

Abstract
The role(s) of the novel stargazin-like γ-subunit proteins remain controversial. We have shown previously that the neuron-specific γ7 suppresses the expression of certain calcium channels, particularly CaV2.2, and is therefore unlikely to operate as a calcium channel subunit. We now show that the effect of γ7 on CaV2.2 expression is via an increase in the degradation rate of CaV2.2 mRNA and hence a reduction of CaV2.2 protein level. Furthermore, exogenous expression of γ7 in PC12 cells also decreased the endogenous CaV2.2 mRNA level. Conversely, knockdown of endogenous γ7 with short-hairpin RNAs produced a reciprocal enhancement of CaV2.2 mRNA stability and an increase in endogenous calcium currents in PC12 cells. Moreover, both endogenous and expressed γ7 are present on intracellular membranes, rather than the plasma membrane. The cytoplasmic C terminus of γ7 is essential for all its effects, and we show that γ7 binds directly via its C terminus to a heterogeneous nuclear ribonucleoprotein (hnRNP A2), which also binds to a motif in CaV2.2 mRNA, and is associated with native CaV2.2 mRNA in PC12 cells. The expression of hnRNP A2 enhances CaV2.2 I Ba, and this enhancement is prevented by a concentration of γ7 that alone has no effect on I Ba. The effect of γ7 is selective for certain mRNAs because it had no effect on α2δ-2 mRNA stability, but it decreased the mRNA stability for the potassium-chloride cotransporter, KCC1, which contains a similar hnRNP A2 binding motif to that in CaV2.2 mRNA. Our results indicate that γ7 plays a role in stabilizing CaV2.2 mRNA.
Introduction
Voltage-dependent calcium channels (CaV) are heteromultimers consisting of a pore-forming α1 subunit, assembled with auxiliary β, α2δ, and possibly γ subunits (for review, see Catterall, 2000; Dolphin, 2003a,b). The role(s) of the γ subunits in relation to calcium channel function remains unclear. The first CaVγ subunit to be identified was γ1, which copurifies with the skeletal muscle calcium channel complex (Jay et al., 1991; Powers et al., 1993). In skeletal muscle, the γ1 subunit appears to have a suppressive effect, because γ1 knock-out mice exhibit increased skeletal muscle calcium currents (Freise et al., 2000). After the identification of stargazin (γ2) (Letts et al., 1998), subsequent studies have identified six additional putative γ subunits (γ3–γ8) (Black and Lennon, 1999; Burgess et al., 1999, 2001; Klugbauer et al., 2000; Moss et al., 2002). However, it is unclear whether any of these novel stargazin-like γ proteins (γ2–γ8) play any role as subunits of voltage-gated calcium channels. All members of this family are thought to possess four transmembrane-spanning domains with intracellular N and C termini. The γ2, γ3, γ4, and γ8 subunits form a subfamily exclusively localized to the CNS (Letts et al., 1998; Klugbauer et al., 2000; Sharp et al., 2001; Moss et al., 2003) whose interaction and functional modulation of CaV channels has been investigated in several studies (Letts et al., 1998; Klugbauer et al., 2000; Kang et al., 2001; Rousset et al., 2001; Sharp et al., 2001; Moss et al., 2003) but which are now thought primarily to represent trafficking proteins for the AMPA subtype of glutamate receptors (TARPS) (Tomita et al., 2003, 2004). However, they might also provide a bridge between calcium channels and AMPA receptors (Kang et al., 2006). Furthermore, γ7, despite not having a classical C-terminal PDZ (postsynaptic density-95/Discs large/zona occludens-1) binding domain, has also been shown recently to have effects on AMPA receptor trafficking (Kato et al., 2007).
The γ7 and γ5 proteins are predicted to represent a distinct subfamily of stargazin-related proteins (Burgess et al., 2001; Chu et al., 2001; Moss et al., 2002), with extremely low sequence identity to γ1 and ∼25% identity to γ2, mainly in the transmembrane domains. We showed that coexpression of the γ7 subunit with CaV2.2 almost abolished the functional expression and markedly suppressed the level of CaV2.2 α1 subunit protein (Moss et al., 2002). It also had smaller suppressive effects on CaV2.1 and CaV1.2 currents (Moss et al., 2002). Our conclusion was that γ7 was not a subunit of these calcium channels. Nevertheless, because of the marked effect of γ7 on calcium channel expression and because both N-type calcium channels and γ7 are specifically expressed in neuronal tissue, we have now examined the mechanism of action of γ7 to probe its physiological function.
The present results indicate that γ7 is involved in regulating the stability of specific mRNAs, including CaV2.2. We propose that the mechanism may involve γ7 sequestering a specific mRNA binding protein, thus compromising the stability of CaV2.2 mRNA.
Materials and Methods
cDNA constructs.
The following cDNAs were used: CaV2.2 (GenBank accession number D14157) and CaV2.2 Δ3′ untranslated region, CaV3.1 (GenBank accession number AF027984), β1b (GenBank accession number X61394; from Dr. T. P. Snutch, University of British Columbia, Vancouver, British Columbia, Canada), α2δ-2 (GenBank accession number AF247139, common brain splice variant), γ7 (GenBank accession number NM031896), mut-3b green fluorescent protein (GFP) (GenBank accession number M62653, except S72A and S65G; from Dr. T. E. Hughes, Montana State University, Bozeman, MT), mouse KCC1 (GenBank accession number AF121118), KV3.1b (GenBank accession number M68880), heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) (GenBank accession number BC070315), and hnRNP A2–ΔRGG (Nichols et al., 2000). Truncated and tagged γ7 constructs and hemagglutinin (HA)-tagged hnRNP A2 were generated using standard molecular biological techniques and confirmed by DNA sequencing. All constructs were cloned into the pMT2 expression vector (Swick et al., 1992), except γ7–HA and γ7–cyan fluorescent protein (CFP), hnRNP A2–ΔRGG, and KCC1, all of which were in pcDNA3.1, except γ7–CFP in pECFP–N1, Kv3.1b in pRC–CMV, and hnRNP A1 in pCGT7. pDsRed2–endoplasmic reticulum (ER) plasmid was used in some experiments (Clontech).
Short-hairpin RNA design and expression plasmid.
siSearch (http://sonnhammer.sbc.su.SE/databases.html) and Jura (http://jura.wi.mit.edu/siRNAext/) software was used to design 19-nucleotide sequences corresponding to human and rat γ7 genes. Databases were searched to ensure that these sequences were not homologous to any other known genes. Rat γ7 (GenBank accession number AF361345) targets are as follows: γ7 96 (5′-CTGGCTGTATATGGAGGAG-3′), γ7 285 (5′-GACAGTACGCACGGCTACA-3′), and γ7 500 (5′-CTGAGCAATACTTTCACTA-3′). Human γ7 (GenBank accession number AF458897) targets are as follows: γ7 96 (5′-CTGGCTGTACATGGAAGAA-3′), γ7 285 (5′-GACAGTGCGCACGGCCACC-3′), and γ7 107 (5′-TGGAAGAAGGCACAGTGCT-3′). Then, for each target, two oligonucleotides (A and B) were synthesized (Invitrogen). Oligonucleotide A contains a 5′ overhang (TTTG) for ligation into a BpiI (BbsI) site, the sense and the antisense of these sequences linked by a hairpin loop of 9 bases [TTCAAGAGA (Brummelkamp et al., 2002)], and a TTTTT sequence corresponding to a termination for transcription of small RNAs by RNA polymerase III; oligonucleotide B contains a 5′ overhang (CTAG) for ligation into an XbaI site and 52 nucleotides corresponding to the reverse complement of the last 52 nucleotides of oligonucleotide A. Forward and reverse strands were annealed and subcloned downstream a U6 promoter into pG418–shRNA–Empty linearized with XbaI and BpiI. pG418–shRNA–Empty is a pBSII plasmid containing a mouse U6 promoter and a neomycin (G418) resistance cassette. A negative control short-hairpin RNA (shRNA), directed against Drosophila gene gnu (Xu and Shrager, 2005) and a positive control shRNA directed against c-jun (Lingor et al., 2005), were also synthesized. Correct orientation and location of oligonucleotide cloning were confirmed by sequencing the plasmids. Validation of the plasmid in PC12 cells was determined by its ability to knockdown c-jun (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Antibodies against γ7.
The γ7 tail polyclonal rabbit antibody (Ab) raised against the C-terminal peptide YPPAIKYPDHLHIS has been described previously (Moss et al., 2002). A γ7 loop Ab was also raised against the peptide VASEYFLEPEINLVTEN, in the loop between transmembrane segments 1 and 2 of γ7. These Abs do not recognize any of the other γ proteins tested (γ2–γ4) (supplemental Fig. 2A–C, available at www.jneurosci.org as supplemental material).
Cell culture and transfection.
COS-7 cells were cultured as described previously (Campbell et al., 1995). The tsA 201 cells were cultured in DMEM with 10% fetal bovine serum (FBS) and 1% l-glutamine. Cells were transfected using either Geneporter (Qbiogene) or Fugene6 (Roche Diagnostics), with equivalent results. The cDNAs (all at 1 μg/μl) for CaVα1, α2δ-2, β1b, γ7, and GFP when used as a reporter of transfected cells, were mixed in a ratio of 3:1.5:2:1.5:1:0.2, unless otherwise stated. When particular subunits were not used, the volume was made up with Tris–EDTA (10 mm Tris and 1 mm EDTA, pH 7), or blank vector, or the volume of transfection reagent was reduced, with equivalent results. In some experiments, cDNA for the nonconducting potassium channel Kir2.1–AAA (Tinker et al., 1996) was used as control for the presence of γ7, also with equivalent results to the use of blank vector.
PC12 cells were grown in DMEM, 7.5% FBS, and 7.5% horse serum. For electrophysiology, PC12 cells were transiently transfected with cDNAs for GFP and/or γ7, using Fugene6. Differentiation was with serum-free medium containing NGF (100 ng/ml murine 7s NGF; Invitrogen), replenished every 48 h. Cells were used for recording after 5–7 d of differentiation. For the generation of stable cell lines, PC12 cells were transfected with γ7–HA or γ7–CFP, and clonal cell lines were established by standard techniques, using 400 μg/ml Geneticin (Invitrogen) for selection. The culture medium was subsequently supplemented with 400 μg/ml Geneticin (Invitrogen).
To obtain high transfection rates with shRNAs, PC12 cells were transfected using an Amaxa Nucleofector, according to manufacturer instructions. The DNA mix contained 2 μg of shRNA plasmid and 0.5 μg of GFP or yellow fluorescent protein (YFP) as a reporter of transfected cells. Cells were used 4–5 d after transfection with shRNA.
For primary culture of superior cervical ganglion (SCG) neurons, rats were killed by either CO2 inhalation or cervical dislocation, according to United Kingdom Home Office Schedule 1 Guidelines. SCGs were dissected from rats at postnatal day 17. Ganglia were desheathed and lightly gashed before successive collagenase (Sigma) and trypsin (Sigma) treatment, both at 3 mg/ml. To produce a single-cell suspension, ganglia were dissociated by trituration and centrifugation. Dissociated cells were plated onto glass-bottomed plates (MatTek) precoated with laminin (Sigma), using one ganglion per five plates. Cells were maintained with Liebovitz L-15 medium (Sigma), supplemented with 24 mm NaHCO3, 10% FBS (Invitrogen), 33 mm glucose (Sigma), 20 mm l-glutamine, 1000 IU of penicillin, 1000 IU of streptomycin (Invitrogen), and 50 ng/ml NGF.
Microinjection.
cDNAs were injected into SCG neurons 18–24 h after they were placed in culture. Microinjection was performed using an Eppendorf microinjection system on a Carl Zeiss Axiovert 200M microscope using the following settings: 100–150 hPa injection pressure, an injection time of 0.2 s, and constant pressure of between 40 and 50 hPa. The cDNAs were injected at 50 ng/μl diluted in 200 mm KCl.
Measurement of mRNA levels by quantitative PCR.
RNA was isolated using RNeasy columns (Qiagen), including an on-column DNase step. Reverse transcription (RT) was performed using random hexamer primers and Moloney murine leukemia virus reverse transcriptase (Promega) at 37°C for 2 h. The quantitative PCR (q-PCR) was performed with an iCycler (Bio-Rad) using the iQ SYBR supermix. For each set of primers and for every experiment, a standard curve was generated using a serial dilution of reverse-transcribed RNA combined from several samples. For q-PCR in PC12 cells, the following primers were used: rat CaV2.2 (GenBank accession number NM147141), 5′-GGCAAGAAGGAGGCAGAG-3′ and 5′-GCAGAAGCGACGGAGTAG-3′; rat γ7 (GenBank accession number AF361345), 5′-CTACTCGGGCCAGTTTCTGC-3′ and 5′-GCCGGAGGGTAATTTTGC-3′; and rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (GenBank accession number AF106860), 5′-ATGACTCTACCCACGGCAAG-3′ and 5′-CATACTCTGCACCAGCA-TCTC-3′. Data were normalized for expression of GAPDH mRNA.
For measurement of mRNA degradation rates, q-PCR of transcript levels was performed in Xenopus oocytes. Plasmid cDNAs were injected intranuclearly for CaV2.2, β1b, and α2δ-2, either with or without γ7. After 24 h, oocytes were incubated for the stated times with actinomycin D (50 μg/ml). RNA extraction, RT, and q-PCR were performed as described above. The following primers were used: CaV2.2, 5′-CTCTGCGCTTACTGAGAATC-3′ and 5′-AACAGGAAGAGCAGGAAGAG-3′; 18S, 5′-TGACTCAACACGGGAAACCT-3′ and 5′-AATCGCTCCACCAACTAAGAAC-3′; α2δ2, 5′-GGTATTTGCTGCCACTGATG-3′ and 5′-AGGCTGCGACGGTAGAAG-3′; and KCC1, 5′-AGCATAAGGTTTGGAAGAAGTG-3′ and 5′-CAGGCGGAGGTGATACAG-3′. Data were normalized for expression of 18S ribosomal RNA.
Oligoribonucleotide binding to hnRNP A2.
This was performed as described previously (Hoek et al., 1998), with the exception that mouse brain, rather than rat brain, was used as a source of hnRNP A2. The following RNA oligonucleotides were used: A2 response element (A2RE) 5′ biotin, GCCAAGGAGCCAGAGAGCAUG-3′; CaV2.2 5′ biotin, GCCAAGGAGCGGGAGCGAGUC-3′; and nonspecific 5′ biotin, CAAGCACCGAACCCGC-AACUG-3′. A2RE and nonspecific sequences are identical in base composition (Hoek et al., 1998). Proteins were taken up in 200 μl of SDS sample buffer and heated at 65°C for 10 min. Twenty microliters of each was run on a 4–12% Bis–Tris gel, and Western blotting and immunodetection was performed with anti-hnRNP A2 Ab (Autogen Bioclear, 1:200).
Yeast two-hybrid assay.
Assays were performed using the MATCHMAKER GAL4 two hybrid system (Clontech). Fragments of hnRNP A2 (amino acids 1–177 or 178–342), the γ7 C terminus (201–275), the CaV2.2 I–II loop (360–483), and CaVβ1b were generated by PCR and subcloned in-frame into the vectors pACT2 and pAS2-1. Plasmids were cotransformed into the yeast strain Y190, and transformants were selected by plating onto minimal selective dropout (SD) -Leu, -Trp agar. Protein interactions were identified by restreaking colonies onto SD -Leu, -Trp, -His plates and performing colony-lift β-galactosidase assays or growth assays in the presence of 10 mm 3-amino-1,2,4-triazole, according to the supplied protocol.
Immunoprecipitation.
γ7–HA was immunoprecipitated from stably transfected PC12 and transiently transfected tsA 201 cells. Endogenous γ7 and hnRNP A2 proteins were immunoprecipitated from untransfected PC12 cells. The following general method was used. Cells were washed with ice-cold PBS (Sigma-Aldrich) and harvested in PBS with 10 mm EDTA and protease inhibitor cocktail (Roche Diagnostics). Cells were lysed and protein solubilized by agitation with extraction buffer (1% Igepal, 20 mm Tris, 150 mm NaCl, 1 mm EDTA, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail, pH 7.4) for 30 min at 4°C. Insoluble material was removed by centrifugation at 40,000 × g for 1 h at 4°C. The supernatant was cleared with 50 μg of protein G linked to agarose beads (Sigma) for 2 h at 4°C. The supernatant was incubated with 2 μg of high-affinity anti-HA Ab (clone 3F10; Roche Diagnostics), γ7 C-terminus Ab, or hnRNP A2 monoclonal Ab EF-67 (Santa Cruz Biotechnology), overnight at 4°C with constant agitation. An additional 20 μg of protein G linked to agarose beads was added and incubated for 2 h at 4°C with constant agitation. Beads were washed twice with a high detergent buffer (1% Igepal, 20 mm Tris, 150 mm NaCl, 1 mm EDTA, and protease inhibitor cocktail, pH 7.4), twice with a high-salt buffer (0.1% Igepal, 20 mm Tris, 500 mm NaCl, 1 mm EDTA, and protease inhibitor cocktail, pH 7.4), and twice with a low-salt buffer (0.1% Igepal, 20 mm Tris, 1 mm EDTA, and protease inhibitor cocktail, pH 7.4). Bound protein was removed from the beads by the addition of lauryl dodecyl sulfate (LDS) sample buffer with reducing agent (Invitrogen) and heating to 65°C for 10 min. Samples containing immunoprecipitated endogenous γ7 were treated with LDS buffer as above and eluted proteins concentrated by precipitation with ice-cold acetone before resuspension in fresh LDS buffer for PAGE.
For protein sequencing, samples were separated on 4–12% Bis–Tris gels (Invitrogen) and stained with Coomassie blue (Simply Safe Blue stain; Invitrogen). Bands of interest were excised from the gel, and protein identification was performed by the Imperial College Proteomics Facility by tryptic mass fingerprinting, confirmed by tandem mass spectrometry. The controls were from nontransfected cells, treated identically.
Coimmunoprecipitation of RNAs associated with hnRNP A2.
PC12 cells were lysed and protein solubilized by agitation with extraction buffer containing 1% Igepal, 20 mm Tris, 150 mm NaCl, 1 mm EDTA, 0.5% sodium deoxycholate, and protease inhibitor cocktail, pH 7.4, supplemented with 5 U/ml RNAguard (GE Healthcare), for 30 min at 4°C. Samples were centrifuged (50,000 × g, 1 h at 4°C), and the corresponding supernatant was then precleared with 50 μg of protein G–Sepharose. The supernatant was divided into two and either incubated with 2 μg/ml high-affinity anti-HA Ab or an equivalent volume of PBS overnight at 4°C with constant agitation. An additional 20 μg of protein G–Sepharose was added and incubated for 2 h at 4°C with constant agitation. Beads were washed six times by centrifugation as described in the previous section, except that the wash buffers were supplemented with 5 U/ml RNAguard. A small aliquot of beads was removed for protein analysis. The RNA on the remaining beads was extracted with Trizol (Invitrogen). Total RNA was reverse transcribed with SuperScript III (Invitrogen) using random primers. PCR was performed using the primers described above.
Western blotting.
Cells were processed for SDS-PAGE as described previously (Raghib et al., 2001). For detection of endogenous γ7 in PC12 cells, cells were lysed on ice by sonication three times for 10 s and centrifuged at 3000 × g for 3 min. This supernatant was decanted and centrifuged at 50,000 × g for 4 h at 4°C. The high-speed supernatant and membrane-containing pellet were then taken up in SDS buffer. Samples (50 μg of cell lysate protein per lane) or immunoprecipitation samples prepared as above were separated using Novex 4–12% Tris–glycine or 4–12% Bis–Tris NuPAGE gels (Invitrogen) and transferred electrophoretically to polyvinylidene fluoride membranes. The membranes were blocked with 3% BSA/0.02% Tween 20 and then incubated overnight at room temperature with the relevant primary Ab: rabbit anti-hnRNP A/B Ab (H-200; 1:1000; Autogen Bioclear or Santa Cruz Biotechnology); anti-hnRNP A/B monoclonal Ab (F16; 1:1000; Autogen Bioclear); anti-hnRNP A2 monoclonal Ab [EF-67; 1:1000 (Nichols et al., 2000)]; rabbit anti-HA Ab (1:1000; Sigma), affinity-purified anti-γ7 Ab (0.4 μg/ml), or anti-α2δ-2(102–117) (1 μg/ml) (Brodbeck et al., 2002). The γ2–γ4 Abs have been described previously (Moss et al., 2003). Detection was performed using anti-rabbit or anti-mouse secondary Ab conjugated to HRP (1 μg/ml; Bio-Rad), and bound Abs were detected using enhanced chemiluminescence (ECL) or ECLPlus reagents (GE Healthcare). Chemiluminescence or fluorescence was detected using a Typhoon 9410 Variable Mode Imager (GE Healthcare), set in chemiluminescence or fluorescence mode, respectively. Protein bands were quantified using Imagequant 5.2, on nonsaturated images.
Immunocytochemistry.
Cells were fixed and permeabilized for immunocytochemistry essentially as described previously (Brice et al., 1997). The primary Abs used were affinity-purified anti-γ7 loop or tail Ab (as stated, 0.8 μg/ml) and mouse monoclonal anti-protein disulfide isomerase (1:100; Abcam). Alexa Fluor 594–phalloidin (Invitrogen) was also used. Secondary Texas Red, FITC, or biotin-conjugated goat anti-mouse (Invitrogen) or goat anti-rabbit (Sigma) Abs were applied at 10 and 5 μg/ml, respectively. When used, Texas Red- or FITC-conjugated streptavidin were applied at 3.33 μg/ml. A cyanine 3- tyramide signal amplification kit (PerkinElmer Life and Analytical Sciences) was used to detect γ7 in SCG neurons. In some experiments, the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI) (300 nm; Invitrogen) was also used to visualize the nucleus. Cells were mounted in Vectashield (Vector Laboratories) to reduce photobleaching and examined on a confocal laser scanning microscope (Leica TCS SP or Carl Zeiss LSM), using a 40× (1.3 numerical aperture) or 63× (1.4 numerical aperture) oil-immersion objective. Optical sections were 1.5 μm. Photomultiplier settings were kept constant in each experiment, and all images were scanned sequentially. In some experiments, when stated, a conventional fluorescence microscope (Axiovert 200; Carl Zeiss) and CCD camera was used; images were captured using Volocity software (Improvision).
Image processing was performed using NIH ImageJ (http://rsb.info.nih.gov/ij/). Colocalization analysis was performed using the colocalization plugin on images converted to 8 bit. Fluorophores are considered as colocalized in each pixel when their respective intensities are higher than the threshold (50) of their channels and when their intensity ratio is >50%.
Electrophysiology.
Xenopus oocytes were prepared, injected, and used for electrophysiology as described previously (Cantí et al., 1999), with the following exceptions. Plasmid cDNAs for the different voltage-dependent calcium channel subunits α1, α2δ-2, β1b, and other constructs such as γ7 were mixed in equivalent weight ratios at 1 μg/μl, unless otherwise stated, and 9 nl was injected intranuclearly, after appropriate dilution. The recording solution for CaV2.2-injected oocytes contained the following (in mm): 10 Ba(OH)2, 80 tetraethylammonium-OH, 2 CsOH, and 5 HEPES, pH 7.4 with methanesulfonic acid.
Whole-cell patch-clamp recording using tsA 201 or PC12 cells was performed and analyzed as described previously (Meir et al., 2000), with 1 mm Ba2+ as charge carrier (unless stated) and a holding potential of −100 mV. Currents were measured 10 ms after the onset of the test pulse, and the average over a 2 ms period was calculated and used for analysis. Data are expressed as mean ± SEM, and I–V plots were fit with a modified Boltzmann equation as described previously (Cantí et al., 2001).
Results
The reduction of functional CaV2.2 protein by γ7 is mediated via the C terminus of γ7
These experiments were initiated in the light of our previous finding that coexpression of γ7 reduced CaV2.2 protein level, as well as its functional expression (Moss et al., 2002). We have now dissected the region of γ7 responsible for the inhibitory effect by making constructs lacking most or part of the cytoplasmic C terminus, γ7(1–217) and γ7(1–238) (Fig. 1 A). After cDNA injection in Xenopus oocytes, when full-length γ7 was coexpressed with CaV2.2, it produced ∼90% suppression of CaV2.2 currents (Fig. 1 B,C). In contrast, the shorter γ7(1–217) transmembrane construct had very little influence on the expression of CaV2.2 currents (Fig. 1 B,C), whereas the γ7(1–238) construct produced a partial reduction of the CaV2.2 current (Fig. 1 C). The C-terminal motif (T/S-SPC) is conserved between γ7 and γ5. Although this does not represent a classical PDZ binding motif, we investigated the importance of this epitope in mediating the effects of γ7 on CaV2.2 currents. A construct lacking these four amino acids [γ7(1–271)] was as effective as γ7 in reducing CaV2.2 I Ba (Fig. 1 C), indicating that this motif is not involved in the effect of γ7. Furthermore, addition of a C-terminal tag, such as HA or CFP, did not affect the ability of γ7 to suppress the expression of CaV2.2 currents. For γ7–HA, 72.5 ± 10.1% (n = 5) inhibition of CaV2.2 currents was observed. These results indicate that a region of the γ7 C terminus between R217 and S271 is responsible for inhibiting CaV2.2 expression.

Effect of C-terminal truncation of γ7 on CaV2.2/β1b/α2δ-2 and CaV3.1 currents. A , Linearized diagram of γ7, indicating the approximate positions of the four transmembrane (TM) segments (black bars), the N-glycosylation site (V) at N45, and the position of the truncations at amino acids 217, 238, and 271. B , Left, Example traces elicited after cDNA injection into Xenopus oocytes by 100 ms step depolarizations to between −40 and 0 mV from a holding potential of −100 mV for CaV2.2/β1b/α2δ-2 (top), plus γ7 (middle) and plus γ7 (1–217) (bottom). The charge carrier was 10 mm Ba2+. The symbols beside the traces refer to the relevant data in the current–voltage relationship (right); CaV2.2/β1b/α2δ-2 (■; n = 19) plus γ7(1–217) (▵; n = 18) and plus γ7 (□; n = 8). Data are fit by a modified Boltzmann function as described in Materials and Methods, with V 50, act of −9.9, −9.8, and +1.2 mV, respectively, and G max of 19.6, 16.6, and 5.9 μS, respectively. C , Mean percentage of control peak I Ba for CaV2.2/β1b/α2δ-2 currents (black bar; n = 29) when coexpressed with γ7 (white bar; n = 31) or its truncated constructs, γ7(1–217) (hatched bar; n = 15), γ7(1–238) (gray bar; n = 12), and γ7(1–271) (striped bar; n = 16). The statistical significances compared with control are **p < 0.01. There was no effect of any of the constructs on the voltage for 50% steady-state inactivation, which from combined experiments was −60.2 ± 0.8 mV for controls (n = 16), −58.6 ± 1.0 mV in the presence of γ7 (n = 7), −61.7 ± 3.6 mV for γ7(1–217) (n = 5), and −59.8 ± 1.0 mV for γ7(1–271) (n = 5). D , Representative traces of peak Ba2+ currents in tsA 201 cells, cotransfected with CaV2.2, β1b and α2δ-2 with pMT2 as control (con) compared with γ7 (panel 1), γ7(1–217) (panel 2), and γ7(stop) (panel 3). Panel 4 shows CaV3.1 expression with pMT2 as control (con) compared with γ7. Currents were elicited by depolarization to +5 mV (CaV2.2, 1 mm Ba2+) or −10 mV (CaV3.1, 10 mm Ba2+), from a holding potential of −100 mV. Calibration bars refer to all traces for CaV2.2. E , Mean inhibition of Ba2+ currents (expressed as percentage of control ± SEM) induced by coexpression of CaV2.2 with γ7 (white bar; n = 21), γ7(1–217) (black bar; n = 15), γ7(stop) (hatched bar; n = 28), or CaV3.1 with γ7 (cross-hatched bar; n = 29). Statistical significance compared with the current size without γ7, **p < 0.01, Student's t test.
The role of the C terminus of γ7 was confirmed in tsA 201 cells. Here again the truncated γ7(1–217) produced no inhibition of CaV2.2 currents (5.1 ± 18.6%) (Fig. 1 D,E), whereas full-length γ7 produced 94.9 ± 18.6% inhibition (Fig. 1 D,E). To confirm that the γ7 protein was responsible for this effect, we showed that a γ7 construct containing a stop codon before the first transmembrane domain produced no inhibition (Fig. 1 D,E). Furthermore, in contrast to the effect of γ7 on CaV2.2 currents, it had no significant effect on CaV3.1 currents (Fig. 1 D,E).
We raised two antipeptide Abs against γ7, to unique peptides in the linker between transmembrane segments I and II, and in the C terminus. Neither Ab recognized any protein bands in untransfected COS-7 cells (Fig. 2 A), and neither Ab cross-reacted with other γ proteins tested (supplemental Fig. 2A–C, available at www.jneurosci.org as supplemental material). We then confirmed that the truncated γ7 constructs were all expressed at a similar level to full-length γ7 and that the truncated constructs were of the expected size (Fig. 2 A). Next, we examined the effect of γ7 and its C-terminal truncations on the level of CaV2.2 protein. Both γ7 and γ7(1–271) produced ∼70% inhibition of CaV2.2 protein, whereas γ7(1–217) caused no reduction (Fig. 2 B). The extent to which CaV2.2 protein expression was suppressed by γ7 and its truncated constructs was closely correlated with the degree of inhibition of CaV2.2 I Ba observed in Xenopus oocytes (Fig. 2 C). As a control, we showed that expression of an unrelated protein of similar size to γ7 (KV3.1b) had no effect on CaV2.2 protein expression (Fig. 2 B).

Effect of C-terminal truncation of γ7 on CaV2.2 protein. A , γ7 and truncated γ7 constructs were expressed at the expected sizes in COS-7 cells (top, γ7 I–II loop Ab; bottom γ7, C-terminal tail Ab). In each lane, the lower band corresponds to the expected protein molecular weight, and the upper band(s) correspond to either the mature glycosylated form or intermediate glycosylated species. As expected, γ7(1–217) and γ7(1–238) were not detected by the γ7(C-terminal tail) Ab. The specificity of the Abs is indicated by the lack of immunostaining in the absence of transfected constructs (−). Similar results were obtained in Xenopus oocytes (data not shown). The same amount of total protein was loaded in each lane (25 μg). B , Examples of Western blots showing the effect of γ7 ( i ) and γ7(1–271) ( ii ) and the lack of effect of γ7(1–217) ( iii ) and KV3.1b ( iv ) on the level of CaV2.2 protein expressed in COS-7 cells. con, Transfection with Kir–AAA cDNA, in place of γ7 (see Materials and Methods). The same amount of total protein was loaded in each pair of lanes (25 μg). C , Correlation between the effect of the γ7 and its various C-terminal truncated constructs on the level of CaV2.2 protein with their effect on CaV2.2 I Ba shown in Figure 1 C. The effect of γ7, γ7(1–217), γ7(1–271), and γ7(1–238) on CaV2.2 protein levels represents the mean inhibition observed in 3–10 experiments. The linear fit has a correlation coefficient, r, of 0.936. All error bars are SEM.
γ7 markedly reduces the stability of CaV2.2 mRNA
Several mechanisms could underlie the suppression of CaV2.2 currents and CaV2.2 protein by γ7, including suppression of channel translation, more rapid protein degradation, suppression of channel transcription, or increased mRNA breakdown. We addressed this question by examining the effect of γ7 on the level and stability of CaV2.2 mRNA. We found that coexpression of γ7 reduced CaV2.2 mRNA levels in two expression systems examined. In COS-7 cells, there was a 64% reduction in CaV2.2 mRNA level in the presence of γ7, 48 h after transfection (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In agreement with this, when CaV2.2 and γ7 were coexpressed in individual Xenopus oocytes, there was a 66% reduction in CaV2.2 mRNA expression after 24 h (Fig. 3 A, see time 0) when compared with control. We then examined whether this was attributable to an increase in the rate of degradation of CaV2.2 mRNA. The transcription inhibitor actinomycin D was applied 24 h after injection of the relevant cDNAs into Xenopus oocytes (at time T 0), and mRNA levels were then determined in individual oocytes at times up to 24 h thereafter. The half-life of CaV2.2 mRNA was 7.8 ± 1.5 h under control conditions (in agreement with Schorge et al., 1999), and it showed more than a twofold decrease to 3.5 ± 1.1 h in the presence of γ7 (Fig. 3 A,B). Importantly, the truncated γ7(1–217) construct, lacking the C terminus, had no effect on CaV2.2 mRNA degradation rate (Fig. 3 A,B), in agreement with its lack of effect on CaV2.2 currents or CaV2.2 protein (Figs. 1, 2).

Effect of γ7 and C-terminally truncated γ7(1–217) on CaV2.2 mRNA stability. A , Effect of γ7 on CaV2.2 mRNA degradation rate. Constructs were expressed in Xenopus oocytes either without (■) or with γ7 (▵) or with γ7(1–217) (□). In the absence of a γ7 construct, an equivalent amount of a similar sized transcript, a nonfunctional K+ channel Kir–AAA cDNA was used. After 24 h (T 0), actinomycin D (50 μg/ml) was added to the medium, and the CaV2.2 mRNA levels were measured at the times shown after this. The numbers of determinations from individual oocytes are between 6 and 12 for each data point; *p < 0.05 compared with CaV2.2 (Student's t test). The lines are apparent linear fits using errors as weight. B , Bar chart showing the percentage of CaV2.2, α2δ-2, and KCC1 mRNA present at time t after actinomycin D addition at time T 0. For CaV2.2 mRNA turnover, t = 9 h. Bars 1–3 are control (black bar; n = 11); + γ7 (white bar, n = 7) and + γ7(1–217) (hatched bar; n = 8). Bars 4–7 show the percentage of α2δ-2 mRNA and KCC1 mRNA present 9 and 24 h, respectively, after actinomycin D addition for the control condition (black bar; n = 8 and 9), + γ7 (white bar; n = 9 and 9). These times were chosen as the percentage of mRNA remaining in control conditions was similar for all mRNA species. *p < 0.05 compared with control, Student's t test. C , Degradation rate for endogenous CaV2.2 in the γ7–CFP PC12 cell line (○; n = 3) compared with control pcDNA3.1-transfected PC12 cell line (■; n = 5), after differentiation with NGF. The CaV2.2 mRNA level is expressed as percentage of time 0 (T 0), when actinomycin D was added. The half-life for endogenous CaV2.2 mRNA was 19.5 h in the pcDNA3.1-transfected PC12 cell line. In γ7-transfected PC12 cells, the half-life was 9.1 h (**p < 0.01, Student's t test). Inset, Reduction of endogenous CaV2.2 mRNA level in γ7–CFP PC12 cell line compared with control pcDNA3.1-transfected PC12 cell line. ***p < 0.001 versus control.
The relative mRNA concentrations at 24 h in the absence and presence of γ7 are in agreement with an effect only on the degradation rate of CaV2.2 mRNA and not on its synthesis rate. Fitting the data to the equation d[RNA]/dt = kf − kd × [RNA], where [RNA] is the concentration of RNA at time t, and kf and kd are the dominant rate constants for RNA synthesis and degradation, respectively, then [RNA] at time t = (kf /kd )(1 − exp(−kd × t)). From the calculated values of kd , kf = 10.1 and 8.8%/h in the absence and presence of γ7, respectively, relative to the control CaV2.2 mRNA level at 24 h, immediately before actinomycin D treatment. Thus, the data provide no evidence of a marked effect of γ7 on the synthesis rate (kf ) of CaV2.2 mRNA, despite a more than twofold increase in the measured degradation rate.
The effect of γ7 on mRNA stability does not affect all calcium channel subunits, because the degradation rate of α2δ-2 mRNA was not affected by γ7 coexpression (half-life of ∼25 h) (Fig. 3 B) (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). In agreement with this, the level of α2δ-2 protein was little affected by γ7 (14.9 ± 2.8% reduction; n = 6) (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material).
We also investigated the effect of γ7 on the stability of KCC1 mRNA (a potassium chloride cotransporter), because it is a membrane protein of similar mass to CaV2.2, whose mRNA has some common sequence motifs (see below). Although KCC1 mRNA was much more stable than that of CaV2.2 (half-life of 50.5 h), γ7 decreased its half-life to 18.3 h, thus increasing its mRNA turnover from 28 ± 11 to 63 ± 5% in 24 h (n ≥ 9) (Fig. 3 B). Using the method described above, again we found no evidence for any effect on the synthesis rate. The kf was calculated to be 5.0 and 4.1%/h in the absence and presence of γ7, respectively, relative to the control KCC1 mRNA level at 24 h. These results indicate that the effect of γ7 on mRNA degradation rate is selective for certain mRNAs but not selective for CaVα1 mRNAs.
In addition, we demonstrated that γ7 had the same effect on endogenous CaV2.2 mRNA, because, 9 h after actinomycin D treatment, the CaV2.2 mRNA level in a PC12 cell line stably transfected with γ7–CFP was reduced by 74% compared with a control PC12 cell line stably transfected with pcDNA3.1 (Fig. 3 C, inset), and the endogenous CaV2.2 mRNA turnover rate was correspondingly enhanced (Fig. 3 C). The half-life of CaV2.2 mRNA was 19.5 h in the pcDNA3.1-transfected PC12 cell line, which was not significantly different from that in control PC12 cells (16.9 h; data not shown). In γ7-transfected PC12 cells the half-life was 9.1 h (Fig. 3 C).
We found no evidence that the effect of γ7 involves the induction of ER stress or the unfolded protein response, although this has been postulated as a mechanism of action of TARP γ2 (Sandoval et al., 2007). Coexpression of CaV2.2 with γ7 in tsA 201 cells, at a high transfection efficiency using Amaxa nucleofection, did not increase the editing of the X binding protein XBP or induce the C/EBP homologous protein CHOP expression, both of which are markers of the induction of the unfolded protein response (Harding et al., 2002), whereas these responses were evoked by exposure of cells to tunicamycin or dithiothreitol, both of which are well known activators of ER stress (D. J. Cox and A. C. Dolphin, unpublished results).
Knockdown of γ7 increases endogenous CaV2.2 mRNA level in PC12 cells
To study the physiological role of endogenous γ7, we chose to silence its expression using RNA interference. We first examined the presence and localization of native γ7 in PC12 cells. Immunocytochemical localization showed that endogenous γ7 is expressed in these cells (Fig. 4 A) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). We therefore used this cell line to examine the effect of knockdown of γ7. We designed three shRNAs specifically complementary to sequences in either rat or human γ7 mRNA. To confirm the effectiveness of the γ7 shRNAs, we made a PC12 cell line overexpressing human γ7–CFP and showed that, 5 d after transfection, a mix of three shRNAs directed against human γ7 mRNA reduced the γ7–CFP protein level in this cell line, by 75% compared with control transfected cells (Fig. 4 B,C). We also found the three corresponding rat γ7 shRNAs reduced endogenous γ7 mRNA levels in PC12 cells, either individually or when mixed, whereas the control Drosophila gnu shRNA did not (Fig. 4 D). This would represent a larger reduction in individual transfected cells, taking into account the incomplete transfection efficiency (∼30%). Furthermore, the native γ7 protein present endogenously in PC12 cells (Fig. 4 A,E) was depleted by 70 ± 6% in individual PC12 cells, after transfection with the rat γ7 shRNAs (Fig. 4 E,F).
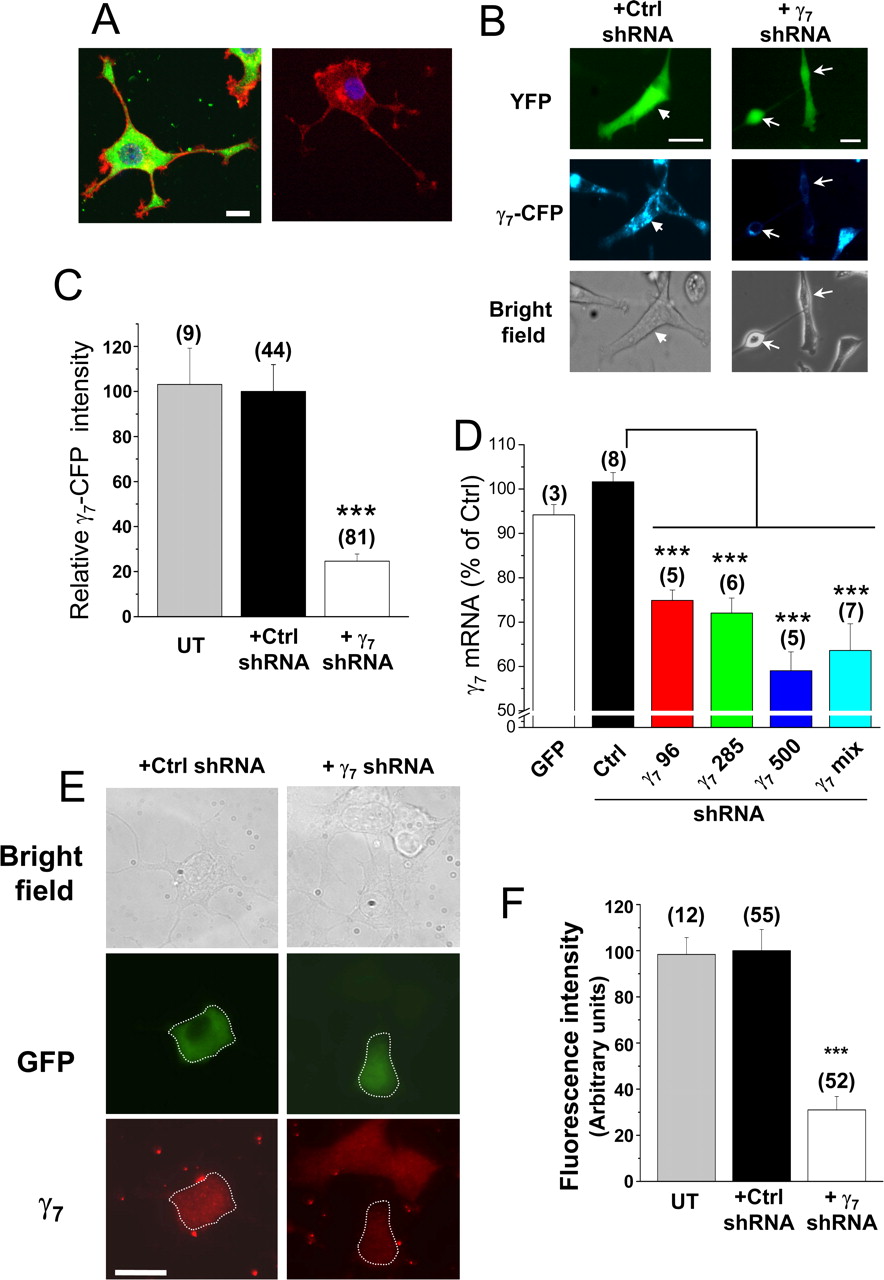
Short-hairpin RNA constructs knockdown γ7 levels in PC12 cells. A , Left, Endogenous γ7 (detected using γ7 I–II loop Ab; green) in a differentiated PC12 cell, together with F-actin (Alexa Fluor 594–phalloidin; red) and nuclear staining (DAPI; blue). Right, Control in the absence of primary γ7 Ab. Scale bar, 10 μm (applies to both images). The images are a Z-stack of five to eight confocal images. B , Fluorescence microscopy of PC12 cells stably transfected with a human γ7–CFP construct. These cells were transiently transfected with YFP and either negative control gnu shRNA (Ctrl; left) or a mixture of three human γ7 shRNAs (γ7; right) and examined after 5 d. Top row, Transfected cells are identified with YFP fluorescence. Middle row, CFP fluorescence is almost abolished in cells transfected with γ7 shRNA. Arrowhead in left indicates a cell transfected with the control shRNA that shows γ7–CFP fluorescence. Arrows in right indicate two cells transfected with γ7 shRNA that do not show γ7–CFP fluorescence. Bottom row, Bright field. Scale bars, 30 μm. C , Bar chart of the mean percentage of CFP fluorescence in PC12 γ7–CFP cells after transfection with γ7 shRNA (white bar) compared with transfection with control shRNA (black bar) or untransfected PC12 cells (UT, gray bar). ***p < 0.001 vs control. D , Effect of rat γ7 shRNA on γ7 mRNA level in PC12 cells. γ7 mRNA was quantified by q-PCR in PC12 cells transfected with GFP (white bar), negative control shRNA (Ctrl, black bar), or human γ7 shRNAs: γ7 96 (red bar), γ7 285 (green bar), γ7 500 (blue bar), or γ7 mix (cyan bar). ***p < 0.001 versus control. E , Effect of transfection with γ7 shRNA on endogenous γ7 protein levels in PC12 cells. Left, Cells transfected with control shRNA and GFP; right, cells transfected with rat γ7 shRNA and GFP. Top row, Bright-field image; middle row, GFP, with transfected cell outlined; bottom row, endogenous γ7 visualized by immunocytochemistry (red), showing reduced fluorescence in γ7 shRNA-transfected cell (outlined). Scale bar, 20 μm (applies to all images, which were taken on a conventional fluorescence microscope). F , Quantification of data including that given in E , showing reduction in mean fluorescence intensity (measured as fluorescence density in arbitrary units) in γ7 shRNA-transfected cells (white bar) compared with control shRNA (Ctrl, black bar) or untransfected cells (UT, gray bar); ***p < 0.001 compared with control.
If γ7 is playing a physiological role in controlling mRNA stability, its knockdown might be expected to have an effect opposite to γ7 overexpression, to increase the endogenous CaV2.2 mRNA level and enhance its stability. This was indeed the case, because CaV2.2 mRNA levels were increased by ∼30% after rat γ7 shRNA transfection (Fig. 5 A), which would represent a larger enhancement in individual transfected cells, taking into account the transfection efficiency. An expected physiological correlate of this would be an increase in calcium channel currents in differentiated PC12 cells. In agreement with this, we recorded a 33.1 ± 4.1% (n = 48) increase in peak somatic calcium channel current density in PC12 cells transfected with rat γ7 shRNAs over the same timescale (Fig. 5 B).

Short-hairpin RNA constructs enhance endogenous CaV2.2 mRNA levels and calcium channel currents in PC12 cells. A , Effect of rat γ7 shRNA on endogenous Cav2.2 mRNA level in PC12 cells. Cav2.2 mRNA was quantified by q-PCR in PC12 cells transfected with GFP (white bar), negative control shRNA (Ctrl, black bar), or γ7 shRNAs: γ7 96 (gray bar), γ7 285 (hatched bar), γ7 500 (cross-hatched bar), or γ7 mix (striped bar). ***p < 0.001 versus control. B , Top, Representative traces of peak calcium channel currents recorded from differentiated PC12 cells transfected with control shRNA (left) and γ7 shRNA (right). Currents were elicited by a 100 ms depolarization step to +10 mV from a holding potential of −100 mV. Bottom, Current–voltage relationships for the two conditions (■, Ctrl shRNA; ○, γ7 shRNA). The charge carrier was 10 mm Ba2+. The knockdown of γ7 induces an increase of the peak current (−16.1 ± 0.9 pA/pF; n = 48) compared with control (−12.5 ± 0.9 pA/pF; n = 17; p < 0.05).
γ7 exists in a complex with RNA binding proteins
Because PC12 cells contain endogenous γ7, they represent a suitable model cell type in which to search for protein complexes containing γ7. For this study, we used a PC12 cell line stably expressing γ7 with a C-terminal HA tag. After immunoprecipitation of γ7–HA from the γ7–HA PC12 cell line and extensive washing, the presence of coimmunoprecipitating proteins, representing proteins in a complex with γ7, was examined by SDS-PAGE (Fig. 6 A). Bands of interest, which were present in the γ7–HA immunoprecipitate but absent from the control, nontransfected PC12 cells (Fig. 6 A, left, arrows, representative of 3 experiments), were excised and identified, after tryptic digestion, by peptide mass fingerprinting. The bands labeled (1, 36 kDa) and (2, 37 kDa) were both identified, with 34 and 38% peptide coverage, respectively, to be hnRNP A2, which has a number of splice variants of 33–38 kDa (Hatfield et al., 2002). Band 2 also contained hnRNP A3 (40% coverage), which shares extensive sequence homology and is found in a complex with hnRNP A2 (Ma et al., 2002). The presence of γ7–HA was confirmed by immunoblotting (Fig. 6 A, right). Both hnRNP A2 and hnRNP A3 are members of the hnRNP A/B subfamily.

Identification of proteins interacting in a complex with γ7. A , Proteins coimmunoprecipitated with γ7–HA from stably transfected PC12 cells. Left, Coomassie blue staining. Right, Western blotting and immunodetection with anti-HA Ab of immunoprecipitated and control samples separated by SDS-PAGE. Solid arrow indicates γ7–HA. Small arrows indicate proteins coimmunoprecipitated with γ7–HA. Bands 1 and 2 were identified by peptide mass fingerprinting to contain hnRNP A2. Band 2 also contained hnRNP A3. The control lane is untransfected PC12 cells. Position of molecular weight markers is shown on the left. Representative of three experiments. B , Endogenous hnRNP A2 coimmunoprecipitates with transiently transfected γ7–HA but not with γ7(1–217)–HA in tsA 201 cells. Western blotting and immunodetection with anti-hnRNP A/B Ab (H-200, top row) and anti-HA Ab (bottom row) of input (left) and HA-immunoprecipitated samples (after 500 mm NaCl wash, right) separated by SDS-PAGE. Position of molecular weight markers is shown on the left. Blots are representative of three to five independent experiments, using anti-hnRNP A/B Abs from two different sources. C , CaVβ1b with a C-terminal HA tag was expressed and immunoprecipitated as described for γ7–HA. Its presence in the precipitate is confirmed in the top blot. The control lane is from cells not transfected with CaVβ1b–HA. Immunoblotting for endogenous hnRNP A2 shows it is present in the input lanes but absent from the immunoprecipitate. D , Coimmunoprecipitation of endogenous hnRNP A2 and γ7 proteins from PC12 cells. Western blotting and immunodetection with anti-hnRNP A2 Ab (EF-67, left) and γ7 C-terminus Ab (right) of input (left lane of both panels) and γ7 C-terminus Ab-immunoprecipitated samples (right lane of both panels) separated by SDS-PAGE. Control immunoprecipitations in which γ7 Ab was omitted are shown in the middle lane of each panel. Position of molecular weight markers is indicated between the panels. Blots are representative of two independent experiments. E , The interaction of the C terminus of hnRNP A2 with the cytoplasmic C-terminal tail of γ7 was independently confirmed by yeast cotransformation tests. Lane 1, Positive control (blue reaction product) showing CaV2.2 I–II loop (BI-II) pACT2 and β1b pAS2–1. Lane 2, Interaction between hnRNP A2 C terminus (hn-C) in pACT2 and γ7 C terminus (γ7-C) in pAS2–1. Lanes 3 and 4, Negative controls showing that the hnRNP A2 C terminus and the γ7 C terminus do not interact with β1b in the alternative vector. The filter was incubated with the X-gal (5-bromo-4-chloro-3-indolyl-b-d-galactopyranoside) substrate for 5 h. F , Regions of colocalization of hnRNP A2 with γ7–CFP (top row) or endogenous γ7 (bottom row) in SCG neurons. i , Immunolocalization of endogenous hnRNP A2 in SCG neuron cell bodies (top, red; bottom, green). ii , Localization of γ7–CFP (top, blue) or immunolocalization of endogenous γ7 (bottom, red). iii , Merger of images of i and ii , with the colocalized regions shown in white (top) or yellow (bottom). iv , Colocalized γ7 and hnRNP A2 pixels are also shown separately for clarity (white). Scale bar, 10 μm (applies to all images). No endogenous γ7 staining was observed in the absence of primary Ab (data not shown).
We then confirmed that immunoprecipitation of γ7–HA was able to pull down endogenous hnRNP A/B proteins in another system, tsA 201 cells transiently transfected with γ7-HA (Fig. 6 B). Three bands were detected (36–42 kDa), using hnRNP A/B Abs, which, according to the antibody specificity and molecular weights, are likely to represent one or more of hnRNP A1, A2, and A3, all of which also have splice variants. No hnRNP A/B immunoreactivity was immunoprecipitated by γ7(1–217)–HA, under the same conditions (Fig. 6 B), indicating that these hnRNPs use the C terminus of γ7 for this interaction. This is in agreement with our previous finding that the C terminus of γ7 is essential for its functional effects. No hnRNP A/B immunoreactive proteins were coimmunoprecipitated with an unrelated HA-tagged protein, HA–CaVβ1b (Fig. 6 C), indicating that the HA tag is not responsible for this binding.
We also found that pull down of endogenous γ7 from untransfected PC12 cells, with the γ7 C-terminal Ab, was able to immunoprecipitate endogenous hnRNP A2 (Fig. 6 D).
The C terminus of hnRNP A2 interacts directly with the C terminus of γ7
To determine whether the interaction between γ7 and hnRNP A2 was direct, we used the yeast two-hybrid technique. When the γ7 C terminus (residues 201–275) was used as bait, we found an interaction with the C terminus of hnRNP A2 (residues 178–342) (Fig. 6 E, lane 2). Because of our previous work in this area, we used the high-affinity (∼10 nm) (Leroy et al., 2005) interaction between the I–II linker of CaV2.2 and the CaVβ1b subunit as a positive control (Fig. 6 E, lane 1). According to the relative time taken for colonies to turn blue in a colony-lift filter assay, the interaction between hnRNP A2 and γ7 C terminus is weaker than that between CaV2.2 and CaVβ1b (data not shown). As negative controls, we showed that there was no interaction between the C termini of either hnRNP A2 or γ7 and CaVβ1b (Fig. 6 E, lanes 3, 4). Furthermore, the N terminus of hnRNP A2 (amino acids 1–86) was found not to interact with the C terminus of γ7 (data not shown). All these data indicate that γ7 is likely to be present in a complex with hnRNP A2. Nevertheless, we cannot rule out that hnRNP A3, which shows marked homology to hnRNP A2, also interacts with γ7, either directly or by binding to hnRNP A2.
hnRNP A2 colocalizes with γ7 in neuronal cytoplasm
We used immunocytochemistry to examine whether γ7 and hnRNP A2 are colocalized in SCG neurons. Although, as expected, most hnRNP A2 is localized in the nucleus, it is also observed in small cytoplasmic granules in SCG neurons (Fig. 6 Fi). In the somatic cytoplasm, there are clear regions of colocalization of both transfected γ7–CFP and endogenous γ7 with hnRNP A2 (Fig. 6 Fii,Fiii). This is in agreement with our suggestion that γ7 may sequester free hnRNP A2, as outlined in Figure 9, A and B.
CaV2.2 mRNA binds to hnRNP A2
The hnRNP A2 proteins are highly expressed in brain and are primarily present in the nucleus, in which they bind to particular nucleotide sequences in mRNA. The combination of mRNA with its many bound proteins, including hnRNP A2, then exits the nucleus as ribonucleoprotein particles (Piñol-Roma, 1997). The interaction with hnRNP A2 has been found to stabilize certain mRNAs for trafficking to a remote site of protein synthesis (Hoek et al., 1998) and also to enhance translation (Kwon et al., 1999). In neurons and oligodendrocytes, hnRNP A2 has been implicated in the transport of mRNAs containing an A2 response element (A2RE) sequence, for local protein synthesis (Hoek et al., 1998; Shyu and Wilkinson, 2000).
We therefore examined whether CaV2.2 mRNA could be present in a complex with hnRNP A2. We found that the CaV2.2 mRNA used in this study contains at least two highly conserved sequences predicted to bind hnRNP A2 (Table 1), from the consensus sequence identified in myelin basic protein (MBP) mRNA (Ainger et al., 1997). These two sequences are in constitutive exons, present in all splice variants of CaV2.2. The sequences preserve A and G at positions 8 and 9, shown previously to be essential for hnRNP A2 binding and function (Ainger et al., 1997; Shan et al., 2003), and the sequences are highly conserved in CaV2.2 between species (Table 1), with related sequences being found in CaV2.1 (data not shown). We found that a biotinylated 21 base ribonucleotide corresponding to sequence 1 from CaV2.2 (Table 1) binds endogenous hnRNP A2 from a mouse brain lysate, to a similar extent to the sequence originally described from MBP (Hoek et al., 1998) (Fig. 7 A). We further found that an anti-HA Ab coimmunoprecipitated N-terminally HA-tagged hnRNP A2, both with overexpressed full-length rabbit CaV2.2 mRNA in tsA 201 cells (data not shown) and also with endogenous CaV2.2 mRNA in PC12 cells (Fig. 7 B). Furthermore, an anti-hnRNP A2 monoclonal Ab coimmunoprecipitated endogenous hnRNP A2 together with endogenous CaV2.2 mRNA from PC12 cells (Fig. 7 C).
hnRNP A2 mRNA binding motifs in CaV2.2 mRNA
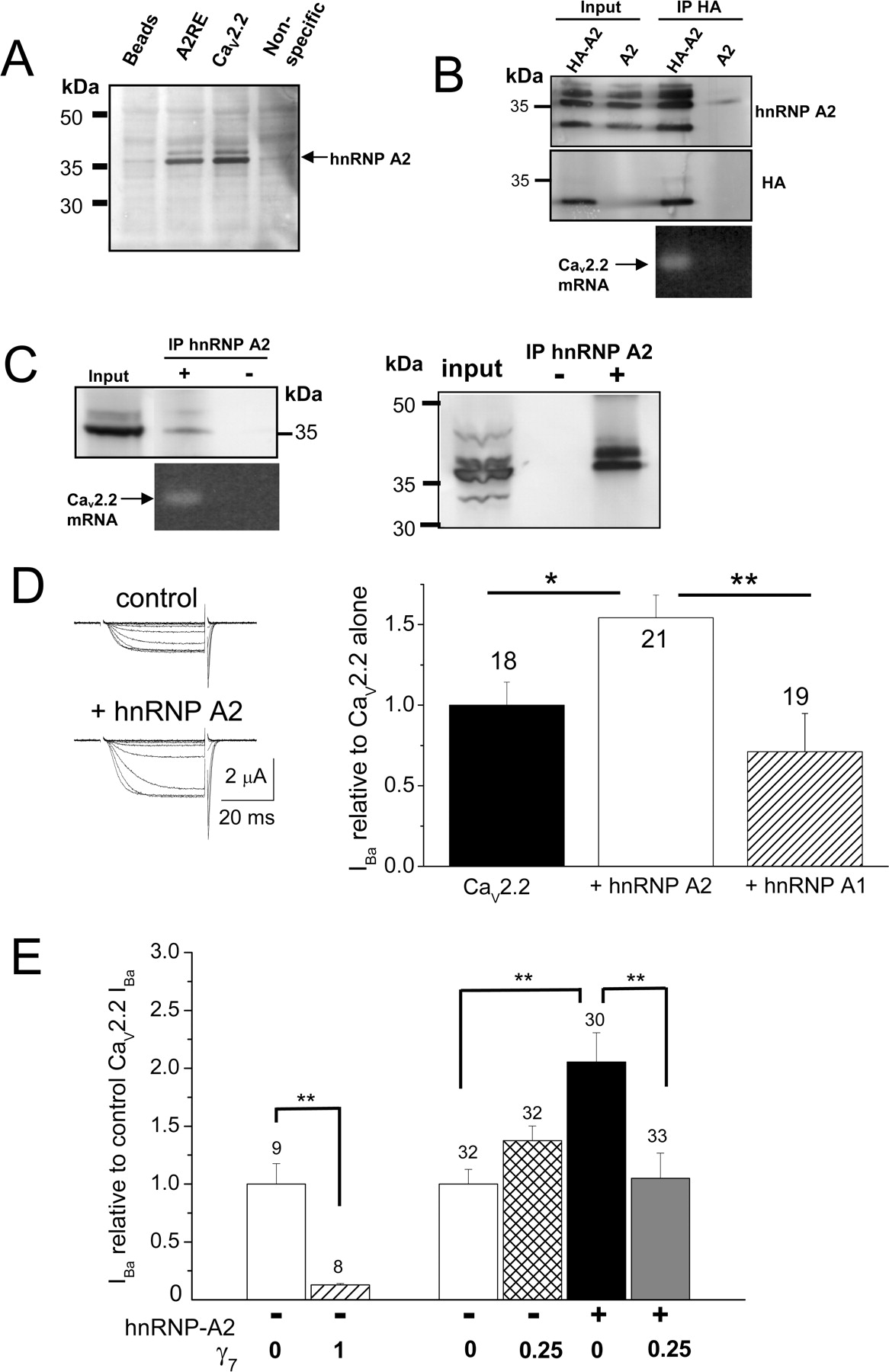
Binding of hnRNP A2 to CaV2.2 mRNA and effect of hnRNP A2 and γ7 on CaV2.2 currents. A , Evidence that one of the two potential A2RE sites in CaV2.2 mRNA binds to hnRNP A2. Endogenous hnRNP A2 (indicated by the arrow) from mouse brain lysate was pulled down by biotinylated oligonucleotides containing the consensus A2RE site (lane 2) and the potential site in CaV2.2 (lane 3) but not by beads alone (lane 1) or by a nonspecific sequence with the same composition as the consensus A2RE site (lane 4). The hnRNP A2 was detected by immunoblotting with anti-hnRNP A2 Ab (F16). Position of molecular weight markers is shown on the left. Representative of two experiments. B , Coimmunoprecipitation of endogenous CaV2.2 mRNA associated with hnRNP A2 in PC12 cells. Two days after transfection with HA–hnRNP A2 (HA-A2, lanes 1 and 3) or hnRNP A2 (A2, lanes 2 and 4), hnRNP A2 protein was immunoprecipitated (IP) from the whole-cell lysate (lanes 1 and 2) with HA antibody (lanes 3 and 4). Immunoblots show that HA Ab pulled down hnRNP A2 [top row, anti-hnRNP A2 (EF-67 Ab); middle row, anti-HA]. Coimmunoprecipitated RNA was extracted, reverse transcribed, and amplified by PCR. A specific PCR product corresponding to the endogenous CaV2.2 mRNA was amplified (35 cycles) only in the condition in which hnRNP A2 protein was pulled down (bottom row, lane 3). C , Left, Endogenous hnRNP A2 proteins were immunoprecipitated from PC12 cells with anti-hnRNP A2 Ab EF-67 (top row, lane 2). In this condition, a specific PCR product corresponding to the endogenous CaV2.2 mRNA was also amplified (35 cycles, bottom row). CaV2.2 mRNA was not detected in the control in which antibody was omitted (lane 3). Right, The immunoprecipitation was repeated, including an acetone precipitation step, to confirm the presence of endogenous hnRNP A2. D , Enhancement by hnRNP A2 but not hnRNP A1 of CaV2.2 currents recorded from Xenopus oocytes. Left, Example traces elicited by 50 ms step depolarizations to between −40 and 0 mV in 10 mV steps, from a holding potential of −100 mV for CaV2.2/β1b/α2δ-2 (top traces) or CaV2.2/β1b/α2δ-2 plus hnRNP A2 (bottom traces). The charge carrier was 10 mm Ba2+. Right, Bar chart shows the effect of hnRNP A2 coexpression with CaV2.2/β1b/α2δ-2 on peak CaV2.2 I Ba amplitude, expressed relative to the mean peak control current in each experiment. Control (black bar; n = 18), for both groups taken from experiments directly comparing hnRNP A1 and hnRNP A2: + hnRNP A2 (white bar; n = 21) and + hnRNP A1 (hatched bar; n = 19). These data are pooled from two separate experiments showing similar results. Statistical significance, *p < 0.05, **p < 0.01 (one-way ANOVA and Bonferroni's post hoc test). E , Inhibition of the hnRNP A2-mediated enhancement of I Ba in Xenopus oocytes by a low concentration of γ7. Bar chart compares the effect of two concentrations of γ7 cDNA (γ7: CaV2.2 ratio 1 and 0.25) and also shows the lack of enhancement by hnRNP A2 on peak CaV2.2 I Ba amplitude, when coexpressed with the lower concentration of γ7 (γ7: CaV2.2 ratio 0.25). Data are expressed as a percentage of the mean peak control CaV2.2 current in each experiment, and all data were recorded 2 d after cDNA injection. Control in the absence of γ7 or hnRNP A2 (white bars); + γ7 (ratio 1; hatched bar), + γ7 (ratio 0.25; cross-hatched bar), + hnRNP A2 (black bar, from data obtained in the same experiments), + hnRNP A2 and γ7 (ratio 0.25; gray bar). Number of determinations given above each bar. These data are pooled from five different batches of oocytes, in all of which similar results were observed. Statistical significance, **p < 0.01 (one-way ANOVA and Bonferroni's post hoc test).
hnRNP A2 enhances the expression of CaV2.2 currents, and this is counteracted by γ7
Because hnRNP A2 binds to CaV2.2 mRNA, we wanted to examine the consequences on calcium channel expression of coexpression with hnRNP A2. When this construct was coexpressed with CaV2.2/β1b/α2δ-2 in Xenopus oocytes, it produced a significant enhancement of the peak calcium current amplitude, without affecting the current kinetics or voltage dependence (Fig. 7 D). In experiments performed in parallel, an unrelated hnRNP (A1), which is involved in mRNA processing and export but does not bind to A2RE sequences (Cullen, 2000), did not enhance CaV2.2 calcium currents (Fig. 7 D). Thus, hnRNP A2 may enhance CaV2.2 mRNA stability in Xenopus oocytes or increase its transport to sites of translation. This is compatible with the role previously attributed to hnRNP A2 that it enhances both transport and translation of specific mRNAs (Kwon et al., 1999). We also found that the effect of hnRNP A2 was able to generalize to other CaV2 channels, because enhancement of CaV2.1/β4/α2δ-2 currents was also observed, the peak I Ba at +5 mV being increased to 194.8 ± 19.9% of control (n = 28; p = 0.0004). CaV2.1 mRNA contains A2RE sequences that are homologous to those of CaV2.2 (data not shown).
We then investigated whether there was an interaction between the effects of hnRNP A2 and γ7 on calcium channel current expression. The inhibitory effect of γ7 on CaV2.2 calcium channel currents, shown in Figure 1 B, was found to be concentration dependent (data not shown), and a low concentration of γ7 was chosen (1:4 dilution of γ7 cDNA), which did not inhibit CaV2.2 currents in the Xenopus oocyte expression system (Fig. 7 E), to examine its interaction with the effect of hnRNP A2. We found that the enhancement of CaV2.2 currents by hnRNP A2 was prevented by coexpression of this low concentration of γ7 (Fig. 7 E). This indicates that γ7 opposes the effect of hnRNP A2, supporting the hypothesis that the binding of hnRNP A2 to its binding site on the C terminus of γ7 reduces the amount available to interact with CaV2.2 mRNA in the cytosol. In agreement with this interpretation, expression of the cytosolic C terminus of γ7 [γ7(201–275)], which our yeast two-hybrid data show to be able to bind hnRNP A2 (Fig. 6 E), was itself able to reduce the amplitude of CaV2.2 currents. In experiments similar to those shown in Figure 1 B, the peak CaV2.2 I Ba current amplitude was reduced by 70.6 ± 4.5% (n = 19) relative to control currents, in the presence of the C terminus of γ7 (data not shown) compared with an 87.4% reduction by full-length γ7 in the same experiment.
Investigation of the subcellular localization of γ7
The topology of γ1 and the TARP γ proteins indicates that the linker between the first and second transmembrane domains is extracellular (Chu et al., 2001), and the same might be anticipated for γ7, which has a predicted glycosylation site in this loop (Fig. 1 A). We obtained mutational evidence that γ7 is N-glycosylated on N45 in the I–II loop, supporting the proposed topology (Fig. 8 A). However, heterologously expressed γ7 is not inserted in the plasma membrane of tsA 201 cells, because it was not detected with an Ab to this I–II loop in nonpermeabilized cells (Fig. 8 B).

Subcellular localization of γ7. A , Western blotting of a γ7 mutant in which the potential glycosylation site, N45 is mutated to A, and immunodetection with anti-γ7 I–II loop Ab. Lane 1, γ7; lane 2, γ7 N45A. The reduction in mass and sharpening of the band indicates that γ7 is normally glycosylated at this site. Positions of molecular weight markers are shown on the left. The data are representative of two independent experiments. B , Immunodetection of transiently transfected γ7 (green) in nonpermeabilized (left) and permeabilized (right) tsA 201 cells, using γ7 I–II loop Ab. The nuclear stain DAPI (blue) was used as a cell marker. Scale bar, 100 μm (applies to both images). No immunostaining was observed in any field in the absence of permeabilization. C , Immunostaining for γ7 using I–II loop Ab (Texas Red secondary Ab, left) colocalizes completely with γ7–CFP (middle), as shown by yellow regions in merged image (right), in permeabilized (top row) but not nonpermeabilized (bottom row) SCG neurons. Scale bar, 40 μm (applies to all images). D , Partial colocalization of γ7–CFP and an ER marker in SCG neurons. Left, γ7–CFP; middle, ER marker (ER-DsRed); right, overlay showing colocalization of DsRed with γ7–CFP (white). Scale bar, 10 μm (applies to all images).
We have observed that γ7–CFP, when heterologously expressed in cultured SCG neurons, is present, in part, in motile intracellular vesicles (Fig. 8 C and data not shown). These colocalize with γ7 I–II loop Ab immunoreactivity only when the cells are permeabilized (Fig. 8 C). No plasma membrane staining was detected in the absence of cell permeabilization (Fig. 8 C). We also found that neither endogenous γ7 (Fig. 4 A) nor γ7–CFP (Fig. 4 B and data not shown) localized to the plasma membrane in PC12 cells. The localization of γ7 is therefore on intracellular membranes in all the cells we have examined. We found that the distribution of γ7–CFP partially overlapped with that of an ER marker in the somata of SCG neurons microinjected with γ7–CFP and pDsRed2–ER (Fig. 8 D). It was also found in vesicular structures, which were negative for the ER marker (Fig. 8 D). Similar results were obtained in PC12 cells using a different ER marker, protein disulfide isomerase (data not shown).
Discussion
We have described previously the identification of two genes that encode γ5 and γ7, by their homology with the mouse stargazin gene (cacng2) and have cloned and expressed the cDNA for both human and mouse γ7 (Moss et al., 2002). The γ7 protein contains 275 amino acids with an estimated protein mass of 31 kDa and has four predicted transmembrane-spanning domains with intracellular N and C termini and a consensus N-glycosylation sequence in the loop between the first two transmembrane segments (Fig. 1 A). Together with the other γ-like proteins, it belongs to the claudin superfamily, members of which play diverse roles in cellular physiology (Sanders et al., 2001). Initially, the novel γ1-like proteins (γ2–γ8) were investigated as potential calcium channel subunits (Letts et al., 1998; Klugbauer et al., 2000; Kang et al., 2001; Rousset et al., 2001; Sharp et al., 2001; Moss et al., 2003). However, we have shown that, although the human γ2 and γ4 TARPS are expressed in Purkinje neurons, there was no effect of these proteins on calcium currents composed of CaV2.1/β4/α2δ-2, a combination that mimics the major calcium channel complement in Purkinje cells (Moss et al., 2003). More recently, these proteins have been shown to have roles in trafficking and localization of AMPA glutamate receptors (Tomita et al., 2003, 2004; Fukata et al., 2005; Kato et al., 2007). Together with the data presented here, these findings suggest that γ proteins may play diverse and possibly multiple roles in intracellular trafficking.
In our original study, we showed that coexpression with γ7 almost completely abolished Ba2+ current through N-type CaV2.2 channels expressed from cDNA in both Xenopus oocyte and COS-7 cell expression systems (Moss et al., 2002). Several mechanisms could underlie the suppression of CaV2.2 currents by γ7 protein, but we have now identified that a major mechanism of suppression of CaV2.2 currents by γ7 results from a decrease in CaV2.2 mRNA stability. This process requires the cytoplasmic C terminus of γ7, because all its effects, to inhibit CaV2.2 current and protein expression and to increase the rate of mRNA degradation, are completely prevented by the removal of most of the C terminus of γ7. It is well known that the effect of overexpression of a protein does not necessarily reflect its physiological function, because an overexpressed protein may act to sequester interacting partners. However, our evidence indicates that the regulation of the stability of CaV2.2 mRNA, and potentially other mRNAs, represents an important role for native γ7. This evidence stems from the finding that knockdown of endogenous γ7 in PC12 cells, using γ7 shRNA, substantially enhances their endogenous CaV2.2 mRNA level and enhances endogenous somatic calcium currents after differentiation.
We have subsequently identified by coimmunoprecipitation from a PC12 cell line stably transfected with γ7–HA, that the RNA binding protein hnRNP A2 is coimmunoprecipitated with γ7. hnRNP A2 has been found to be involved in the stability, trafficking, and localization of particular mRNAs and has been identified to bind to several different mRNA sequences, including CGG repeats (Sofola et al., 2007), AU-rich elements (Hamilton et al., 1999), and a well characterized binding motif termed A2RE (Ainger et al., 1997; Shan et al., 2003; Fähling et al., 2006). Most hnRNP A2 is localized in the nucleus, in which it binds to specific sequences in transcribed mRNA, and is involved in mRNA export into the cytoplasm (for review, see Shyu and Wilkinson, 2000) and subsequently in trafficking and enhancement of translation for those mRNAs containing an A2RE sequence (Kwon et al., 1999). Our yeast two-hybrid data indicates that γ7 C terminus binds directly to hnRNP A2, and our subcellular localization data for γ7 indicate that this interaction will occur on intracellular membranes within the cytoplasm (as depicted in Fig. 9 B).

Diagram of proposed function of γ7. A , The physiological distribution of hnRNP A2 and γ7. The hnRNP A2 (red circles) is localized primarily in the nucleus in which it binds to A2RE sequences on mRNAs and exits to the cytoplasm with these mRNAs, including that of CaV2.2 (green). This is thought to stabilize certain mRNAs, reducing degradation and therefore enhancing expression. Our results suggest this may be the case for CaV2.2. hnRNP A2 may also stabilize the mRNAs for transport. γ7 (dark blue) is present on the ER and is also associated with motile vesicles. B , After overexpression of γ7, it may sequester cytoplasmic hnRNP A2 and therefore increase degradation of CaV2.2 mRNA.
There are two highly conserved consensus hnRNP A2-binding A2RE motifs in the CaV2.2 mRNA used in this study (Table 1) that contain the conserved A and G at positions 8 and 9, which have been found to be a prerequisite for binding of hnRNP A2 (Ainger et al., 1997). There are also several other, less well conserved, A2RE motifs in the CaV2.2 mRNA sequence that may also be functional (data not shown). Here we have demonstrated that hnRNP A2 binds to one of the conserved sequences (Table 1, sequence 2) and, by homology, is highly likely to bind to the other sequence. Furthermore, hnRNP A2 coimmunoprecipitates endogenous CaV2.2 mRNA from PC12 cells. We suggest that hnRNP A2 normally binds to sequences on CaV2.2 mRNA in the nucleus, and, when the ribonucleoprotein particle so formed exits the nucleus, it is transported to its site of translation and also protected from degradation (as depicted in Fig. 9 A). A similar mechanism of interaction with hnRNP A2 has been proposed for MBP mRNA that contains a canonical A2RE sequence (Hoek et al., 1998). It has also been shown that hnRNP A2 itself is subject to transport (Brumwell et al., 2002). Indeed, the hnRNP A/B proteins have been identified as components in isolated RNA-transporting granules (Kanai et al., 2004). Furthermore, the Drosophila homolog Hrp48 is involved in the localization of oskar mRNA (Huynh et al., 2004; Yano et al., 2004). Other RNA binding proteins, such as Staufen, have been shown to have an effect on both localization and decay of specific mRNAs (Broadus et al., 1998; Kim et al., 2005).
Our results provoke the hypothesis that γ7 is involved in the regulation of hnRNP A2 function, as depicted in Figure 9 B. γ7 may sequester hnRNP A2, and this may be the mechanism whereby the stability of specific mRNAs, including that of CaV2.2, are compromised. Furthermore, our results indicate that native, as well as heterologously expressed, γ7 both influence the physiological regulation of CaV2.2 mRNA stability, because knockdown of native γ7 increased endogenous CaV2.2 mRNA and CaV2.2 current levels. This indicates that endogenous γ7 may function to limit the availability of cytoplasmic hnRNP A2. A related pathological mechanism of hnRNP A2 sequestration by binding to expanded CGG repeats of FMR1 mRNA has been proposed recently to occur in fragile X-associated tremor/ataxia syndrome (Sofola et al., 2007). It will be of interest to examine whether a similar interaction occurs with other triplet repeat diseases.
The expression of CaV2.2 channel proteins may be particularly vulnerable to a reduction in available hnRNP A2 because the mRNA degradation rate of CaV2.2 is relatively high. Nevertheless, other mRNAs are also likely to be similarly affected, and we show that the degradation of KCC1 mRNA, which also contains an A2RE sequence, is also enhanced by γ7.
Both CaV2.2 (Mori et al., 1991) and γ7 (Moss et al., 2002) are selectively expressed in neurons. CaV2.2 channel expression and function is dynamically regulated both physiologically (Pravettoni et al., 2000; Inchauspe et al., 2004) and in pathology (Hendriksen et al., 1997). It is of interest that CaV2.2 channels are functionally most important in early development, and, in many instances, their role is later substituted by P/Q-type calcium channels (CaV2.1), in terms of both somatic currents (Salgado et al., 2005) and synaptic transmission (Iwasaki et al., 2000). Over a similar time period, we found that γ7 expression is strongly upregulated (M. Nieto-Rostro and A. C. Dolphin, unpublished results). It is now recognized from many studies that regulation of mRNA stability is a very important part of the posttranscriptional control of expression of numerous genes (Wilusz and Wilusz, 2004).
There are indications that CaV2.2 mRNA is subject to transport, because it has been identified in dendritic growth cones (Crino and Eberwine, 1996) and in processes of motor neurons (Jablonka et al., 2007). Furthermore, N-type calcium channels are localized in the presynaptic terminals of peripheral neurons, such as DRG neurons, and local synthesis of transmembrane proteins has been demonstrated recently in axons and axonal growth cones (Brittis et al., 2002). It will be of great interest in the future to examine whether hnRNP A2 affects the transport of CaV2 family mRNAs, to regions distinct from the cell soma.
Footnotes
-
This work was supported by the Wellcome Trust, the Biotechnology and Biological Sciences Research Council (BBSRC), and the Medical Research Council for support. L.F. held a fellowship from Fondation pour la Recherche Medicale, and J.L. held a Wellcome Trust International fellowship. D.J.C. was supported by a BBSRC PhD studentship and D.W. by a British Heart Foundation PhD studentship. We are grateful to Dr. W. J. Frith for mathematical advice, Kanchan Chaggar for technical assistance, Dr. R. Nichols for the hnRNP A2 constructs, Dr. J. Caceres for hnRNPA1 cDNA, Dr. S. Alper for KCC1 cDNA, Dr. T. J. Shafer for PC12 cells, and Drs. A. Cahill and A. Fox for pG418–shRNA–Empty vector.
- Correspondence should be addressed to Annette C. Dolphin, Laboratory of Cellular and Molecular Neuroscience, Andrew Huxley Building, Department of Pharmacology, University College London, Gower Street, London WC1E 6BT, UK. a.dolphin{at}ucl.ac.uk
This article is freely available online through the J Neurosci Open Choice option.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}