
Abstract
Preferential enhancement of dopamine transmission within the nucleus accumbens (NAc) shell is a fundamental aspect of the neural regulation of cocaine reward. Despite its importance, the nature of this effect is poorly understood. Here, we used fast-scan cyclic voltammetry to examine specific transmission processes underlying cocaine-evoked increases in dopamine transmission within the NAc core and shell. Initially, we examined altered terminal dopamine concentrations after global autoreceptor blockade. This was the first examination of autoreceptor regulation of naturally occurring phasic dopamine transmission and provided a novel characterization of specific components of dopamine neurotransmission. Comparison of increased dopamine signaling evoked by autoreceptor blockade and cocaine administration allowed robust resolution between increased frequency, concentration, and duration of phasic dopamine release events after cocaine delivery. Cocaine increased dopamine transmission by slowed uptake and increased concentration of dopamine released in the core and shell. However, an additional increase in the number phasic release events occurred only within the NAc shell, and this increase was eliminated by inactivation of midbrain dopaminergic neurons. This represents the first evidence that cocaine directly increases the frequency of dopamine release events and reveals that this is responsible for preferentially increased dopamine transmission within the NAc shell after cocaine administration. Additionally, cocaine administration resulted in a synergistic increase in dopamine concentration, and subregion differences were abolished when cocaine was administered in the absence of autoregulation. Together, these results demonstrate that cocaine administration results in a temporally and regionally specific increase in phasic dopamine release that is significantly regulated by dopamine autoreceptors.
- in vivo voltammetry
- neurotransmission
- carbon-fiber microelectrode
- drug abuse
- addiction
- reward
Introduction
The reinforcing properties of cocaine are significantly mediated by enhanced dopamine transmission (Kelley, 2004; Wise, 2004; Everitt and Robbins, 2005), and cocaine exerts its greatest increase in extracellular dopamine concentration ([DA]) within the shell subregion of the nucleus accumbens (NAc) (Di Chiara and Bassareo, 2007). Cocaine increases [DA] by slowing uptake via blockade of dopamine transporters (DATs) (Giros et al., 1996) and by increasing the amount of dopamine exocytosed through mobilization of vesicles normally unavailable for release (Venton et al., 2006). However, neither mechanism can account for preferential enhancement of dopamine transmission within the NAc shell, because both are mediated through terminal DATs and DAT expression is significantly lower in the NAc shell compared with the core (Nirenberg et al., 1997).
It has been suggested that this paradox may be explained by an increased number of dopamine release events within the shell after cocaine administration (Di Chiara and Bassareo, 2007). However, this hypothesis cannot be confirmed using microdialysis because it measures general increases in [DA] without resolving specific transmission processes, such as release and uptake (Wightman et al., 1988). It is often suggested that dopamine release can be inferred from electrophysiological assessment of dopaminergic neuron activity (Schultz, 2002). However, dopamine signaling is significantly modulated by terminal transmission (Cragg, 2006; Britt and McGehee, 2008), and extracellular electrophysiological identification of a dopaminergic phenotype can be unreliable (Margolis et al., 2006). Therefore, unambiguous assessment of terminal dopamine release events requires real-time measurements of [DA].
This is robustly achieved, in freely moving subjects, with fast-scan cyclic voltammetry (FSCV) (Wightman et al., 2007). FSCV uses subsecond measurements that resolve specific components of phasic dopamine transmission: the occurrence of a release event, the concentration of dopamine released (i.e., exocytosed), and the duration of increased [DA] (Wightman et al., 1988). Although these features have been characterized using electrically stimulated dopamine release (Wightman and Zimmerman, 1990), the present study required distinction between transmission components after cocaine administration. Thus, it was necessary to provide pharmacological characterization of drug-evoked alterations in naturally occurring phasic dopamine release events, i.e., dopamine “transients” (Wightman and Robinson, 2002).
Here, transient characterization was facilitated by manipulating dopamine autoreceptors because their regulation of dopamine transmission is extremely well understood (White, 1996). Comparisons of real-time measurements after cocaine administration and systemic blockade of dopamine autoreceptors provided reliable resolution between increases in the frequency of dopamine release events and the concentration of dopamine released. Using these distinctions, we show that preferential enhancement of dopamine transmission within the NAc shell evoked by cocaine is attributable to a greater number of phasic release events originating from midbrain dopaminergic neuron activity. This subregion difference was abolished by autoreceptor blockade before cocaine administration. Finally, cocaine administration in the absence of autoregulation resulted in a synergistic increase in [DA]. Together, this study provides novel characterization of the distinct processes that encompass extracellular dopamine transmission, shows the first evidence that cocaine directly increases dopamine release events in a temporally and regionally specific manner, and demonstrates the significance of autoregulation in cocaine-evoked dopamine transmission.
Materials and Methods
Animals and surgery.
Male Sprague Dawley rats purchased with implanted jugular vein catheters (n = 30; ∼325 g; Charles River Laboratories). Rats were anesthetized with ketamine hydrochloride (100 mg/kg, i.p.) and xylazine hydrochloride (20 mg/kg, i.p.). Ag/AgCl reference electrodes (placed in contralateral cortex) and bipolar stimulating electrodes (Plastics One) [placed in the ventral tegmental area (VTA); 5.2 mm posterior, 1.0 mm lateral, 7.5 mm ventral relative to bregma (Paxinos and Watson, 1998)] were secured as described in detail previously (P. E. Phillips et al., 2003; Wightman et al., 2007). Guide cannulas (Bioanalytical Systems) were aimed at the NAc core (1.3 mm anterior, 1.3 mm lateral, −2.5 mm ventral) or shell (1.7 mm anterior, 0.8 mm lateral, −2.5 mm ventral relative to bregma). These placements are within rostral portions of the NAc shell, which has greater [DA] (Heidbreder and Feldon, 1998) and negligible noradrenergic input (Delfs et al., 1998; Baldo et al., 2003) compared with caudal portions. After the experiment, an electrolytic lesion was made at the microdrive setting used during the experiment and verified histologically. All experiments were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Fast-scan cyclic voltammetry.
After 5–7 d recovery from surgery, glass-encased carbon-fiber electrodes were lowered using a locally constructed microdrive (University of North Carolina at Chapel Hill, Department of Chemistry Instrument Shop) and positioned where both electrically evoked (biphasic pulses, 2 ms/phase, 24 pulses, 60 Hz, 120 μA) and transients were detected (Wightman et al., 2007). Waveform generation and processing, current transduction, and data collection and filtering have been described in detail previously (Wightman et al., 2007). To ensure that the signal of interest was attributable to dopamine and not other analytes, current was converted to [DA] using principal component regression as described previously (Heien et al., 2005). Consistent with previous studies (Heien et al., 2005), a dopamine transient was defined as a fivefold or greater surge in [DA] relative to the root-mean-square noise value taken from the same electrode. Events that met this criterion were rarely below 30 nm, and transients below 30 nm were not reliably detected across electrodes and were therefore excluded from the analysis. The conservative criterion used ensured that all measurements occurred beyond the electrochemical baseline associated with FSCV that primarily comprises undifferentiated chemical and electrical noise (Wightman et al., 2007) and digitally subtracted from the signal of interest (Heien et al., 2005). Transient amplitude and half-width were determined with Mini Analysis (Synaptosoft) with the first minima 300–600 ms before the peak serving as the baseline (Cheer et al., 2007b). Transients above 30 nm were accurately identified by the “differential method” for transient detection (Heien et al., 2005), and this analysis was therefore used to determine transient frequency. For the differential method, cyclic voltammograms were compared with that of electrically evoked dopamine release from the same subject (Stuber et al., 2005). Cyclic voltammograms with a correlation coefficient >0.86 were assigned to dopamine, and a 40% decay defined transient termination (Heien et al., 2005; Stuber et al., 2005). Mean increases in [DA] relative to the zero point established by background subtraction applied to the raw data after drug treatment are obtained from averaging [DA] traces resulting from principal component regression. Change in mean [DA] was restricted to a 90 s sampling period because electrode drift prevents reliable analysis beyond this point for most electrodes (Heien et al., 2005). After the experiment, electrodes were calibrated as described previously (Wightman et al., 2007). All chemicals were purchased from Sigma-Aldrich.
VTA microinfusions.
Microinfusions were made into a cannula attached to the stimulating electrode (Plastics One) directed at the VTA using a syringe pump with a flow rate of 0.5 μl/min. A within-subjects design (n = 5) was used in which vehicle (0.5 μl of sterile saline) or the GABA agonists (50 ng baclofen/5 ng muscimol) were delivered in a counterbalanced manner with the second infusion occurring after the baseline frequency of dopamine transients was reestablished, ∼45 min later. Each microinfusion was followed 1 min later by a slightly lower dose cocaine (coc) infusion (2 mg/kg, i.v.) compared with that used in the primary experimental design (3 mg/kg, i.v.). This lower dose resulted in a statistically equivalent increase in cocaine-evoked transient frequency compared with 3 mg/kg infusion. The lower dose was chosen to minimize recovery time between infusions in an attempt to reduce the length of the experiment. A minimum duration is desired for a within-subjects design because it increases the likelihood of equivalent electrode stability across time. After the experiment, 0.5 μl of a 2% Chicago Sky Blue solution was infused into the VTA immediately before the animal was perfused. Subjects showed dye covering ∼70% of the VTA [including cellular subpopulations that project to the shell (Ikemoto, 2007)] with minimal staining of structures outside of the VTA.
Statistics.
Changes in [DA], transient frequency, and transient amplitude after raclopride (rac) and cocaine administration were assessed using linear mixed models, which were chosen based on their ability to properly handle data in which observations are not independent (such as repeated measures data), correctly model correlated error terms, and incorporate random-subject effects. To determine mean changes of [DA], measurements were averaged into 2.5 s time bins and treated as the dependent variable, with time serving both as a repeated measure and fixed-effect variable. Estimation of time bins at which concentration differed from predrug baselines was achieved by construction of simple slopes and comparison of corresponding critical T values to the reference period (10 s before drug infusion; “predrug”). Core and shell differences were assessed by including region as a fixed-effect variable, enabling investigation of location effects and time × location interactions. Transient frequency and amplitude followed Poisson and β distributions, respectively. For transient frequency, time (in 1 min bins) was treated as a repeated and fixed effect, and region was treated as a fixed effect. Time points during a given experimental condition in which transient frequency differed from the preceding condition were determined by comparing all time points of the session of interest to a fixed baseline value of the preceding session that was based on the mean transient frequency over the last 2 min of the recording session (which never differed between subregions). Although there were no statistical differences between core and shell, frequency within the shell was consistently higher after saline administration in coc–rac subjects. Therefore, transient frequency after saline in this group of subjects was included as a covariate for analysis after drug administration. For transient amplitude, both region (core vs shell) and amplitude distribution (in 10 nm bins) were treated as fixed effects, enabling analysis of subregion and drug-induced differences in transient concentration. Changes in mean cocaine-induced transient frequency after VTA inactivation were assessed during the first 3 min after cocaine infusion, using a one-way repeated measures ANOVA with Student–Newman–Keuls post hoc tests. Mean transient half-width over the entire duration of each recording session was compared using a one-way ANOVA with Student–Newman–Keuls post hoc test. Statistical significance was designated at α = 0.01 for mixed-model analyses and at α = 0.05 for ANOVA and post hoc comparisons. All statistical analyses were performed in SPSS version 14 for Windows (SPSS) or SAS for Windows (SAS Institute).
Results
Design and histology
Carbon-fiber microelectrodes (Wightman, 2006) were secured in discrete locations (6 × 100 μm) that supported electrically stimulated and naturally occurring dopamine release (Wightman et al., 2007). Real-time fluctuations in [DA] were measured during a series of 15 min recording sessions (Fig. 1A,B). At the completion of each session, electrode stability was tested by stimulation of dopaminergic neurons (60 Hz, 24 pulses, 120 μA) (Fig. 1A,B).

Experimental design and histology. A, B, Recording sessions (indicated by arrows) were followed by electrical stimulation of dopaminergic cell bodies (indicated by boxes after the arrows). Subsequent recording sessions began 5 min after stimulation offset. Specific manipulations are color coded throughout the study, with baseline (no manipulation) recordings coded as white and saline control sessions as black. A, rac/coc subjects received raclopride infusions (1 mg/kg; 6 s) (green) followed by cocaine (3 mg/kg; 6 s) (gray). B, coc/rac subjects received cocaine (orange) followed by raclopride (blue). C, For rac/coc subjects, dopamine measurements were conducted in either the NAc core (n = 7) or shell (n = 6). D, Measurements for coc/rac subjects were also conducted in either the core (n = 7) or shell (n = 5).
Experiments began with a baseline recording session (Fig. 1A,B) followed by a control session that tested the effects of intravenous saline infusion (0.2 ml; 6 s) (Fig. 1A,B). At the beginning of the next recording session, rac/coc subjects received intravenous infusion of the D2-type dopamine receptor antagonist raclopride (1 mg/kg), followed 20 min later by intravenous infusion of cocaine (3 mg/kg) (Fig. 1A). A separate set of subjects, referred to as coc/rac, received the reverse order of drug administration: cocaine followed 20 min later by raclopride (Fig. 1B). Data from the NAc core and shell (Fig. 1C,D) were analyzed separately.
Phasic dopamine transmission
Neurochemical changes at all potentials of the measurement scan (oxidative scan, −0.4 to 1.3 V; reductive scan, 1.3 to −0.4 V) were visualized by plotting current changes in false color (Michael et al., 1999) (Fig. 2A). The dopamine oxidation peak occurs at ∼0.65 V and the reduction peak at −0.2 V (Fig. 2A). The cyclic voltammagram is converted to [DA] using principal component regression (Heien et al., 2005) (Fig. 2B). Consistent with previous studies (Gonon, 1988), the color plot (Fig. 2A) and corresponding concentration trace (Fig. 2B) show that stimulation of dopaminergic neurons that mimics synchronous burst firing (20 Hz, 4 pulses, 120 μA) (Grace and Bunney, 1984b) results in phasic dopamine release. In contrast to interpretations suggesting that only a small amount of phasic dopamine release escapes the synapse (Floresco et al., 2003), the extracellular [DA] resulting from this release event was ∼150 nm (Fig. 2B).

Analysis of dopamine transients. A, The holding potential applied to the carbon-fiber electrode (−0.4 V) was ramped to 1.3 V and back 10 times per second. Oxidation and reduction reactions at the carbon surface result in changes in current that are plotted in false color. Dopamine release evoked by electrical stimulation (white dashed line) and naturally occurring dopamine transients (inverted white triangles) appear as brief green events at the potential of peak dopamine oxidation (∼0.65 V). B, Current was converted into [DA] and amplitudes for stimulated release (boxes on the trace indicate stimulation onset and offset), and transients are displayed above each event (indicated by orange circles). The point at which half-width was calculated is shown by the orange dashed lines, and the half-width values are given below.
FSCV also detects naturally occurring phasic dopamine release events that are termed dopamine transients (Wightman and Robinson, 2002). In the seconds following electrical stimulation, two transients were detected that had no apparent environmental cause (Fig. 2A,B). Concentrations of dopamine transients are consistent with the contents of dopamine vesicles (∼0.5 m) (Sulzer and Pothos, 2000) being released and diluted in the extracellular volume (Garris et al., 1994; Cragg and Rice, 2004). Typical transient magnitudes result in a twofold to fivefold phasic increase over basal [DA] (∼20 nm) (Shou et al., 2006) and are within the range of binding affinities for dopamine receptors (Richfield et al., 1989). The physiological relevance of concentrations measured by FSCV is also attributed to the minimal tissue damage caused by the microsensors used (Peters et al., 2004).
Critically, measuring dopamine transients allows the investigation of specific components of phasic dopamine signaling. Transient detection reveals the occurrence of a dopamine release event, transient amplitude reflects the amount of dopamine exocytosed, and transient half-width represents the duration of a surge in [DA] (Robinson and Wightman, 2007) (Fig. 2B). The goal of the present study was to use these measurements (transient frequency, amplitude, and half-width) to assess the contribution of phasic dopamine transmission to increased [DA] induced by autoreceptor blockade and cocaine administration. However, it was first necessary to quantify drug-evoked increases in [DA] within the NAc core and shell.
Real-time fluctuations in dopamine concentration
Quantification of drug-evoked increases in [DA] (which includes tonic and phasic contributions) was accomplished by continuous measurement of concentration fluctuations over a 90 s sampling window encompassing intravenous drug infusion (Heien et al., 2005). Measurements began 10 s before drug infusion and continued as initial drug effects were observed (∼10–20 s after infusion) and 1 min thereafter. Representative examples after administration of raclopride demonstrate robust increases in [DA] in the NAc core (Fig. 3A) and shell (Fig. 3B). Although these increases partly comprised a gradual rise in [DA], increased dopamine transmission was primarily driven by a robust increase in the frequency of phasic release events. Insets allow closer inspection of individual transients and emphasize the high amplitude and long duration of these phasic release events (Fig. 3A,B).

Representative examples of increases in dopamine concentration during drug delivery. A–D, Individual examples of 90 s [DA] traces across intravenous drug infusion (indicated by yellow boxes; 6 s; 0.2 ml). Insets provide increased resolution of naturally occurring transient events. A, B, Raclopride-evoked changes in [DA] within the NAc core (A) and NAc shell (B). C, Cocaine-evoked changes in [DA] within the NAc core (C) and NAc shell (D).
In the NAc core, cocaine induced a gradual increase in [DA] indicative of slowed uptake of ongoing release events (Fig. 3C) (Venton et al., 2003; Wightman et al., 2007). The inset shows that dopamine transients remained superimposed on the gradual rise (Fig. 3C). However, their initial magnitude was low compared with that after autoreceptor blockade. Conversely, within the NAc shell, cocaine caused a more rapid and robust increase in [DA] (Fig. 3D). Dopamine transients superimposed on the gradual rise (Fig. 3D, inset) achieved concentrations and durations similar to those seen after autoreceptor blockade, and slowed uptake of these events (mediated by cocaine binding to terminal DATs) facilitated the accumulation of dopamine in the extracellular space.
Quantification of concentration changes
For quantification of changes in [DA], traces were averaged across subjects. Saline infusion did not alter [DA] in the NAc core (see Figs. 4A, 5A) or shell (see Figs. 4B, 5B). However, raclopride administration evoked robust increases in [DA] in both subregions (Fig. 4A,B). For statistical analysis, concentration data were binned every 2.5 s. Raclopride significantly increased [DA] compared with predrug levels beginning 17.5 and 25 s after infusion onset in the shell and core, respectively (Fig. 4C). [DA] was significantly greater in the shell compared with the core soon after infusion onset (17.5–30 s) but not thereafter (Fig. 4C). Subregion differences were not attributable to electrode placement or sensitivity because electrically stimulated dopamine release after raclopride administration did not differ between subregions (supplemental Figs. S1A,B, S2A,B, available at www.jneurosci.org as supplemental material).

Average drug-evoked increases in dopamine concentration and core–shell comparisons: rac/coc subjects. A, B, Mean concentration traces (solid lines) and mean plus SE (dashed lines) for saline (black), raclopride (rac; green), and cocaine in the presence of raclopride (rac-coc; gray) are superimposed on one another. Yellow boxes indicate the time of drug infusion. A, B, Drug-evoked [DA] changes within the NAc core (A) and NAc shell (B). C, D, [DA] was binned every 2.5 s for comparisons between the core and shell after raclopride (rac) administration C and cocaine administration in the presence of raclopride (rac-coc; D). Data from the NAc shell are displayed by open circles, and data from the core are shown as filled circles. Error bars indicate SEM. > signs indicate statistical significance (p < 0.01) between groups identified by their appropriate color-coded symbol. An asterisk inside the symbol indicates when drug-evoked [DA] is significantly greater compared with [DA] before drug infusion.
Consistent with previous studies (Stuber et al., 2005), cocaine-evoked increases in [DA] within the NAc core began to rise ∼30 s after drug infusion (Fig. 5A) but were not significantly increased over predrug levels until 50 s after infusion onset (Fig. 5C). Within the shell, cocaine-evoked increases in [DA] were evident ∼10 s after infusion onset (Fig. 5B) and were significantly increased over predrug levels beginning at 17.5 s (Fig. 5C). Furthermore, [DA] was significantly greater within the shell compared with the core from 17.5 s after infusion onset and remained higher throughout the sampling period (Fig. 5C). Subregion differences were not attributable to differences in terminal release potential because electrically stimulated dopamine release did not differ between the core and shell before or after cocaine administration (supplemental Fig. S1C,D, S2C,D, available at www.jneurosci.org as supplemental material).

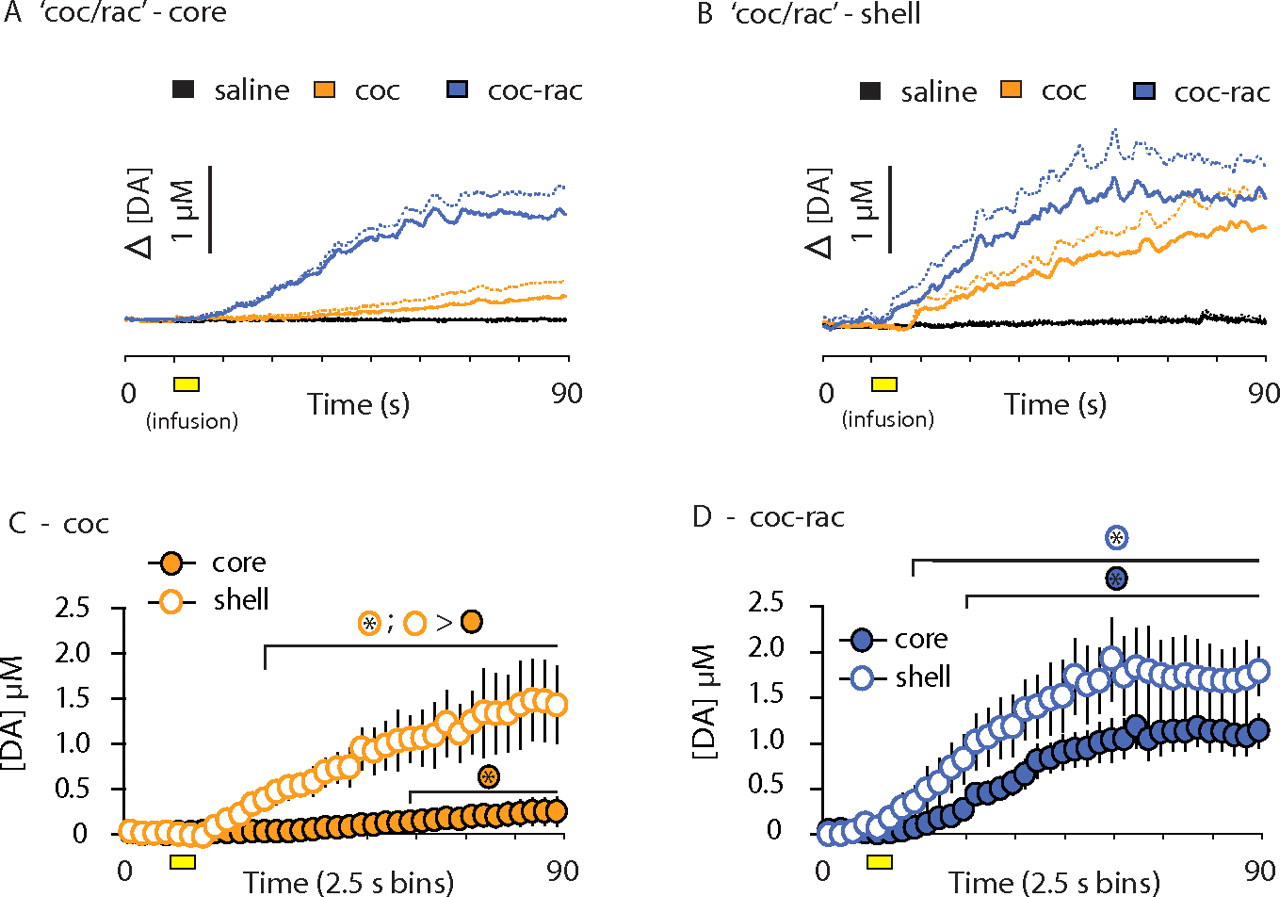
Average drug-evoked increases in dopamine concentration and core–shell comparisons: coc/rac subjects. Mean concentration traces (solid lines) and mean plus SE (dashed lines) for saline (black), cocaine (coc; orange), and raclopride in the presence of cocaine (coc-rac; blue) are superimposed on one another. Yellow boxes indicate the time of drug infusion. A, B, Drug-evoked [DA] changes within the NAc core (A) and NAc shell (B). C, D, [DA] was binned every 2.5 s for comparisons between the core and shell after cocaine (coc) administration (C) and raclopride administration in the presence of cocaine (coc-rac; D). Data from the NAc shell are displayed by open circles, and data from the core are shown as filled circles. Error bars indicate SEM. > signs indicate statistical significance (p < 0.01) between groups identified by their appropriate color-coded symbol. An asterisk inside the symbol indicates when drug-evoked [DA] is significantly greater compared with [DA] before drug infusion.
Consistent with microdialysis studies (Rouge-Pont et al., 2002), cocaine administration in the absence of autoregulation (via raclopride pretreatment) induced a synergistic increase in [DA] (Fig. 4A,B). [DA] was significantly greater than predrug levels at 7.5 and 12.5 s after infusion onset in the core and shell, respectively (Fig. 4D). Cocaine-evoked increases in the presence of raclopride did not differ across subregions (Fig. 4D), suggesting that autoreceptors mediate subregion differences evoked by cocaine alone (Fig. 5C).
Similar increases were observed when raclopride was administered in the presence of cocaine (Fig. 5A,B). Raclopride-evoked increases in [DA] occurred sooner in the shell (10 s) compared with the core (20 s). Although there was a trend for greater raclopride-evoked [DA] (in the presence of cocaine) within the shell early in the sampling period (Fig. 5D), this did not reach statistical significance. Importantly, such robust increases within the NAc core demonstrate that the modest increase in [DA] within the core after cocaine administration alone (Fig. 5A,C) was not attributable to a lower release potential within this subregion.
Finally, the magnitude of synergistic increases differed depending on drug order. In both the core and shell, peak [DA] was significantly higher when cocaine was given in the presence of raclopride (5.09 ± 0.61 μm) (Fig. 4D) compared with raclopride administration in the presence of cocaine (1.80 ± 0.24 μm; p < 0.001) (Fig. 5D). This is most likely attributable to the diminished effects of cocaine in 20 min, whereas raclopride effects are stable for >1 h.
Transient frequency
Although real-time assessment of changes in [DA] provided unprecedented information regarding the timing and magnitude of drug-evoked increases in dopamine signaling, this analysis alone does not directly address the underlying nature of enhanced dopamine transmission. To determine the contribution of specific transmission processes to increases in [DA], dopamine transients were analyzed. Data were background subtracted every 30 s (Cheer et al., 2007b; Wightman et al., 2007) to permit transient analysis across recording sessions, and transient frequency was quantified to determine drug-evoked alterations in phasic dopamine release events.
Control infusions of saline did not significantly increase transient frequency compared with the baseline session (baseline data not shown) (Fig. 6A,B). However, raclopride administration increased transient frequency to a similar degree in both subregions for the entire recording session (Fig. 6A). This demonstrates that global autoreceptor blockade results in a stable increase in phasic dopamine release events. Given that transient frequency did not differ between the core and shell, subregion differences in raclopride-evoked increases in [DA] concentration (Fig. 4C) are not attributable to differences in the number of release events. Importantly, with phasic release events increased by raclopride administration, cocaine administration equally increased transient frequency (relative to the end of the raclopride session) within the NAc core and shell (Fig. 6A).

Transient frequency. A, Transients per minute after saline (black), raclopride (rac; green), and cocaine administration in the presence of raclopride (rac-coc; gray). B, Transients per minute after saline (black), cocaine (coc; orange), and raclopride administration in the presence of cocaine (coc-rac; blue). A, B, Data from the NAc shell are displayed by open circles, and data from the core are shown as filled circles. > signs indicate statistical significance (p < 0.01) between groups identified by their appropriate color-coded symbol. Error bars indicate SEM. C, Illustration of the approximate spread of microinfusions (based on dye infusion) into the VTA and location of carbon-fiber placements within the NAc shell. D, Mean baseline and cocaine-evoked (obtained from the first 3 min after cocaine infusion) transient frequency after either vehicle infusion (0.5 μl of saline) or vehicle containing the GABA agonists baclofen and muscimol (Bac/Mus) into the VTA. Error bars indicate SEM. *p < 0.05.
Within the NAc core, cocaine administration induced a significant (but modest compared with raclopride) increase in transient frequency compared with the saline condition for the entire recording session (Fig. 6B). Raclopride administration (in the presence of cocaine) resulted in a robust increase in transient frequency compared with cocaine administration alone (Fig. 6B). Because transient detection is maximal after DAT blockade (Robinson and Wightman, 2004), this raclopride-evoked increase represents a true increase in the number of release events and is therefore a reliable index of a neurochemical consequence of increased dopaminergic neuron firing induced by autoreceptor blockade (White, 1996). Furthermore, it confirms that the NAc core has the capacity for robust increases in phasic release events but simply did not respond in this manner after cocaine administration alone.
Within the shell, cocaine increased transient frequency for 6 min compared with saline (Fig. 6B). Transient frequency was significantly greater in the shell compared with the core for the first 3 min after cocaine administration (Fig. 6B). Raclopride administration in the presence of cocaine increased transient frequency to the same level as raclopride administration alone (Fig. 6B). Although cocaine never induced this magnitude within the NAc core, cocaine-evoked transient frequency within the shell was not statistically different from the raclopride condition for 2 min after cocaine administration, a duration consistent with the time of peak [DA] assessed by microdialysis using 60 s temporal resolution (Wise et al., 1995). These data reveal that subregion differences in cocaine-evoked dopamine transmission (Fig. 5C) are attributable to a greater number of phasic release events within the NAc shell immediately after cocaine administration.
Given that the magnitude of raclopride-evoked transients is a result of increased dopaminergic neuron firing and that this magnitude was achieved in the shell after cocaine, these data suggest that cocaine may briefly increase the firing of dopaminergic neurons projecting to the shell (Ikemoto, 2007). To test this hypothesis, an additional experiment was conducted (n = 5) in which cocaine administration was preceded by infusion of saline (0.5 μl) or a combination of GABA agonists (50 ng of baclofen and 5 ng of muscimol) (McFarland and Kalivas, 2001) directly into the VTA (Fig. 6C). Intra-VTA infusion saline did not influence dopamine transmission (Fig. 6D). However, infusion of GABA agonists eliminated cocaine-evoked increases in transient frequency within the NAc shell (Fig. 6D) and, as a result, attenuated cocaine-evoked elevation in [DA] (0.274 ± 0.053 μm after intra-VTA saline; 0.032 ± 0.030 μm after intra-VTA GABA agonists; p < 0.01; values obtained from the 90 s sampling window after cocaine infusion). These data demonstrate that cocaine-evoked increases in phasic release events within the shell originate from increased activity of midbrain dopaminergic neurons.
Transient amplitude
Transient amplitude is an index of the concentration of dopamine released (Wightman et al., 1988; Schmitz et al., 2001, 2002). A representative color plot (Fig. 7A) and concentration trace (Fig. 7B) show that, although the majority of transients are of lower concentration (30–60 nm), higher concentration transients (>100 nm) also occur (Fig. 7A,B). For amplitude quantification, transient histograms were organized into 10 nm bins (Fig. 7). Amplitude histograms were compared using a linear mixed model with rightward shifts in distribution representing increased transient concentration.

Transient amplitude distributions. A, B, A color plot and corresponding [DA] trace from a baseline condition demonstrate the variability of transient amplitudes. Symbols are described in Figure 2. C–H, All dopamine transients within the 15 min recording sessions were grouped in 10 nm bins beginning with transients between 30 and 39 nm (30) and ending with transients between 150 and 159 nm (150). C–E, Core–shell comparisons of amplitude histograms for saline (from rac/coc subjects; C), raclopride (rac; D), and cocaine administration in the presence of raclopride (rac-coc; E). F–H, Core–shell comparisons for saline (from coc/rac subjects; F), cocaine (coc; G), and raclopride administration in the presence of cocaine (coc-rac; H). C–H, Data from the NAc core are presented as filled circles, and data from the NAc shell are presented as open circles. > signs indicate statistical significance (p < 0.01) between groups identified by their appropriate color-coded symbol. Error bars indicate SEM.
Saline infusion did not alter amplitude compared with baseline sessions (baseline data not shown). After saline administration in rac/coc subjects, the majority of dopamine transients were of lower concentration in both subregions (Fig. 7C). Raclopride increased the concentration of dopamine transients compared with saline in the core and shell (Fig. 7D), and this is consistent with studies showing that autoreceptor blockade increases the concentration of dopamine released (White, 1996; Benoit-Marand et al., 2001; P. E. Phillips et al., 2002). Raclopride-evoked increases in transient concentration were significantly greater in the shell compared with the core (Fig. 7D) and is therefore likely responsible for subregions differences in [DA] after raclopride administration (Fig. 4C). In both subregions, cocaine administration (in the presence of raclopride) significantly increased transient amplitude beyond that of raclopride administration alone (Fig. 7E).
In coc/rac subjects, amplitude distribution after saline administration showed the same pattern compared with rac/coc subjects (Fig. 7, compare C, F), and transient concentration was significantly greater in the shell compared with the core (Fig. 7F). Consistent with cocaine-evoked increases in the amount of dopamine released (Venton et al., 2006), cocaine significantly increased transient amplitude compared with saline in both subregions (Fig. 7G). Thus, increased transient detection (Robinson and Wightman, 2004) and the concentration of dopamine exocytosed (Venton et al., 2006) contribute to cocaine-evoked increases in [DA] (Fig. 5C) and transient frequency (Fig. 6B). However, amplitude histograms after cocaine did not differ between the core and shell (Fig. 7G). This indicates that enhanced dopamine detection and transient amplitude are not responsible for subregion differences in dopamine transmission after cocaine administration (Figs. 5C, 6B). Raclopride administration (in the presence of cocaine) similarly increased transient amplitude compared with cocaine administration alone in both subregions (Fig. 7H).
Transient half-width
Transient half-width measures the duration of a release event, a process governed by uptake (Wightman et al., 1988; Benoit-Marand et al., 2000; Cragg and Rice, 2004). Transient duration was brief after raclopride (Fig. 8A) compared with the very broad and long-lasting transients evoked by cocaine (Fig. 8B). This is consistent with uptake being directly influenced by DAT blockade (Giros et al., 1996) and indirectly influenced by autoreceptor blockade (Cass and Gerhardt, 1994; Wu et al., 2002) [via their interaction with DATs (Lee et al., 2007)].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transient half-width. A, B, Representative color plot and corresponding [DA] trace after raclopride (A) and cocaine (B) administration. A, B, Examples are representative of ongoing transients between 5 and 15 min after drug delivery (i.e., after the initial drug effects described in Figs. 3⇑–5). C–F, Half-width data were taken from all dopamine transients within the 15 min recording sessions; solid bars represent data from the NAc core, and striped bars represent data from the shell. C, D, Mean transient half-width after saline (sal; black), raclopride (rac; green), and cocaine administration in the presence of raclopride (rac-coc; gray) within the core (C) and within the shell (D). E, F, Mean transient half-width after saline (black), cocaine (coc; orange), and raclopride administration in the presence of cocaine (coc-rac; blue). C–F, Error bars indicate SEM. *p < 0.05.
Saline infusion did not significantly alter transient half-width compared with baseline measurements (baseline data not shown). Raclopride administration significantly increased transient half-width over saline levels in both the NAc core (Fig. 8C) and shell (Fig. 8D). Administration of cocaine in the presence of raclopride further increased transient half-width compared with raclopride treatment alone (Fig. 8C,D). Cocaine administration increased transient half-width to its maximum mean value within the NAc core (Fig. 8E) and shell (Fig. 8F). Administration of raclopride in the presence of cocaine did not further increase half-width (Fig. 8E,F). Because transient half-width did not differ between the core and shell after any drug treatment, increases in the duration of phasic dopamine transmission contributes to drug-evoked increases in [DA] but not the subregion differences observed.
Discussion
Extracellular [DA] and phasic dopamine signaling were predictably manipulated by experimenter delivery of an autoreceptor blocker (raclopride) as well as cocaine. Comparison of real-time dopamine transmission after these manipulations allowed neurochemical characterization of distinct aspects of dopamine neurotransmission. Specifically, the occurrence of dopamine release events was revealed by detection of dopamine transients. The concentration of dopamine released and the duration of release events were examined by analyzing transient amplitude and half-width, respectively. Analysis of these specific transmission components provided the first description of alteration in phasic dopamine signaling after autoreceptor blockade and permitted the determination that cocaine slowed uptake and increased the concentration of ongoing release events within both the NAc core and shell. However, cocaine administration caused an additional brief and robust increase in phasic release events only within the NAc shell, and this effect was eliminated by inactivation of dopaminergic neurons. Subregion differences in [DA] after cocaine delivery were abolished by raclopride pretreatment, and cocaine administration resulted in a synergistic increase in [DA] in the absence of autoregulation. Together, these data demonstrate that subregion differences in [DA] after cocaine administration are attributable to temporally and regionally specific dopamine release that is significantly regulated by dopamine autoreceptors.
Autoreceptor regulation of phasic dopamine transmission
This study provides the first assessment of dopamine transients after autoreceptor blockade. Autoreceptors are located on cell bodies and terminals of dopaminergic neurons (White, 1996; Cragg and Greenfield, 1997; Centonze et al., 2002). Blockade of somatodendritic autoreceptors increases dopaminergic neuron firing (Bunney et al., 1973; Grace and Bunney, 1984a; White, 1996), and the dose of raclopride used in the present study (1 mg/kg, i.v.) robustly increases burst firing (Andersson et al., 1995). Because burst firing results in phasic dopamine release (Gonon, 1988), raclopride-evoked transients reported in the present study likely originate from burst firing induced by blockade of somatodendritic autoreceptors. Terminal autoreceptors regulate dopamine release and synthesis (White, 1996; Benoit-Marand et al., 2001; P. E. Phillips et al., 2002). Therefore, raclopride-evoked increases in transient amplitude are likely mediated by autoreceptors located at dopaminergic terminals.
Studies of the time course of electrically evoked dopamine release show that autoreceptor blockade slows uptake (Cass and Gerhardt, 1994; Wu et al., 2002). In the present study, raclopride administration caused a gradual rise in [DA] that is attributable in part to slowed uptake. Furthermore, raclopride administration resulted in greater transient amplitude within the shell, and this is therefore likely responsible for subregion differences in raclopride-evoked increases in [DA]. This apparent autoreceptor imbalance between subregions is mostly likely attributable to less efficient uptake within the shell compared with the core (Nirenberg et al., 1997). Indeed, raclopride increased mean transient half-width over control levels but not if subjects were pretreated with cocaine, which confirms that autoreceptors influence uptake through interaction with DATs (Lee et al., 2007).
Because autoreceptor regulation of dopamine transmission has been extensively characterized (White, 1996), raclopride-evoked alterations of transient frequency, amplitude, and half-width provide reliable distinctions between phasic release events, the concentration of dopamine released, and the duration of increased [DA], respectively. This novel characterization of pharmacologically evoked dopamine transmission allowed for unprecedented interpretation of cocaine-evoked increases in [DA].
Cocaine-evoked dopamine release in the nucleus accumbens shell
It is known that cocaine-evoked increases in [DA] are action potential dependent (Nomikos et al., 1990). Within the NAc core, cocaine induced a gradual increase in [DA] indicative of slowed uptake of ongoing release events (Venton et al., 2003; Wightman et al., 2007). Cocaine also increased transient amplitude, which is consistent with increased dopamine exocytosed per release event (Venton et al., 2006). Thus, enhanced dopamine detection after DAT blockade (Robinson and Wightman, 2004) and increased release magnitude (per event) (Venton et al., 2006) allow a greater number of transients to reach detection limits (Wightman et al., 2007) and are responsible for increased transient frequency in the core. However, despite maximum detection after this high dose of cocaine (3 mg/kg, i.v.), increased transient frequency within the core did not reach levels produced by autoreceptor blockade. This suggests that cocaine did not substantially increase the activity of dopaminergic neurons projecting to the core, which is consistent with studies showing that cocaine does not typically increase firing of dopaminergic neurons (Einhorn et al., 1988; Shi et al., 2004) projecting to this region (Ikemoto, 2007).
Consistent with previous studies (Pontieri et al., 1995; Lecca et al., 2007), cocaine-evoked a substantially greater increase in [DA] within the NAc shell compared with the core. In addition to the processes described above, cocaine administration increased the number of phasic release events within the shell. Cocaine-evoked transient frequency within this subregion achieved the same magnitude as that reached after raclopride administration, a manipulation known to increase dopaminergic neuron firing (Andersson et al., 1995; White, 1996). This suggests that greater transient frequency within the shell is attributable to a brief increase in dopaminergic neuron firing.
In support of this, VTA inactivation eliminated the cocaine-evoked increase in transient frequency within the shell, indicating that the preferential dopamine transmission within the shell is attributable to increased activity of dopaminergic neurons. Furthermore, because this manipulation did not completely eliminate baseline transients, the shell-specific increase in transient frequency would have been detected if this effect were mediated at the terminal level. Therefore, anatomical differences between the core and shell (Carboni et al., 2006) and differential afferent regulation known to influence terminal dopamine transmission (Howland et al., 2002; A. G. Phillips et al., 2003) do not significantly contribute to preferential enhancement of dopamine transmission with the NAc shell. Although this initially appears inconsistent with electrophysiology studies showing that cocaine primarily decreases the firing rate of dopaminergic neurons (Einhorn et al., 1988; Shi et al., 2004), these studies primarily targeted neurons projecting to the NAc core and caudate–putamen but not the NAc shell (Ikemoto, 2007). Electrophysiological assessment of cocaine administration on shell-projecting neurons would be valuable, but dopamine release undergoes significant modulation within terminal fields (Cragg, 2006; Britt and McGehee, 2008). Therefore, real-time electrochemical assessment is required for unambiguous conformation of terminal dopamine transmission after cocaine administration.
Real-time measurements allow reliable interpretation of the origin of increased [DA]. The current study demonstrates that phasic dopamine release is a consequence of electrical stimulation that mimics bursting (Gonon, 1988) and raclopride treatment known to increase bursting (Andersson et al., 1995). Therefore, cocaine-evoked increases in phasic release events, beyond that attributed to enhanced dopamine detection and exocytosis, may be regarded as originating from increased burst firing. Burst firing is regulated by glutamate (White, 1996) and cocaine increases glutamate within the VTA, via somatodendritic dopamine release (Bradberry and Roth, 1989) activating D1-type dopamine receptors (Kalivas and Duffy, 1995). Therefore, increased glutamate transmission is likely responsible for direct increases in phasic release events by cocaine. The VTA contains glutamatergic neurons (Yamaguchi et al., 2007) and receives glutamatergic afferents from areas ranging from the brainstem to the cortex (Geisler et al., 2007), and it will be important for future studies to identify the source of cocaine-evoked glutamate release within the VTA. Additionally, subregion differences in transient frequency were abolished by autoreceptor blockade, suggesting differences in somatodendritic autoregulation between core and shell projection systems (Ikemoto, 2007; Lammel et al., 2008). Thus, although specific mechanistic details require future investigation, the current study provides the working hypothesis that reduced autoregulation (Lammel et al., 2008) of cocaine-evoked bursting (Shi et al., 2004) among shell-projecting neurons (Ikemoto, 2007) is responsible for preferentially increased [DA] by cocaine within the NAc shell.
Conclusion
Dopamine transmission mediates a wide range of adaptive behaviors (Becker et al., 2001; Champagne et al., 2004; Roitman et al., 2004; Aragona et al., 2006; Day et al., 2007), and dopamine signaling within the NAc shell is critical for potentiated seeking of natural rewards (Kelley and Berridge, 2002; Everitt and Robbins, 2005). However, intake of artificially purified drugs of abuse (Nesse and Berridge, 1997; Panksepp et al., 2002) results in a pathological enhancement of dopamine signaling within the NAc shell (Di Chiara et al., 2004). Cocaine-evoked dopamine transmission can result in maladaptive increases in incentive motivation for drug (Robinson and Berridge, 2003) and extremely strong learned associations to drug-related stimuli (Berke, 2003; Kelley, 2004). Importantly, drug-related learning is mediated through activation of low-affinity dopamine receptors within the shell (Di Chiara and Bassareo, 2007), and high [DA] events are required to activate these receptors (Richfield et al., 1989; Reynolds et al., 2001; Cheer et al., 2007a). Therefore, the shell-specific increase in phasic release events described in the present study are likely essential for primary drug reinforcement (Wise, 2004; Di Chiara and Bassareo, 2007). Furthermore, the duration of increased phasic release events closely matches the time of peak subjective pleasure expressed by humans after experimenter-delivered intravenous cocaine (Volkow et al., 2000), suggesting the possibility that cocaine-evoked dopamine release may also be associated with the hedonic impact after cocaine intake (Risinger et al., 2005; Wheeler et al., 2008). Thus, by resolving distinct transmission processes that modulate extracellular [DA], the current study provides insight into a neural consequence of cocaine intake that may be essential for multiple aspects of drug reward and abuse.
Footnotes
- Received May 16, 2008.
- Revision received July 10, 2008.
- Accepted July 24, 2008.
-
This work was supported by National Institutes of Health Grants F32 21489 (B.J.A.), DA 17318 (R.M.C., R.M.W.), and DA 10900 (R.M.W., R.M.C.). We thank Rita Fuchs Lokensgard, Donna R. Ramirez, and Mark Stuntz for technical assistance, Daniel Serrano and Chris Wiesen for statistics consulting, Collin McKinney, John Peterson, and Larry George for instrumentation expertise, and Joshua L. Jones and Robert A. Wheeler for critical reading of this manuscript.
- Correspondence should be addressed R. Mark Wightman, Department of Chemistry, Campus Box 3290, Venable and Kenan Laboratories, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290. rmw{at}unc.edu
- Copyright © 2008 Society for Neuroscience 0270-6474/08/288821-11$15.00/0