescence microscopy at 4× magnification.
Cresyl violet staining and DiI labeling.
Brains were fixed in 4% PFA by transcardial perfusion. For cresyl violet, 50 μm sections were cut using a vibratome, mounted, and stained. For DiI labeling, 200 μm sections were cut and labeled “diOlistically,” as previously described (Gan et al., 2000; Hung et al., 2008). The 1.7 μm tungsten beads (Bio-Rad) were coated with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate crystals (DiI) (Invitrogen) and shot into fixed sections with a Helios gene gun (Bio-Rad) through a 3 μm pore membrane (Nalge Nunc International). Sections were postfixed in 4% PFA at RT overnight and mounted.
DiI-labeled neurons were imaged using a LSM510 confocal microscope system (Carl Zeiss) and an Axioplan microscope (Carl Zeiss), using a 100× oil-immersion lens. Each image consisted of a z-stack of pictures taken at an interval of 0.5–0.8 μm. Three 50–70 μm segments from CA1 secondary apical dendrites were examined for spine morphological analysis. MetaMorph software was used to measure spine length, width, and density. Spine length was defined as the distance from the base to the tip of the spine; width was defined as the maximum distance perpendicular to the long axis of the spine. All imaging and quantification were performed blind to genotype.
Electron microscopy.
Mice were anesthetized with Avertin (0.5 mg/g, i.p.) and perfused through the heart with heparinized saline followed by 100 ml of 2% paraformaldehyde/2% glutaraldehyde in phosphate buffer (PB) (0.1 m), pH 7.4. Brains were postfixed for 2 h in the same fixative and sectioned on a vibratome at 50 μm. CA1 hippocampus was dissected and prepared for electron microscopy according to two protocols. For standard procedure, sections were treated for 1 h at RT in 1% osmium tetroxide in PB, rinsed in maleate buffer, pH 6.0, incubated 1 h in 1% uranyl acetate, dehydrated in graded alcohols, infiltrated, and flat-embedded in Epon-Spurr plastic. A second procedure was also used, designed to optimize contrasting of the postsynaptic density; this involved treating in 1% tannic acid in maleate buffer for 1 h, and then 1% CaCl2 for 30 min. Sections were incubated for 1 h in uranyl acetate; subsequent processing was as described previously. Chips from stratum radiatum were cut and glued to plastic blocks; 70–80 nm thin sections were cut on an ultramicrotome, collected on 300 mesh copper grids, and poststained with uranyl acetate and Sato's lead. Random images including well defined synapses were collected at 15,000× on a Philips Tecnai microscope operating at 80 kV; blinded data were analyzed as previously described (Hung et al., 2008).
Electrophysiology.
Transverse hippocampal slices (400 μm thickness) were prepared from 3- to 5-week-old mice in ice-cold dissection buffer (in mm: 238 sucrose, 2.5 KCl, 1 CaCl2, 5 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, gassed with 5% CO2/95% O2, pH 7.4). Slices were incubated in an interface incubation chamber containing extracellular artificial CSF (aCSF) (in mm: 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, gassed with 5% CO2/95% O2, pH 7.4) for 60 min at 30°C, and maintained at room temperature (24–26°C) for at least 2 h. Slices were then transferred to a submerged recording chamber and continuously perfused with aCSF. The recording pipettes (1.5–2 MΩ; 3 m NaCl) for field recording were placed in the stratum radiatum. The tungsten bipolar electrode (FHC) was placed in the stratum radiatum, and the Schaffer collateral/commissural fibers were stimulated at 0.1 Hz. For LTP or LTD experiments, the stimulus strength was adjusted to evoke an EPSP of slope value between 0.11 and 0.15 mV/ms or 0.13 and 0.17 mV/ms, respectively. All experiments were performed at 28°C, using a temperature controller (TC-324B; Warner Instruments). Results are reported as mean ± SEM. Statistical significance was evaluated by Student's t test. Statistical significance was set at p < 0.05. All experiments and the analysis of data were performed in a blind manner. Recordings were performed using a MultiClamp 700B amplifier and Digidata 1320B, and the signal was filtered at 1 kHz and digitized at 10 kHz, and data were acquired and analyzed using Clampex 9.2 and Clampfit 9.2 (Molecular Devices).
Behavioral analysis.
For all behavioral assays, only male littermates between 2 and 5 months of age were used. Experiments and analyses were performed blind to genotype. Open-field test and rotarod were performed as described previously (Crawley, 2007).
Fear conditioning and extinction.
Mice were trained in a standard fear conditioning apparatus (MED Associates). On day 0, they were allowed to explore for 3 min before the onset of a 30 s white noise tone [conditioned stimulus (CS)] that coterminated with a 1 s, 0.75 mA footshock [unconditioned stimulus (US)]. After an additional minute, a second CS–US pair was delivered, and the mice remained in the box for 30 s longer before being removed. Twenty-four hours after training, contextual fear memory was tested by exposing the mice to the original training chamber for 3 min. Cued memory was tested 48 h after training by placing the mice in a new context with a different smell, floor, and walls. Mice were allowed to explore for 2 min before the onset of a 3 min white noise tone.
For fear extinction assays, the training protocol remained the same as before. Every 24 h after training, mice were exposed to either the same context (contextual fear extinction) or to a novel context with tone (cued fear extinction). Separate cohorts of mice were used for the contextual and cued fear extinction assays.
The percentage of time spent freezing was used as an index of fear and was measured automatically using Video Freeze software (MED Associates), with freezing thresholds set by an observer blind to the genotype.
Morris water maze.
Mice were trained in the water maze four times per day over a period of 14 consecutive days. The intertrial interval for each mouse was ∼30 min. Each trial ended either when the mouse found the platform or after 60 s had elapsed. A minute-long probe trial in which the platform was removed was conducted on day 15. The swimming traces were digitized and analyzed using HVS Image 2020 Plus Tracking software (HVS Image).
Results
Generation of transgenic mice expressing constitutively active or dominant-negative Rap2 in postnatal forebrain
To examine the role of Rap2 in vivo, we generated transgenic mice expressing either dominant-negative (Rap2N17) or constitutively active Rap2 mutants (Rap2V12). These transgenes were driven by the αCaMKII promoter, which expresses in forebrain from postnatal stages to adult (Fig. 1A) (Lerosey et al., 1991; Tsien et al., 1996). The Rap2N17 mutant (Ser-to-Asn substitution at position 17) has reduced affinity for GTP and should sequester GEFs that normally activate Rap2. The Rap2V12 mutant (Gly-to-Val substitution at position 12) hydrolyzes GTP more slowly than wild type and therefore spends more time in the active state. SV40 intron/poly(A) sequence was added at the 3′ end, and a HA epitope tag was fused to the N terminus of each transgene.

Generation and characterization of forebrain-specific constitutively active and dominant-negative Rap2 transgenic mice. A, Schematic of Rap2V12 and Rap2N17 transgene constructs. αCaMKII promoter was used to drive expression. HA, Hemagglutinin epitope tag. SV40 intronic sequence and a poly(A) tail were added to the 3′ end of the transgene. B, Brain regions from 2-month-old Rap2V12, Rap2N17, and wild-type littermate mice were immunoblotted with HA or Rap2 antibodies. Last four lanes, Extracts from HEK293T cells expressing HA-Rap2V12 or HA-Rap2N17 were titrated and run alongside the brain extracts. The arrows indicate transgenic Rap2 protein; the arrowheads indicate endogenous Rap2. CTX, Cortex; HIP, hippocampus; CBL, cerebellum; BS, brainstem. C, Immunohistochemistry of brain sections from 2-month-old Rap2V12, Rap2N17, and wild-type littermate mice using antibody against HA. HIP, Hippocampus; STR, striatum; AMYG, amygdala; CBL, cerebellum. Scale bar, 500 μm. D, Rap2 activity assay. Forebrains from 2-month-old Rap2V12, Rap2N17, or wild-type littermate mice were solubilized and incubated with GST-RalGDS RBD. As positive control, WT lysate was preincubated with nonhydrolyzable GTPγS. As negative control, WT lysate was preincubated with GDP. Proteins precipitated by GST-RalGDS RBD were immunoblotted for Rap2, HA, and Rap1. The arrows indicate transgenic Rap2 protein; the arrowheads indicate endogenous Rap2. E, Biochemical fractionation of endogenous and transgenic Rap2V12. Forebrains from 2-month-old Rap2V12 mice were fractionated by differential centrifugation and immunoblotted with Rap2 antibody. Twenty micrograms of protein were loaded for each fraction. F, Developmental expression of Rap2V12 transgenic protein. Forebrain extracts from 1-d-old and from 0.5-, 1-, 2-, 3-, 4-, and 6-month-old mice (Rap2V12 vs wild-type controls) were immunoblotted using HA, Rap2, and tubulin antibodies.
At least 10 founder lines were created for both Rap2N17 and Rap2V12 transgenes. Expression in these founder lines was assayed by Western blotting and immunohistochemistry on brains from 2-month-old mice. On immunoblots using either HA or Rap2 antibody, Rap2V12 mice showed expression of the transgenic protein in cortex and hippocampus, but not in cerebellum and brainstem (Fig. 1B). The transgenic protein (Fig. 1B, arrow) was expressed at approximately fourfold to fivefold higher levels than endogenous Rap2 (arrowhead). The mobility of the Rap2V12 protein from transgenic brain was slightly retarded compared with the endogenous Rap2, but it comigrated with HA-Rap2V12 heterologously expressed in HEK293 cells (Fig. 1B). Rap2N17 founder mice showed only weak expression of the transgene in hippocampus and cortex by immunoblotting (transgenic protein level <10% of endogenous Rap2) (Fig. 1B).
Immunohistochemistry using HA antibody confirmed that Rap2V12 was robustly expressed throughout the forebrain, including hippocampus, cortex, striatum, and amygdala, but not in the cerebellum (Fig. 1C). Staining was concentrated in synapse-rich neuropil areas; cell bodies were mostly spared of immunoreactivity. We noted particularly intense staining along the hippocampal mossy fiber pathway. A similar Rap2V12 staining pattern was observed by Rap2 antibodies (data not shown). Consistent with its low expression by immunoblotting, Rap2N17 transgenic protein could not be detected above background by immunohistochemistry using either HA or Rap2 antibodies (Fig. 1C).
To measure levels of active Rap2 in transgenic brains, we performed a pulldown assay with the Ras binding domain (RBD) of Ral-guanine nucleotide dissociation stimulator (GDS), which binds specifically to the active (GTP-bound) form of Ras, Rap1, and Rap2 (Hofer et al., 1994; Kikuchi et al., 1994; Urano et al., 1996; Nancy et al., 1999). Immunoblot analysis of precipitated Rap2 from the Ral-GDS pulldown assay showed that forebrain lysates from Rap2V12 mice contained elevated levels of active Rap2 compared with wild-type animals, all of which migrated at the size of the transgenic protein (Fig. 1D, arrow). Activated endogenous Rap2 was weakly detected in brain extracts from Rap2V12 or wild-type animals, but could be massively increased by addition of GTPγS to the wild-type lysate (Fig. 1D).
Ral-GDS pulldown did not precipitate the transgenic Rap2N17 protein from Rap2N17 lysates, which is not unexpected given that this mutant is inactive (Fig. 1D). However, Rap2N17 mice also did not show any significant decrease in basal levels of active endogenous Rap2. This negative result could be attributable to inadequate expression of the dominant-negative Rap2N17, or an already very low basal level of active endogenous Rap2. In Rap2N17 as well as Rap2V12 lines, we could not detect basally active endogenous Rap1 in forebrain lysates with the Ral-GDS pulldown assay; however, the presence of Rap1 could be revealed by addition of GTPγS (Fig. 1D). Because we could not obtain a Rap2N17 line that expressed the transgene highly and exhibited decreased Rap2 activity, we focused on Rap2V12 transgenic mice in subsequent experiments (in some experiments, Rap2N17 mice were used as negative controls).
We further characterized the subcellular distribution of Rap2V12 by immunoblotting biochemical fractions of forebrain extracts. Both the transgenic Rap2V12 and the endogenous Rap2 were particularly abundant in P2 (crude synaptosomes) and LP1 (synaptosomal membranes) fractions, as well as being present in LP2 (synaptic vesicle enriched) and P3 (light membrane) fractions (Fig. 1E). Rap2V12 and endogenous Rap2 were barely detectable in cytosolic (S3) fractions of the brain. Thus, both endogenous Rap2 and overexpressed Rap2V12 appear to be mostly membrane-associated.
We also examined expression of transgenic Rap2V12 protein during postnatal development by immunoblotting of cerebral cortex and hippocampus from mice of different ages (Fig. 1F). As expected with the CaMKIIα promoter, transgenic protein expression was undetectable at birth, increased between 0.5 and 1 month, and remained relatively constant thereafter. The expression of endogenous Rap2 protein followed a similar developmental profile (Fig. 1F).
Rap2V12 mice have shorter and fewer dendritic spines in hippocampus
We began phenotyping our Rap2V12 and Rap2N17 mice by examining brain anatomy and dendritic spine morphology. By cresyl violet staining, we observed no gross abnormalities in the brain anatomy of 2-month-old transgenic mice; cell density, hippocampal size, thickness and laminar organization of cortex, and white matter tracts all appeared normal in Rap2V12 and Rap2N17 mice (Fig. 2A). To examine spine morphology, CA1 pyramidal neurons were labeled using the “diOlistic” method, in which dye-coated particles are shot into cells by biolistics (Gan et al., 2000). For quantification, we measured dendritic spines from secondary apical dendrites. Rap2V12 mice showed modest but significant decreases in mean spine length and spine density but no change in mean width of spine heads (Fig. 2B,C) (length, p < 0.01, unpaired t test; density, p < 0.01, unpaired t test). Neurons from Rap2N17 mice showed no changes in mean spine length, width, or density compared with wild-type controls (Fig. 2B,C). All morphometric analysis was performed blind to the genotype. Our findings are consistent with active Rap2 being a negative regulator of spine morphogenesis in vivo.

Brain anatomy and spine morphology in Rap2V12 transgenic mice. A, Coronal sections from 2-month-old Rap2V12, Rap2N17, and wild-type littermate mice stained with cresyl violet. Scale bar, 1 mm. B, Spine morphology of CA1 hippocampal neuron apical dendrites in Rap2V12, Rap2N17, and wild-type littermate mice (age, 2 months), visualized by DiI staining. Scale bar, 5 μm. C, Quantification of CA1 spine length, width, and density in Rap2V12 and Rap2N17 transgenic mice versus wild-type littermates. Error bars indicate SEM. *p < 0.05, unpaired t test.
We also analyzed thin-section electron-microscopic (EM) micrographs from 2-month-old Rap2V12 and Rap2N17 mice. In the stratum radiatum, PSDs from Rap2V12 or Rap2N17 mice showed no detectable differences from WT in terms of length or thickness of the PSD (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Rap2V12 mice show reduced ERK signaling
In certain contexts, both Rap1 and Rap2 have been shown to inhibit Ras-mediated ERK signaling (Kitayama et al., 1989, 1990a,b; Ohba et al., 2000). Rap2 can also activate another mitogen-activated protein (MAP) kinase, JNK, and this Rap2–JNK signaling pathway may be involved in synaptic depotentiation (Machida et al., 2004; Zhu et al., 2005). How does Rap2V12 overexpression affect ERK and JNK activity in transgenic neurons? To measure these signaling pathways, we immunoblotted forebrain fractions from 2-month-old mice for activated pERK and pJNK. Two isoforms of ERK, ERK1 and ERK2 (also known as p44 and p42 MAPK, respectively), are known to be activated downstream of Ras (Boulton et al., 1990, 1991). We found that basal pERK2 (p42) levels were reduced in LP1 (synaptosomal membrane-enriched) and LP2 (synaptic vesicle-enriched) fractions from Rap2V12 mice (Fig. 3A,B) (p42 LP1 fraction, p < 0.05, unpaired t test; p42 LP2 fraction, p < 0.05, unpaired t test). In contrast, levels of pERK1 (p44) and pJNK did not differ significantly between Rap2V12 and WT brain in all fractions tested (Fig. 3A–D).

Reduced pERK levels in Rap2V12 brain fractions. A, Forebrains from Rap2V12 and wild-type mice were fractionated and immunoblotted for pERK. Blots were stripped and reprobed with antibody against total ERK. B, Quantification of pERK levels. The ratio of pERK/ERK intensity was calculated for each fraction and normalized to wild type (which is shown in white bars). The top band representing p44 (ERK1) and the bottom band representing p42 (ERK2) were quantified separately. C, As in A but immunoblotted for pJNK and total JNK. D, Quantification of pJNK levels. The ratio of pJNK/JNK intensity was calculated for each fraction and normalized to wild type. For S1, S2, P2, and LP1 fractions, the top band representing p54 and the bottom band representing p48 were quantified separately. For LP2, S3, and P3 fractions, the total intensity in each lane was quantified. Error bars indicate SEM.
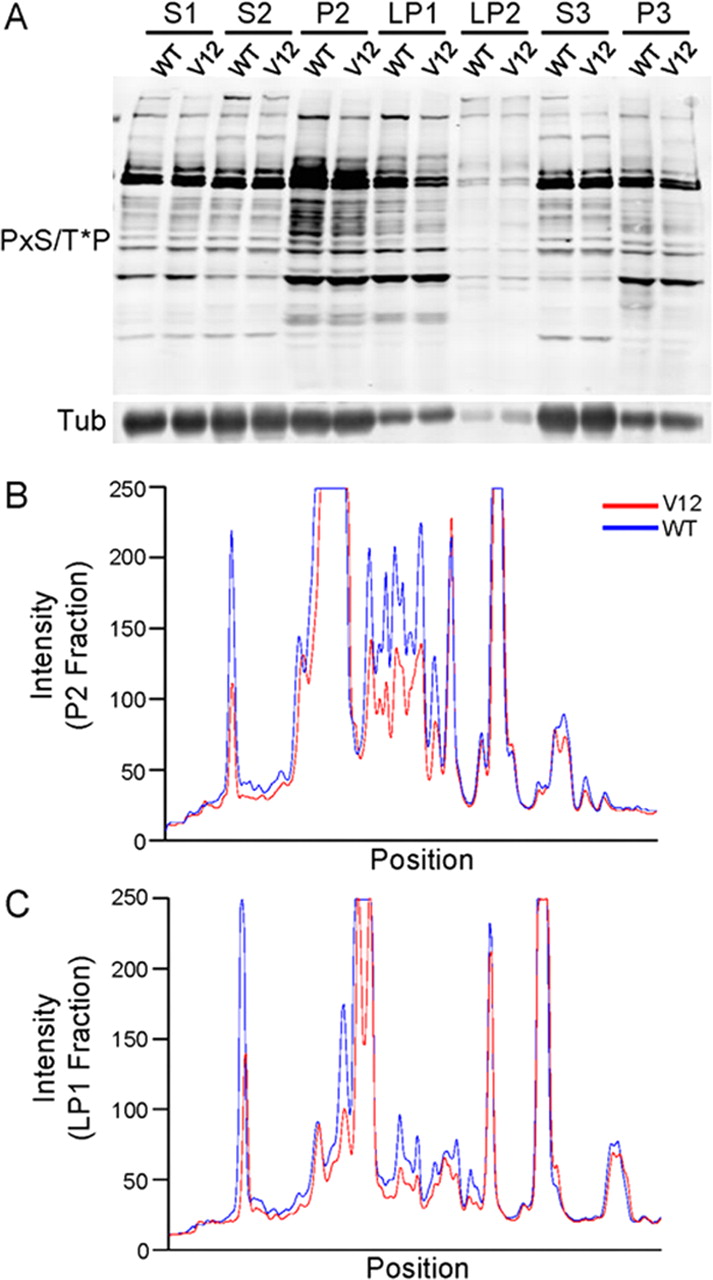
To test further whether MAPK signaling is dysregulated in Rap2V12 mice, we immunoblotted subcellular fractions with a phospho-antibody raised against a MAP kinase phospho-motif peptide library (biased for P-x-S/T-P sequences) that recognizes a large set of phosphorylated MAPK substrates (Fig. 4A) (D. Edbauer and M. Sheng, unpublished data). This MAPK phosphomotif antibody also showed reduced intensity by immunoblotting in a subset of bands in the P2 and LP1 fractions of Rap2V12 mice, corroborating that MAPK signaling is partially inhibited in these fractions (Fig. 4A–C). The decreased levels of some phosphorylated MAPK substrates could be attributable in part to the reduction in ERK2 activity seen in Rap2V12 transgenic brain (Fig. 3).

Reduced phosphorylation of a subset of putative MAPK substrates in Rap2V12 transgenic mice. A, Forebrains from Rap2V12 and wild-type mice were fractionated and immunoblotted with a phospho-antibody raised against the phosphorylated MAPK target motif PxS/T*P. Tubulin was immunoblotted to control for equal loading of wild-type versus Rap2V12 extracts. B, C, Linescan analysis of immunoblot data from A, comparing wild type versus Rap2V12 for P2 fraction and LP1 fraction. Blue trace, WT; red trace, Rap2V12.
Rap2V12 mice show normal basal synaptic transmission and enhanced LTD
To investigate the effects of Rap2 on synaptic function, we measured excitatory synaptic transmission at CA3–CA1 synapses in acute hippocampal slices from 3- to 5-week-old Rap2V12 mice and their wild-type littermates. We first measured AMPA receptor-mediated field EPSPs (fEPSPs) in the stratum radiatum (Fig. 5A). At all stimulation intensities tested, fEPSPs from Rap2V12 slices were similar to those from WT slices, indicating that basal synaptic transmission is normal in Rap2V12 transgenic mice.

Synaptic transmission and plasticity in Rap2V12 hippocampal slices. A, Left, Sample traces (average of 10 consecutive responses) represent the responses evoked with six different stimulus intensities from wild-type or Rap2V12 hippocampal slices. Stimulus artifacts were truncated. Right, Summary graph of the input–output relationships of field EPSPs of wild-type (n = 9 slices from 5 mice) and Rap2V12 mice (n = 9 slices from 5 mice). The symbols indicate the mean ± SEM. FV, fiber volley. B, Top, Sample traces of field EPSPs of wild-type and Rap2V12 mice recorded at the times indicated in summary graph. Bottom, Summary graph of the averaged time course of LTP (WT, n = 7 slices/4 mice; Rap2V12, n = 8 slices/5 mice). Tetanic stimulation (100 Hz; 1 s) was applied at 0 min. Initial EPSP slopes were measured, and the values were normalized to the averaged slope value measured during the baseline period (−30–0 min). C, Sample traces (above) and summary graph (below) of the averaged time course of LTD (WT, n = 11 slices/8 mice; Rap2V12, n = 12 slices/8 mice). A 3 Hz, 5 min stimulation protocol was applied at 0 min. D, Shown is a frequency–response graph of the fEPSP changes induced by four different stimulus frequencies; 0.1 Hz was used to monitor synaptic transmission throughout the experiments. E, Sample traces (left) and summary graph (right) of the averaged time course of depotentiation (WT, n = 11 slices/7 mice; Rap2V12, n = 11 slices/8 mice). After tetanic stimulation (100 Hz; 1 s) at 0 min, low-frequency stimulation (1 Hz; 15 min) was applied at 15 min to induce depotentiation.
We next investigated bidirectional synaptic plasticity at CA3–CA1 synapses in acute hippocampal slices from 3- to 5-week-old mice. The magnitude of LTP induced by high-frequency stimulation (100 Hz; 1 s) was not significantly altered in Rap2V12 slices (WT, 1.34 ± 0.05 of baseline at 58–60 min; Rap2V12, 1.39 ± 0.11; p = 0.717) (Fig. 5B).
Rap GTPases have been implicated in synaptic depression, so we examined LTD more closely over several frequencies of stimulation. Low-frequency stimulation (LFS) at 1 Hz for 15 min induced a slightly larger magnitude of LTD in Rap2V12 slices than WT, although this did not reach statistical significance (WT, 0.89 ± 0.03 of baseline at 73–75 min; Rap2V12, 0.82 ± 0.03; p = 0.100) (Fig. 5D). At a stimulation of 3 Hz for 5 min, LTD was significantly enhanced in Rap2V12 slices compared with WT controls (WT, 0.94 ± 0.02 of baseline at 63–65 min; Rap2V12, 0.84 ± 0.04; p < 0.05) (Fig. 5C,D). A protocol of 5 Hz stimulation for 3 min also induced slightly, but not significantly, larger LTD in Rap2V12 slices (WT, 0.94 ± 0.03 of baseline at 61–63 min; Rap2V12, 0.89 ± 0.03; p = 0.432). The effects of different conditioning protocols are summarized in frequency–response plots, which show the propensity of Rap2V12 mice to develop stronger LTD responses than wild type to low-frequency stimuli (Fig. 5D).
Last, we tested depotentiation in wild-type and Rap2V12 slices by delivering LFS (1 Hz; 15 min) 15 min after tetanic stimulation (100 Hz; 1 s) (Fig. 5E). After LFS, WT and Rap2V12 slices showed similar decreases in synaptic response from their potentiated states (WT, 1.13 ± 0.04 of baseline at 89–90 min; Rap2V12, 1.11 ± 0.08; p = 0.932). Thus, Rap2V12 transgenic expression does not significantly alter the magnitude of depotentiation in the CA1 region.
Rap2V12 mice are hyperactive
The Rap2V12 transgenic mice allowed us to investigate Rap2 effects on behavior. We first examined motor coordination and locomotor activity. In the rotarod test (three training trials per day for 4 consecutive days), Rap2V12 mice showed a mild deficit on the first day, but they learned quickly and performed similarly to their WT littermates on subsequent days (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).
In the open-field test, mice were allowed to explore for 10 min and horizontal activity was measured by an infrared grid. Rap2V12 mice were hyperactive, moving a greater total distance and spending a greater percentage of time in motion than their WT littermates (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material) (total distance, p < 0.01, unpaired t test; move time, p < 0.001, unpaired t test). In contrast, Rap2N17 mice in the open field showed no differences from WT mice in distance moved or time spent moving (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material).
Rap2V12 mice exhibit impaired spatial learning
We next tested spatial learning in Rap2V12 mice using the hidden-platform version of the Morris water maze. Rap2V12 mice and their WT littermates were trained over 14 consecutive days, with each mouse receiving four training sessions per day. Over the course of training, Rap2V12 mice took significantly longer times than WT littermates to find the hidden platform (Fig. 6A) (p < 0.01, repeated-measures two-way ANOVA).

Rap2V12 transgenic mice exhibit impaired spatial learning. A, Hidden-platform Morris water maze. Latency to find hidden platform is plotted for 2- to 4-month-old Rap2V12 or wild-type mice receiving four trials a day for 14 d (each daily block is average of 4 trials). *p < 0.05, repeated-measures two-way ANOVA. B, Probe trial. After 14 d training, the platform was removed. Fraction of time spent in each quadrant was measured. T, Target quadrant; O, opposite quadrant; L, left adjacent quadrant; R, right adjacent quadrant. Error bars indicate SEM.
At the end of the 14 d training session, the platform was removed, and the mice were placed in the maze for a minute-long probe trial. During the probe trial, Rap2V12 mice spent an amount of time in the target quadrant similar to that of their WT littermates (Fig. 6B) (p = 0.46, t test). However, the Rap2V12 mice crossed the exact platform position ∼50% less often than their WT littermates (WT, 3.45 ± 0.74 crossings; Rap2V12, 1.46 ± 0.48 crossings; p < 0.05, t test). Together, these results indicate that Rap2V12 mice are impaired in spatial learning; however, the defect is modest and only picked up by measuring their spatial memory at finer resolution (time to reach platform during training and platform crossings in the probe trial, as opposed to time spent in correct quadrant).
Normal acquisition but impaired extinction of fear memory in Rap2V12 mice
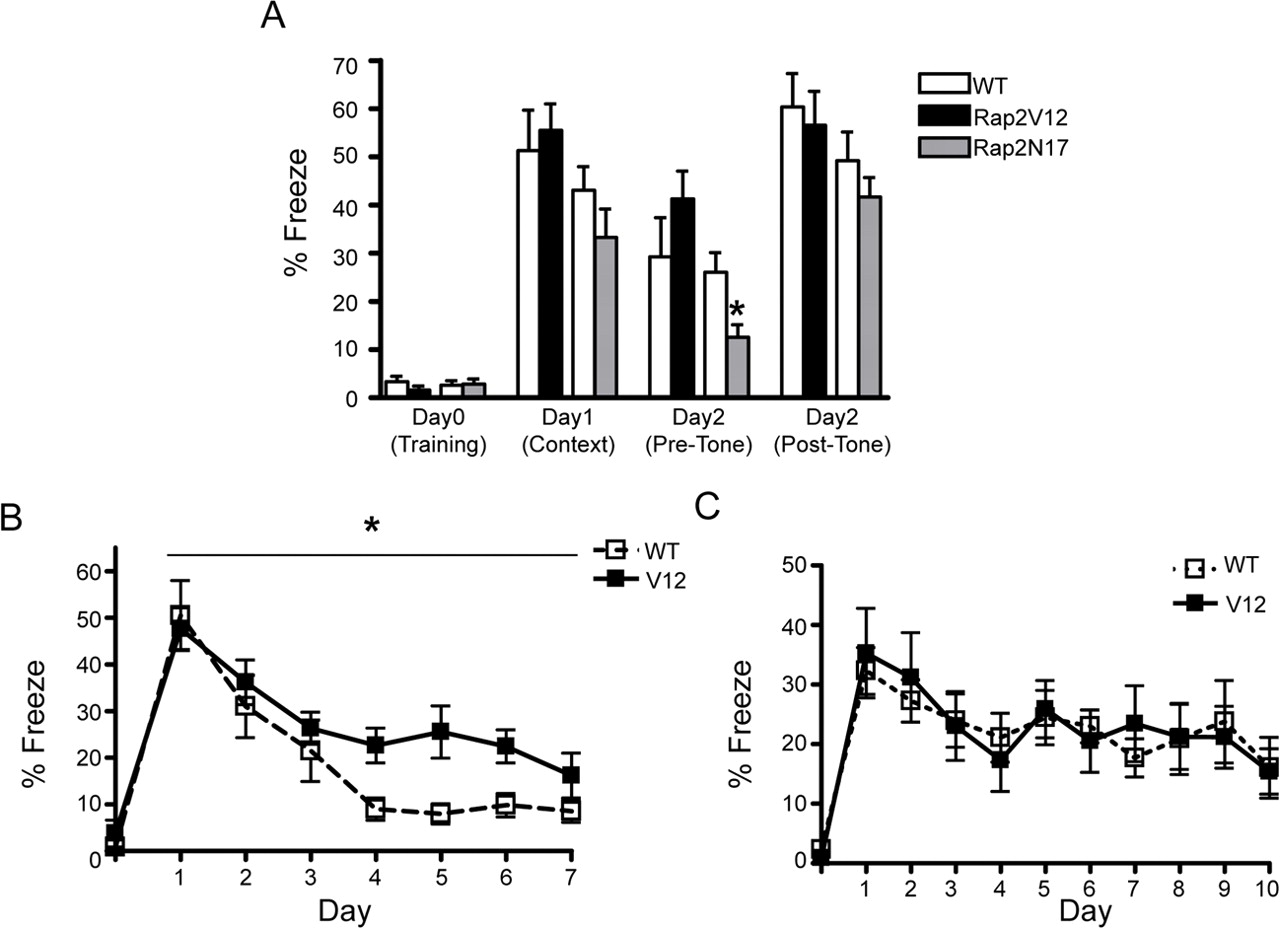
Fear conditioning represents another form of long-term memory, and is thought to involve both the hippocampus and amygdala. Rap2V12 protein is expressed in both these regions (Fig. 1C). For contextual and cued fear conditioning, mice were trained by exposing them to two tone–shock pairings, spaced 1 min apart in a defined environment. Baseline freezing (before training) was similar in Rap2V12 and WT mice (<5%) (Fig. 7A). Twenty-four hours after fear conditioning, freezing behavior was measured in the same context in which training occurred. Rap2V12 and WT mice froze for similar amounts of time (∼50%), indicating that Rap2V12 overexpression does not affect contextual fear learning (Fig. 7A) (p = 0.67, unpaired t test). Forty-eight hours after training, cued fear memory was tested by exposing the mice to the tone in a novel context. Again, the WT and Rap2V12 mice froze for similar amounts of time, both before the tone and in response to the tone (Fig. 7A) (pretone freezing, p = 0.23, unpaired t test; freezing during tone, p = 0.71, unpaired t test). Like Rap2V12 mice, Rap2N17 mice showed no significant abnormalities in either contextual or cued fear conditioning, although we noted a decrease in pretone freezing at 48 h after training (Fig. 7A) (p < 0.05, unpaired t test).

Normal fear acquisition but impaired extinction of contextual fear in Rap2V12 transgenic mice. A, Contextual and cued fear learning. Two- to 4-month-old Rap2V12, Rap2N17, or wild-type littermate mice were subjected to fear conditioning on day 0 and were tested for contextual fear at 24 h and cued fear at 48 h. *p < 0.05, unpaired t test. Percentage of time spent immobile (% Freeze) was measured as index of fear. B, Time course of contextual fear extinction. Two- to 4-month-old Rap2V12 or wild-type mice were trained on day 0 and reexposed to the training context once a day for the following 7 d. *p < 0.05, repeated-measures two-way ANOVA. C, Time course of cued fear extinction. Two- to 4-month-old Rap2V12 or wild-type mice were trained on day 0 and reexposed to the tone in a novel context once a day for the following 10 d. Error bars indicate SEM.
Fear memory is extinguished after repeated exposure to the context or the tone in the absence of footshock. We performed contextual and cued fear extinction in separate cohorts of animals. The training protocol for these extinction experiments was identical with that used for fear conditioning. Starting 24 h after the training session, these mice were exposed to either the training context (contextual fear extinction) or a novel context with tone (cued fear extinction) once a day for 3 min. During both types of extinction training, WT mice eventually reduced their freezing responses (Fig. 7B,C). However, Rap2V12 mice showed a significantly slower time course of contextual fear extinction (Fig. 7B) (p < 0.05, repeated-measures two-way ANOVA). Cued fear extinction was normal in Rap2V12 mice (Fig. 7C) (p = 0.96, repeated-measures two-way ANOVA). Therefore, Rap2V12 mice can acquire contextual and cued fear memories normally, but are specifically impaired in the extinction of contextual fear.
Rap2V12 mice display reduced ERK activation during fear extinction
MEK (mitogen-activated protein kinase kinase) inhibitors have been shown to impair the extinction of fear memories, and ERK activation occurs after fear extinction trials as well as after fear conditioning (Atkins et al., 1998; Fischer et al., 2007). Because Rap2V12 mice showed reduced basal levels of pERK, we hypothesized that reduced ERK signaling might underlie the fear extinction deficit we observed. To test this idea, we measured ERK activation in hippocampal and amygdalar extracts from Rap2V12 mice and their wild-type littermates at multiple stages of fear extinction training: 1 h after fear conditioning, and 1 h after the second, third, and fourth fear extinction trials (Fig. 8A, E2, E3, E4).

Rap2V12 mice show reduced ERK activity during contextual fear extinction. A, Schematic of experiment. Hippocampal and amygdalar extracts were isolated from Rap2V12 mice and wild-type littermates (WT) at the following time points: 1 h after fear conditioning (FC); 1 h after second extinction trial (E2); 1 h after third extinction trial (E3); 1 h after fourth extinction trial (E4). B, C, WT and Rap2V12 pERK/ERK ratios were calculated for each time point and normalized to WT FC time point. Error bars indicate SEM. *p < 0.05; **p < 0.01.
In hippocampus, Rap2V12 and wild-type mice showed similar levels of pERK after fear conditioning (Fig. 8B). In contrast, Rap2V12 transgenic animals showed decreased hippocampal ERK activity relative to WT after the second and third extinction trials (Fig. 8B, E2 and E3) (p < 0.01 for E2; p < 0.05 for E3; unpaired t test). In amygdala, pERK levels were not significantly different between Rap2V12 and wild-type mice after fear conditioning and after each of the extinction trials (Fig. 8C). These data indicate that Rap2V12 transgenic mice have blunted ERK activation during fear extinction, correlating with behavioral impairment in fear extinction.
Discussion
To investigate Rap2 neuronal function in vivo, we constructed transgenic mice expressing either constitutively active or dominant-negative Rap2 in postnatal forebrain. Unfortunately, we were unable to generate a highly expressing Rap2N17 line, perhaps because excessive inhibition of Rap2 function in the brain is lethal. Presumably because of inadequate expression of the dominant-negative construct (<10% of endogenous Rap2 levels), our Rap2N17 lines showed no detectable phenotype in several morphological, biochemical, and behavioral assays. In contrast, Rap2V12 mice expressed the transgene at high levels and displayed significant changes in neuron structure, biochemistry, electrophysiology, and behavior.
Rap2 and MAPK signaling
Rap2V12 transgenic mice showed lower pERK levels and reduced phosphorylation of a subset of MAPK substrates in the basal state, corroborating the idea that Rap2 suppresses the ERK signaling pathway in the brain. Activated ERK (pERK) was specifically reduced in LP1 and LP2 biochemical fractions from Rap2V12 forebrains. These fractions are enriched in synaptosomal membranes and synaptic vesicles, respectively, and thus the data suggest that activated Rap2 can suppress ERK signaling in synaptic compartments. Indeed, endogenous Rap2 and overexpressed Rap2V12 proteins are present in the fractions in which the changes in pERK were observed (LP1 and LP2). However, Rap2V12 is also present in other fractions (e.g., P2 and P3) in which no significant changes in pERK were seen. Therefore, the subcellular fractionation of Rap2V12 does not completely correlate with the sites of changes in pERK levels. It is possible that Rap2V12 suppresses ERK specifically in LP1 and LP2 by antagonizing the Ras–Raf–ERK pathway, which would be expected to be enriched and active mainly at synapses. Recent data show that different ERK signaling pathways can be spatially segregated from one another in neurons using scaffolding proteins (Shalin et al., 2006; Schrick et al., 2007). In P2 and P3 fractions, ERK may be regulated by upstream activators that are unaffected by Rap2.
Rap2 has been shown to bind specifically to Traf2- and Nck-interacting kinase (TNIK) (Machida et al., 2004; Taira et al., 2004; Zhu et al., 2005). TNIK, in turn, can activate the MAP kinase JNK, and in hippocampal slices, Rap2 has been proposed to signal through TNIK and JNK to mediate depotentiation of synaptic strength (Zhu et al., 2005). However, we found no evidence of increased pJNK in Rap2V12 mice and no change in basal synaptic transmission or depotentiation. It is possible that compensatory mechanisms restore normal synaptic function and modifiability in the face of chronically elevated Rap2 activity. For example, the function of PSD-95 in promoting synaptic strength is revealed with acute molecular knockdown in vitro but not in the knock-out mouse (Elias et al., 2006). The magnitude of LTD was enhanced, however, in Rap2V12 mice, supporting the general idea that the function of this Rap GTPase is to promote synaptic weakening, in counterpoint to Ras–ERK signaling (Zhu et al., 2002).
Rap2 and dendritic spines
Overexpression of Rap2V12 causes loss of spines and depletion of PSD-95 clusters in dissociated hippocampal neurons (Fu et al., 2007). Consistent with this, we found that Rap2V12 mice have fewer and shorter spines in CA1 hippocampal neurons, although the effect is much smaller than in culture. How might Rap2 negatively affect spine number and size? Activation of the ERK pathway is necessary for the formation of new spines/filopodia in neurons stimulated by depolarization or NMDAR stimulation (Wu et al., 2001; Goldin and Segal, 2003). By antagonizing the positive role of Ras–ERK signaling in spine morphogenesis, Rap2V12 could suppress spine number and length, as observed in our transgenic mice.
Another pathway that Rap2 might use to regulate dendritic spines is via its effector protein kinase TNIK. In vitro, TNIK can phosphorylate and activate the F-actin-fragmenting enzyme gelsolin, and in cultured cells, TNIK induces actin fiber disassembly (Fu et al., 1999). We found no evidence of altered TNIK expression or distribution in Rap2V12 brain (data not shown). Rap2 could also alter spine structure via integrins and cadherins, two classes of cell adhesion molecules found at synapses (Tashiro et al., 2000; Chan et al., 2003, 2006, 2007; Shi and Ethell, 2006; Webb et al., 2007). Rap1 regulates inside-out signaling to several integrin heterodimers, and loss of Rap1 activity disrupts cadherin localization (Katagiri et al., 2000; Reedquist et al., 2000; Bos, 2005). Although less well characterized in cell adhesion, Rap2 has recently been shown to be necessary for integrin-dependent adhesion in B-lymphocytes (McLeod et al., 2004).
The role of Rap2 in spatial memory and contextual fear extinction
Rap2V12 mice showed impaired spatial learning in the Morris water maze test, as well as impaired extinction of contextual fear. The ERK signaling pathway has been implicated in several different types of learning and memory. Training in the Morris water maze leads to ERK activation in the hippocampus, and pharmacological inhibition of the ERK pathway impairs learning in this assay (Blum et al., 1999; Selcher et al., 1999). Similarly, hippocampal pERK levels are induced by, and are necessary for, contextual fear extinction training in mice (Fischer et al., 2007). It is therefore possible that the impaired spatial learning and defective fear extinction seen in Rap2V12 transgenic mice is attributable to repressed ERK signaling by active Rap2.
Consistent with this idea, Rap2V12 mice showed blunted ERK activation in the hippocampus after extinction training trials (E2 and E3). Interestingly, levels of ERK phosphorylation in both amygdala and hippocampus were normal in Rap2V12 mice after fear conditioning. The impaired biochemical activation of ERK during fear extinction, but not during fear conditioning, correlates with the behavior of Rap2V12 mice, which show normal fear conditioning but impaired fear extinction. Fear conditioning has also been shown to be ERK-dependent, and Rap2V12 mice may be normal in this paradigm because they have wild-type levels of ERK activation at this stage (Atkins et al., 1998). Overall, these results support the idea that Rap2 functions in vivo to suppress Ras–ERK signaling.
Spatial learning in the Morris water maze requires the hippocampus (Morris et al., 1982). Fear extinction is thought to be mediated primarily by the amygdala and the prefrontal cortex (PFC) (Lu et al., 2001; Milad and Quirk, 2002; Myers and Davis, 2002, 2007; Lebrón et al., 2004; Milad et al., 2006, 2007). In addition, the hippocampus seems to be important specifically for contextual modulation of fear extinction (Schimanski et al., 2002; Fischer et al., 2004, 2007; Sananbenesi et al., 2007). The Rap2V12 transgenic protein is highly expressed throughout the forebrain, including the hippocampus, cortex, and amygdala, so it could interfere with biochemical mechanisms of learning in one or all of these critical regions. Because the Rap2V12 mice show normal extinction of cue-dependent fear and also because ERK activation appears to be normal in amygdala during fear extinction, we speculate that the amygdala and PFC are working normally in this process. Therefore, although a more targeted genetic approach is needed to know with certainty, the hippocampus appears to be a major site of Rap2 action underlying the learning and extinction disabilities.
Several psychiatric disorders, including major depressive disorder and PTSD, are thought to occur in part from a failure to extinguish fearful or aversive memories (Rothbaum and Davis, 2003; Bremner et al., 2005; Gillespie and Ressler, 2005). In addition, patients suffering from these diseases have been reported to display deficits in spatial memory as well (Gurvits et al., 2002, 2006). The behavioral phenotype of Rap2V12 mice shares certain characteristics with PTSD, and it would be of interest to explore whether Rap2 signaling might play a role in the molecular mechanisms behind this disease.
Footnotes
-
M.S. is an Investigator of the Howard Hughes Medical Institute. We are grateful to A. Hung for experimental advice and guidance; M. Hayashi, T. McHugh, and A. Govindarajan for assistance with animal behavior and helpful reading of this manuscript; K. Phend and S. Grand for EM processing and analysis; and D. Rooney and X. Zhou for microinjections.
- Correspondence should be addressed to Morgan Sheng, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, 77 Massachusetts Avenue (46-4303), Cambridge, MA 02139. msheng{at}mit.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}