

Abstract
A cardinal feature of inflammation is heightened pain sensitivity at the site of the inflamed tissue. This results from the local release by immune and injured cells of nociceptor sensitizers, including prostaglandin E2, bradykinin, and nerve growth factor, that reduce the threshold and increase the excitability of the peripheral terminals of nociceptors so that they now respond to innocuous stimuli: the phenomenon of peripheral sensitization. We show here that the proinflammatory cytokine interleukin-1β (IL-1β), in addition to producing inflammation and inducing synthesis of several nociceptor sensitizers, also rapidly and directly activates nociceptors to generate action potentials and induce pain hypersensitivity. IL-1β acts in a p38 mitogen-activated protein kinase (p38 MAP kinase)-dependent manner, to increase the excitability of nociceptors by relieving resting slow inactivation of tetrodotoxin-resistant voltage-gated sodium channels and also enhances persistent TTX-resistant current near threshold. By acting as an IL-1β sensor, nociceptors can directly signal the presence of ongoing tissue inflammation.
Introduction
Intense mechanical and thermal stimuli as well as diverse chemical irritants act on the peripheral terminals of nociceptor sensory neurons to initiate nociceptive pain, the acute pain that accompanies noxious stimuli and warns of impending tissue damage. Nociceptor activators, the external stimuli that drive this pain, act on specific high threshold heat-, mechanical- or chemical-sensitive ion channel transducers expressed by the nociceptors, to produce inward currents in their peripheral terminals (Julius and Basbaum, 2001). These inward currents produce generator potentials that activate voltage-gated sodium currents, and if threshold is exceeded, lead to a flow of action potentials from the periphery to the CNS, to signal the presence, location and intensity of noxious stimuli.
Normally, in the absence of any noxious stimuli, nociceptors are silent and there is no pain. When a tissue is injured, however, inflammation occurs, and this results in spontaneous pain in the absence of any external stimulus and also in hypersensitivity to painful stimuli. Pain hypersensitivity at the site of inflammation (primary hyperalgesia) is largely the consequence of the sensitization of the peripheral terminals of nociceptors due to a reduction in their threshold and an increase in their excitability (Hucho and Levine, 2007). Multiple proinflammatory mediators are released soon after injury by damaged tissue and infiltrating immune cells (Hucho and Levine, 2007). These include kinins, amines prostanoids, purines, NGF, and protons that, via their cognate receptors on the nociceptor terminal, rapidly activate intracellular kinases, and thereby produce posttranslational changes in transducer and voltage-gated ion channels, leading to increased insertion of the channels into the membrane, altered thresholds, and modified kinetic properties (Hucho and Levine, 2007). Spontaneous pain during inflammation may arise either from the presence in the inflamed tissue of chemicals like ATP that directly activate nociceptors via P2X ligand-gated ion channels or by indirect increases in membrane excitability, triggered by G-protein coupled or membrane tyrosine kinase receptor mediated activation of intracellular signaling pathways that lead to increased sodium currents; such as occurs for PGE2 via EP receptors and PKA on Nav 1.8 and 1.9 sodium channels (Rush and Waxman, 2004; Baker, 2005).
The two prototypic proinflammatory cytokines IL-1β and TNFα are rapidly released from, and contribute to, the recruitment and activation of immune cells generating inflammation (Verri et al., 2006). These cytokines also contribute to the establishment of peripheral sensitization via an induction of NGF (Safieh-Garabedian et al., 1995) and a production of PGE2, secondary to induction of cyclooxygenase-2 in immune and other local non-neuronal cells (Maier et al., 1990). TNFα has in addition an excitatory action on nociceptor neurons (Jin and Gereau, 2006). We now find that IL-1β also acts directly on primary sensory neurons to substantially increase excitability and consequently their spontaneous and evoked activity. The boost in excitability is mediated via p38 MAP kinase and involves enhancement of two types of TTX-resistant sodium current, low-threshold “persistent” sodium current and higher-threshold “slow” sodium current.
Materials and Methods
Cell culture.
Primary DRG neuron cultures were prepared from 6 to 8-week-old Sprague Dawley (SD) male rats. DRGs were removed and placed into DMEM and 1% penicillin–streptomycin (Sigma), then digested in 5 mg/ml collagenase, 1 mg/ml Dispase II (Roche) and 0.25% trypsin, followed by addition of 2.5% trypsin inhibitor. Cells were triturated in the presence of DNase I inhibitor (50 U) and centrifuged through 15% BSA (Sigma). The cell pellet was resuspended in 1 ml of Neurobasal (Sigma) containing B27 supplement (Invitrogen), penicillin and streptomycin (Sigma), 10 μm AraC, 100 ng/ml 2.5S NGF (Promega) and GDNF (2 ng/ml). Cells were plated onto poly-lysine- (500 μg/ml) and laminin- (5 mg/ml) coated 35 mm tissue culture dishes (BD Biosciences) at 8000–9000 per well, at 37°C, 5% carbon dioxide. Recordings were made <48 h after plating.
Electrophysiology.
Whole-cell transmembrane currents and membrane voltages were recorded in whole-cell and fast current-clamp modes respectively using an Axopatch 200A amplifier (Molecular Devices) at room temperature (22 ± 2°C). Average cell size of recorded neurons was 23 ± 2 μm (n = 175).
Data were low-pass filtered at 2 kHz (−3 dB, 4 pole Bessel filter) and sampled at 5–10 kHz. For whole-cell current-clamp recordings, data were low-pass filtered at 10 kHz (−3 dB, single-pole Bessel filter). Recordings were made within 30 min of obtaining whole-cell configuration to minimize rundown of TTX-r currents. Patch pipettes were pulled from thick-walled borosilicate glass capillaries (1.5 mm outer diameter) on a Sutter Instruments P-97 puller and had a resistance of 1–2 MΩ. For sodium current recordings, either pipettes were coated to within ∼100 μm of the tip with Sylgard (Dow Corning) or shanks were manually wrapped by paraffin film to reduce pipette capacitance.
For voltage-clamp recordings using CsCl-based internal solution, pipette potential was zeroed before seal formation and membrane potential not corrected for the small liquid junction potential (−2.2 mV). For both voltage-clamp and current-clamp recordings, care was taken to maintain membrane access resistance as low as possible (usually 3–7 MΩ and always <10 MΩ). For voltage-clamp recordings, capacity currents were cancelled and series resistance 80% compensated. Linear leakage currents were digitally subtracted on-line using a P/4 procedure. Command voltage protocols were generated on-line with a Digidata 1200 A/D interface (Molecular Devices). Data were digitized on-line using pCLAMP 8 (Molecular Devices). Data averaging and peak detection were made using pCLAMP 8.2 software (Axon Instruments). Data were fitted using Origin 6 (OriginLab). For composition of the extracellular solutions see Table 1.
Extracellular solution composition
Voltage-clamp recordings.
For these only neurons that had a TTX-r sodium current were analyzed.
Intracellular solution for voltage-clamp recordings was as follows (in mm): CsCl, 110; CsOH, 25; MgCl2, 2; CaCl2 1; EGTA 11; HEPES, 10 (pH = 7.4 with CsOH).
Voltage dependence of sodium current activation and current availability.
For total sodium currents, 20 ms depolarizing steps were applied to a range of test potentials in 10 mV increments, from a holding potential of −70 mV. The voltage at which sodium current was half-maximally activated (V 1/2) was estimated by obtaining the macroscopic conductance (G) from peak current amplitudes recorded at different test potentials using the extended Ohm's law: G = I peak/(V test − V rev), where I peak is the amplitude of the peak current, V rev is the apparent reversal potential of sodium, measured for each cell (+47 ± 3 mV, n = 100), and V test is the test potential. For TTX-r sodium current cells were preincubated for 30 min with 300 nm TTX before recording. The stimulation protocol was modified such that test step was prolonged to 120 ms to detect persistent sodium current. To analyze TTX-r-per-mediated sodium current, cells were held at −90 mV and 120 ms test pulses from −100 to +10 mV were applied (at 5 s intervals). To decrease the effect of slow inactivation, neurons were held for 30 s at −100 mV prior the stimulation protocol (120 ms depolarizing steps in 5 mV increments).
Availability curves were generated from a double-pulse protocol consisting of one constant 30 ms pulse to 0 mV after 180 ms prepulses to voltages varying from −120 to 10 mV in steps of 5 or 10 mV. Peak sodium current during the test pulse was normalized and plotted vs prepulse. Data were fit by a Boltzmann relationship: G = G max/(1 + exp[(V 1/2 − V M)/k)), where V 1/2 is the potential at which one-half of the channels are activated or inactivated and k is the slope factor (in mV). Activation rates were quantified as the time to peak current. Inactivation was fit (starting 1–3 ms after peak) with a falling double-exponential function: y = y 0 + A1 × exp(− time/τ1) + A2 × exp(− time/τ2), which gave better fits than single or triple exponential functions. External solution 1 was used for sodium current amplitude analysis; external solution 2 was used for sodium current kinetics analysis (Table 1).
The protocol to study voltage dependence of slow inactivation of TTX-r sodium channels was as follows (Fleidervish et al., 1996; Blair and Bean, 2003): from holding potential of −100 mV, 5 s long conditioning pulses (V cond) were applied. The amplitude of the conditioning pulses varied systematically (in 10 mV steps) between −120 and +20 mV. Then a 20 ms step to −100 mV was applied to remove fast inactivation and was followed by a 10 ms test pulse (V test) to 0 mV. Resulting TTX-r current amplitude was normalized to the maximal current amplitude (fraction available) and plotted (mean ± SEM) versus voltage of conditioning pulses. The data were fitted using Boltzmann equation I test/I max = 1/(1 + exp[(V − V 1/2)/k)), where V is the conditioning pulse potential, V 1/2 is the potential at which one-half of the channels are available and k is the slope factor (in mV). To measure onset of slow inactivation of TTX-r sodium channels cells were held at −100 mV. Conditioning pulses of variable duration (2.5–10,000 ms) to 0 mV were followed by 40 ms step to −100 mV to remove fast inactivation. Then a test pulse (10 ms) to 0 mV was applied. The normalized current amplitude (mean ± SEM) was plotted as a function of conditioning pulse duration and fitted with double-exponential function I test/I max = y 0 + (A1 × exp(− time/τ1) + A2 × exp(− time/τ2)), where I test/I max is a fraction available, A1 and A2 are fractions of channels inactivating with time constants τ1 and τ2 respectively and y 0 is steady state asymptote. The kinetics of recovery from slow inactivation was measured at −70 mV. Slow inactivation was produced by stepping the membrane from V holding of −100 to 0 mV for 5 s. Then the membrane was held at −70 mV for variable recovery periods (2.5–10,000 ms). Recovery from slow inactivation was examined by plotting the peak current in response to a 5 ms step to 0 mV versus duration of recovery period [modified from Choi et al. (2007)]. Data were fitted using single-exponential function I test/I max = y 0 + (A × exp(− time/τ) where I test/I max is the fraction available, A is a fraction of channels recovering with the time constant τ, t is the duration of recovery period and y 0 is the steady state asymptote.
Quasi-steady-state I–V curves were generated from the currents evoked by a slow depolarizing voltage ramp (2.5 s, from −70 to +20 mV). The currents were completely abolished by 5 μm TTX (data not shown). Currents were analyzed after manual off-line or P/4 online leak subtraction. The onset of persistent component of current was detected at the potential at which clear deviation from zero line was observed. The threshold of slow component was measured at the voltage of onset of the second negative slope. External solution 3 was used for quasi-steady-state I–V curve recordings (Table 1).
Current-clamp recordings.
These were only made from small (23 ± 6 μm, n = 45) dissociated rat DRG neurons and single action potentials evoked by 4 ms depolarizing current pulses. Only neurons (n = 35) that had action potential characteristics consistent with nociceptive-like neurons (prominent deflection on the falling phase) were investigated. The AP duration was measured at half-maximal amplitude. Rheobase (minimum current required to evoke AP), threshold and apparent input resistance were measured using a series of 500 ms hyperpolarizing and depolarizing square pulses (1–2 nA in increments of 0.025–0.1 nA each). Input resistance (R in)was determined as the slope of voltage current curve measured in the linear part of hyperpolarizing range.
Series of 4 ms depolarizing steps (25 pA) from 0 to 1 nA were applied to measure the threshold of action potential. Subthreshold passive responses were fitted well by single-exponential. Action potential threshold was determined at the onset of clear sharp deviation from passive response with definite acceleration of dV/dt with subsequent action potential generation. Spike amplitudes were measured from threshold to peak. Maximal upstroke velocity (V/s) was obtained from dV/dt max. Depolarizing (1.5 s, 2 nA) current ramps were applied to examine cumulative spike latency, presented for each spike as ratio of cumulative latency after application of IL-1β versus control. Because in the control condition there were only 6.7 ± 2.8 spikes per ramp, the ratio is presented for the first 5 APs only. AP threshold (only for cells with <2 mV discrepancy between square pulse protocol and ramp protocol), the latency to discharge of the first AP and duration of AP firing (measured from the ramp onset) during the ramp were also analyzed. For current clamp recordings external solution 4 was used (Table 1). Membrane potential was corrected for a liquid junction potential of −15 mV.
Pipette solution contained the following (in mm): K gluconate, 135; MgCl2, 2; KCl, 6; HEPES, 10; Mg ATP, 5; Li2GTP, 0.5 (pH = 7.4 with KOH). In a few experiments, ATP and GTP were omitted with no obvious effects.
IL-1β application.
IL-1β (10 ng/ml) was applied using custom-designed multibarrel fast drug delivery system positioned ∼200–300 μm apart from the recording neuron. Solution exchange was complete in less than a second. Vehicle refers to bath solution applied through drug delivery system. In experiments with SB 203580, neurons were preincubated for 45–60 min with SB 203580 (10 nm, 100 nm, 10 μm) before recording (Hudmon et al., 2008).
Single-fiber electrophysiology.
Skin nerve preparations that contained the saphenous nerve attached to the area of hindlimb skin that it innervates were prepared from f 8 female Sprague Dawley rats weighing between 80 and 300 g, as described previously (Reeh, 1986). The skin was kept in a chamber under laminar perfusion with carbogenized (95% O2-5% CO2) physiologic buffer. The saphenous nerve was kept in an adjacent recording chamber immersed in paraffin oil and split into small filaments until recordings could be performed from single fibers. The receptive fields of single sensory fibers (classified according to heat sensitivity, cold sensitivity, mechanosensitivity, and conduction velocity) were isolated from the surrounding extracellular solution with a thick-walled metal ring (volume 500 μl). After application of physiologic buffer for 5 min (used as control), IL-1β was applied at 20 ng/ml for 5 min and exchanged several times with a pipette. We used a feedback controlled radiant heat device (Hofmann Elektronik) to apply radiant heat to the epidermal side of the receptive field via the translucent bottom of the organ bath. The stimulus lasted 22 s and consisted of a ramp heat stimulus from 32 to 48°C over 20 s followed by a 2 s plateau at 48°C. In the 30 s after the heat stimulation, the receptive field cooled passively to 32°C. The heat stimuli were applied at 5 min intervals. The criterion for assigning heat responsiveness to a fiber (CMH or AMH) was a discharge of at least 5 spikes per stimulus in two repeated responses. The signal was filtered using a differential amplifier (ISO 80, WPI) at 100 Hz and 1 kHz and DAPSYS (Johns Hopkins University) was used to record and analyze the data. The sample comprised 8 Aδ-fibers (cv 0.43 ± 0.12 m/s, median von Frey threshold 9.7 mN) and 18 C-fibers (cv 3 ± 0.68 m/s, median von Frey threshold 11.4 mN). All afferents were mechanosensitive and 6 C- and one Aδ-fiber were also heat-sensitive (CMH, AMH afferents). Mechanosensitivity was assessed using a set of custom-designed gravity-driven von Frey hairs that were applied perpendicularly to the receptive field before the application of the IL-1β.
Immunohistochemistry and Western blot.
For immunohistochemistry animals were perfused with 4% paraformaldehyde, tissues cryoprotected in 20% sucrose and processed for sectioning and immunohistochemistry as described (Amaya et al., 2006). Double labeling was performed using rabbit anti IL-1R type I antibodies (R & D systems, 1:800), mouse anti peripherin antibodies (Millipore, 1:500), rabbit anti phospho-p38 MAP kinase (Cell Signaling, 1:600). Other primary antibodies used in this study were polyclonal antibodies for phospho-JNK (Cell Signaling, 1:1000) and phospho-ERK (Cell Signaling, 1:500). Western blot analysis was conducted using rabbit anti-p-p38 antibody (Millipore, 1:500), and rabbit anti-p38 (Millipore, 1:500) for loading control.
Behavior.
Rats were habituated, and tests were performed blind to the treatment. Intraplantar injections of saline or IL-1β (1 pg, 10 μl of saline) (Amaya et al., 2006) into the left hindpaw were made and mechanical and thermal sensitivities determined using von Frey hairs, hot plate and radiant heat respectively (Amaya et al., 2006) at the times indicated. In another set of experiments, intraplantar injection of 100 μg of ibuprofen (Sigma, in 10 μl of saline) was followed, 20 min later, by saline or IL-1β intraplantar injection as described above.
Statistical analysis.
Statistical analysis was performed using Student's t test or one-way ANOVA, followed by Dunnett's test as appropriate, and data are represented as mean ± SEM.
Results
IL-1β increases the excitability of adult DRG nociceptor-like neurons in vitro
We first investigated what effect IL-1β has on the excitability of small (25 ± 4 μm, n = 35) dissociated rat DRG neurons that had action potential waveforms characteristic of nociceptors (Abdulla and Smith, 2001), with 5.1 ± 1.1 ms duration at half maximal amplitude and a prominent inflection on falling phase (Fig. 1 A, inset). The action potential threshold of the neurons (−21 ± 5 mV, n = 25) was stable over time (25 min of vehicle wash-in) (Fig. 1 C), but was rapidly (< 2 min) and dramatically reduced by IL-1β (10 ng/ml), reaching −39 ± 6 mV at 10 min (Fig. 1 C). This effect reversed fully 45 min after washout (p = 0.1, n = 5) (Fig. 1 C). In the presence of IL-1β, previously subthreshold current steps elicited trains of action potentials (Fig. 1 A) and suprathreshold steps elicited more spikes (1.9 ± 0.1-fold) (Fig. 1 A,D), whereas a long depolarizing current ramp (2 nA, 1.5 s) elicited action potential firing earlier, with a twofold increase in spike number and less accommodation (Fig. 1 B; supplemental Table 1, available at www.jneurosci.org as supplemental material). This increased excitability was not accompanied by changes in single action potential amplitude or upstroke velocity (supplemental Table 1, available at www.jneurosci.org as supplemental material). We used solutions containing ATP and GTP to ensure stable recordings. Given the potential effects of GTP on neuronal excitability (Baker et al., 2003), we monitored AP threshold, spike adaptation and spontaneous firing during prolonged (25 min) application of vehicle solution. This failed to produce changes in neuronal AP threshold and firing properties (Fig. 1 C; supplemental Table 1, available at www.jneurosci.org as supplemental material). In addition, IL-1β was applied only after obtaining a stable baseline for at least 10 min, and we observed no changes over this period. Finally, we were able to reproduce the effects of IL-1β on using an internal solution without GTP (n = 3 of 3, data not shown).

IL-1β increases the excitability of DRG neurons. A , Current-clamp recordings of nociceptor-like DRG neurons before and after IL-1β (10 ng/ml, 10 min). Inset, Expanded traces, Note the broadness of the action potential and the inflection on its falling phase. The dotted lines indicate the action potential threshold obtained as the point of deviation from single exponential fit (blue line) of subthreshold responses. B , IL-1β reduced threshold and decreased spike adaptation during a 1.5 s depolarizing ramp (333 pA/s). C , Time course of IL-1β effects on action potential threshold (n = 25). Time point “-10” refers to data collected shortly (<1 min) after membrane breakthrough; time point “-5” refers to data collected 5 min before the application of IL-1β or vehicle. Black arrow indicates time of application of IL-1β or vehicle, gray arrow indicates start of washout (n = 5 for washout). D , IL-1β (10 min) increased the number of spikes (mean ± SEM) evoked by suprathreshold stimuli. x-axis indicates the current step level normalized to rheobase for each cell tested (n = 25). E , F , Action potential firing elicited by IL-1β at resting membrane potential (V rest, typically −65 to −70 mV) and −30 mV (means ± SEM). Recordings made using extracellular solution 4 (Table 1).
Before IL-1β treatment (vehicle for 15 min), the neurons had a stable resting membrane potential of −67 ± 4 mV with no spontaneous action potential firing (Fig. 1 E). Low-frequency firing was elicited in all recorded cells by steady depolarization to −30 mV (Fig. 1 E; supplemental Table 1, available at www.jneurosci.org as supplemental material). IL-1β did not change the resting membrane potential (−69 ± 4 mV, p = 0.4), but did elicit action potential firing at the resting potential in 25% of the neurons and also dramatically increased firing at −30 mV in all neurons (Fig. 1 E,F; supplemental Table 1, available at www.jneurosci.org as supplemental material).
IL-1β excites and sensitizes nociceptor peripheral terminals
We next analyzed whether IL-1β increases the excitability of nociceptor peripheral terminals by recording from 26 single nociceptors in an isolated rat skin/nerve preparation. IL-1β exerted both a rapid excitatory and heat sensitizing effect in 4 of 7 heat-sensitive fibers (Fig. 2 A,B). Some mechanosensitive C-fibers were also excited by IL-1β (3/12), and in one case sensitized to heat. Across the whole population of C (n = 18) and Aδ (n = 8) fibers, IL-1β produced approximately a threefold increase in ongoing activity (6 ± 3 action potentials measured over a 5 min period of superfusion with a control physiologic buffer solution vs 17 ± 7 action potentials detected during 5 min of the IL-1β application, n = 26, p < 0.05). This matches the decrease in threshold of nociceptors to mechanical and thermal stimulation produced by IL-1β in vivo (Fukuoka et al., 1994), and the increased heat-evoked release of CGRP from nociceptor terminals (Opree and Kress, 2000). We found, moreover, that intraplantar IL-1β (1 pg) in vivo produced, 20 min after injection, a decrease in the mechanical withdrawal threshold and an increased response to noxious heat (Fig. 2 C). The acute IL-1β mediated change in threshold was not reduced by administration of the nonselective cyclooxygenase inhibitor ibuprofen (100 mg) (Fig. 2 C). Moreover, no significant increase in COX-2 mRNA levels was observed at these early time points (<1 h) after injection of IL-1β (1 pg) (data not shown). Together, these data suggest that the short latency IL-1β-mediated increase in nociceptor excitability and decrease in threshold of nociceptors to mechanical and thermal stimulation occurs in a COX2-independent manner.

IL-1β directly activates and sensitizes nociceptors. A , Instantaneous discharge rates (IDR) (each dot represents one action potential) of a saphenous nerve C-fiber (0.39 m/s, 1 mN) recorded in a skin/nerve preparation and its response to radiant heat stimulation (ΔT, narrow gray bars; 32−48°C, 22 s) (see Materials and Methods for description of ramp) applied in 5 min intervals (broad gray bars) before and after IL-1β (20 ng/ml). Arrows represent mechanical stimulation of receptive field. B , IL-1β-sensitized heat responses in CMH and AMH fibers (n = 6 CMH and n = 1 AMC) from 14.3 ± 0.1 to 39.4 ± 15.2 (50 s). (Bins 2 s, subject to two-point averaging.) C , Intraplantar IL-1β (1 pg/10 μl) and combination of IL-1β (1 pg/10 μl) with ibuprofen (100 mg/10 μl) induced mechanical (von Frey threshold) and thermal sensitivity (hot plate latency) in vivo. Mean ± SEM; *p < 0.05.
IL-1β increases sodium currents in nociceptor-like DRG neurons
Because exposure to IL-1β did not affect the membrane input resistance (R in) of small DRG neurons (Fig. 1 A; supplemental Table 1, available at www.jneurosci.org as supplemental material), the increased excitability is likely to result from alterations in voltage-dependent ion channels. Indeed, shortly after exposure to IL-1β (∼2 min), total sodium current assayed in voltage clamp increased nearly twofold (peak current density 302 ± 12 pA/pF vs 173 ± 12, p < 0.05, n = 50) (Fig. 3 A, inset), with a time course similar to the augmented excitability (Fig. 3 B). IL-1β did not dramatically alter the voltage dependence of activation or inactivation of the total sodium current (Fig. 3 C; supplemental Table 2, available at www.jneurosci.org as supplemental material), but rise time was reduced without a change in decay time (supplemental Table 2, available at www.jneurosci.org as supplemental material). During application of the vehicle alone, no increase in sodium current was ever observed; in fact, the current amplitude decreased slowly with time (Fig. 3 B).

IL-1β increases sodium currents in DRG neurons. A , Increase in total sodium current produced by IL-1β (10 ng/ml). Inset, Averaged I–V curves obtained before and 3 min after application of IL-1β (n = 50). B , Time course of IL-1β mediated increase in total sodium current (n = 15, last 2 points n = 3, mean ± SEM). Note gradual decrease of sodium current amplitude when recorded without IL-1β (n = 10, last 3 points n = 3). Arrow indicates time of application of IL-1β or vehicle. Time point “-5” refers to data collected shortly after membrane breakthrough before the application. Peak current normalized to peak current density detected at “0” time point (173 ± 12 pA/pF, n = 50). C , G/G max (activation) and I/I max (availability) curves for total sodium curves reveal no shift in activation and inactivation of the current by IL-1β (p = 0.35, n = 10). D , IL-1β increased the TTX-r sodium current (300 nm TTX) (dark traces indicate TTX-r-per at −30 mV). Inset, Averaged I–V curves (n = 28) obtained before and 3 min after IL-1β application. E , Time course of the effect of IL-1β (mean ± SEM, n = 28 for IL-1β, n = 10 for controls, last 3 points n = 3). Arrow indicates time of application of IL-1β or vehicle. Time point “-5” refers to data collected shortly after membrane breakthrough before the application. Peak current normalized to peak current density detected at “0” time point (135.1 ± 12.1 pA/pF, n = 28). F , G/G max and I/I max curves of TTX-r sodium current shows no change in activation and fast steadystate inactivation (p = 0.5, n = 10). Recordings for A , B , D , and E were made using extracellular solution 1 and those for C and F using extracellular solution 2 (Table 1).
Total sodium current in small DRG neurons is a combination of TTX-resistant current (the major component) and TTX-sensitive (TTX-s) channels (a smaller component). IL-1β increased the peak amplitude of the tetrodotoxin-resistant (TTX-r) sodium current from 135 ± 12 pA/pF to 264 ± 15 (p < 0.05, n = 28) (Fig. 3 D, inset; E). The time to peak of the TTX-r current was reduced by IL-1β (supplemental Table 2, available at www.jneurosci.org as supplemental material), with no major changes in voltage dependence of either activation or inactivation (Fig. 3 F; supplemental Table 2, available at www.jneurosci.org as supplemental material). To assay possible effects on TTX-sensitive (TTX-s) current, we used a protocol that takes advantage of the different voltage dependence of inactivation of the two components. The amplitude of TTX-s current was estimated by digital subtraction of the TTX-r sodium current from the total current, assessed by a series of depolarizing steps after a 500 ms step to either −40 mV (for TTX-r current) or −120 mV (total current) (Gold et al., 1996; Flake et al., 2004). These experiments suggested that IL-1β also increases the amplitude of TTX-s current (peak current density from 47 ± 23 pA/pF to 72 ± 15 pA/pF, p < 0.05, n = 6) (Fig. 4 A,D). However, because of the small size of the TTX-s current relative to TTX-r current, its isolation may be imperfect.

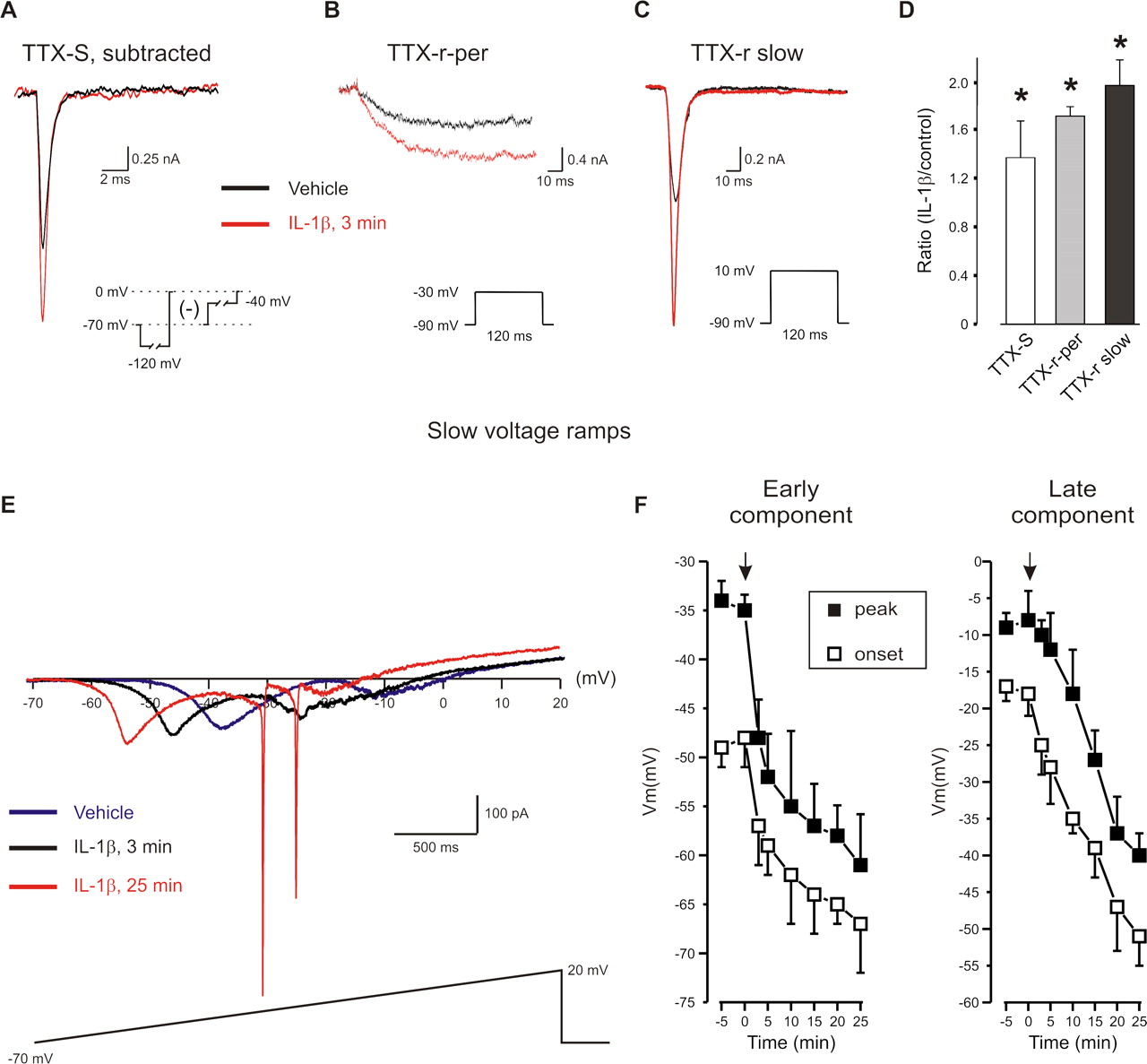
IL-1β increases the TTX-S and the slow and persistent components of the TTX-r sodium current. A , Representative traces of TTX-S before and after IL-1β following a depolarizing step to 0 mV. TTX-S current was assessed by subtraction of the current evoked by a 10 mV depolarization step after a 500 ms step to either −120 mV (for total sodium current) or −40 mV (for TTX-r current). B , C , In cells preincubated with 300 nm TTX, steps to −30 mV evoked TTX-r-per, whereas steps to +10 mV evoked TTX-r slow. D , IL-1β increased the density of TTX-S as well as TTX-r-per and TTX-r slow currents (n = 6 for TTX-S; n = 10 for TTX-R). E , Current–voltage curve during a slow depolarizing ramp (35 mV/s, −70 to +20 mV). Note “W” shaped inward current with clear early and late components. IL-1β increased the amplitude of early and late components of the inward current and shifted their activation and peak to more negative potentials. Fast inward currents after the slow ramp after prolonged (25 min) exposure IL-1β appeared in all recorded cells (n = 15) and reflect lost of voltage-clamp control possibly due to increase in membrane excitability. F , Arrow indicates time of application of IL-1β or vehicle. Time point of “-5” refers to data collected before the application. Recordings for A–D were made using extracellular solution 1, and those for E and F using extracellular solution 3 (Table 1).
IL-1β increases both TTX-r slow and TTX-r-per sodium currents
Two components of TTX-r current can be distinguished. The tetrodotoxin-resistant slow current (TTX-r slow) contributes to the upstroke of action potentials in nociceptor neurons (Renganathan et al., 2001) whereas the small persistent TTX-r current (TTX-r-per) contributes to threshold and excitability (Herzog et al., 2001; Baker et al., 2003). To examine whether IL-1β has an effect on TTX-r-per we used a step protocol that distinguishes this current from TTX-r slow, based on their differential thresholds (Amaya et al., 2006) (Fig. 4 B,C). Depolarization steps from −60 to −30 mV elicited a small TTX-r-per in ∼80% of small DRG neurons (current density at −30 mV = 17 ± 3pA/pF, calculated at the end of a 120 ms step, n = 20). We have shown that this current is absent in Na(v) 1.9 KO mice (Amaya et al., 2006). IL-1β produced a rapid (∼2 min) reversible increase in this current (current density = 27 ± 3, p < 0.05, n = 20) (Fig. 4 B,D), with a 10 mV leftward shift in its activation curve (V 1/2 Vehicle= −29 ± 1 mV; V 1/2 IL-1 β= −38 ± 1 mV, p < 0.05, n = 20) and accelerated activation kinetics (time to peak Vehicle = 26 ± 5 ms vs 12 ± 5 ms 3 min after application of IL-1β (n = 20, p < 0.05)). The TTX-r slow current density also increased in response to IL-1β (Fig. 4 C,D).
We measured quasi-steady-state current-voltage curves using a slow (35 mV/s) 2.5 s voltage ramp (−70 to +20 mV) that inactivates nonpersistent components (Fleidervish et al., 1996). In control conditions a small inward current (100 ± 32 pA, n = 15) with activation at −18 ± 3 mV and a peak at −8 ± 3 mV was detected (Fig. 4 E). IL-1β shifted the activation of the inward current toward the resting potential (−51 ± 5 mV, n = 15, 25 min) and increased its amplitude 2.4-fold (180 ± 56 pA) (p < 0.05) (Fig. 4 F). In 70% of the nociceptor-like cells, a slow ramp current appeared with a “W” shape because of an additional inward component (amplitude 156 ± 34 pA, n = 10) that activated at −48 ± 3 mV and peaked at −35 ± 2 mV (Fig. 4 E). From its voltage dependence, the early component is likely to be the TTX-r-per current, and the late component the TTX-r slow current (see also Maingret et al., 2008). The putative TTX-r-per mediated early component was significantly increased by IL-1β to 220 ± 45 pA (p < 0.05, n = 10), and the onset and peak shifted toward the resting potential (onset = −67 ± 5 mV, peak = −61 ± 5 mV) (Fig. 4 F). This effect can explain the induction of spontaneous firing by IL-1β.
We conclude that an IL-1β-mediated increase in TTX-r currents contributes to the increase in the excitability of nociceptors and underlies the spontaneous firing at the resting potential that is induced by an IL-1β. Supporting this, pain hypersensitivity produced by intraplantar IL-1β is reduced in mice with a null mutation in Nav1.9, the sodium channel α subunit responsible for TTX-r-per (Amaya et al., 2006).
IL-1β changes the voltage dependence of sodium channel slow inactivation
One possible mechanism for the IL-1β enhancement of the TTX-r current could be a reduction of resting slow inactivation of sodium channels (Fleidervish et al., 1996) which is significant in DRG neurons (Ogata and Tatebayashi, 1992). To assess this we studied the effect of IL-1β on the steady state voltage dependence of TTX-r sodium channel slow inactivation. Slow inactivation was assayed using 5 s conditioning prepulses followed by 20 ms at −100 mV (to remove fast inactivation) and a test pulse to 0 mV (Fig. 5 A). In control conditions slow inactivation of TTX-r was strongly voltage-dependent with a midpoint (V 1/2) of −57 ± 3 mV and a slope factor of 17 ± 3 mV (n = 10), saturating at potentials from −20 mV to +20 mV (Fig. 5 B). IL-1β shifted the voltage dependence of slow inactivation of the TTX-r sodium current in the depolarizing direction (V 1/2 = −43 ± 2 mV with slope of 14 ± 2 mV, n = 10, p < 0.05). The IL-1β-mediated decrease in slow inactivation was most prominent at potentials from −70 to −30 mV. In this voltage range the available fraction of TTX-r sodium current increased by ∼22%, shortly (3 min) after application of IL-1β (Fig. 5 A,B). Conditioning pulses to more depolarizing potentials abolished the TTX-r sodium current in the control condition, but not after application of IL-1β (Fig. 5 A,B).

IL-1β increases TTX-r sodium current due to a modulation of the voltage dependence of TTX-r sodium channel slow inactivation. A , Superimposed representative traces of TTX-r sodium currents before (black) and 3 min after (red) application of IL-1β. Currents were elicited by 10 ms test step to 0 mV after 5 s conditioning pulses (V cond) to indicated voltages. A 20 ms step to −100 mV was applied after conditioning pulses to remove fast inactivation. Note that prominent reduction of TTX-r current amplitude after prepulses to −70, −40, and −20 mV was attenuated by application of IL-1β (3 min). Traces (150 μs) were blanked to remove uncompensated capacitance transient currents. B , Voltage dependence of TTX-r sodium channels slow inactivation expressed as I test/I max plotted as a function of V cond before (open circles) and 5 min after (filled circles) IL-1β. Note significant (13.4 mV, p < 0.05, n = 10) rightward shift of midpoint voltage dependence of slow inactivation after application of IL-1β, together with reduction of maximal slow inactivation at depolarized voltages. Inset, Stimulus protocol. V cond was held constant at 5 s and its amplitude was varied between −120 and +20 mV. 20 ms step to −100 mV was applied before V test · I test was evoked by stepping to 0 mV. Membrane was held at −100 mV. Recordings made using extracellular solution 1 (Table 1).
Consistent with the effect of IL-1β being primarily a change in voltage dependence of slow inactivation, its effects were much smaller at steady holding potential of −100 mV, at which slow inactivation is removed (Fig. 5 B) (see also Fleidervish et al., 1996; Blair and Bean, 2003). Control TTX-r current density was substantially increased by a holding potential of −100 mV applied at least 30 s before the step protocol (365 ± 28 pA/pF) compared with −70 mV (148 ± 10.7, n = 10, p < 0.05). At the −100 mV holding potential, however, IL-1β increased the current only moderately (406 ± 23 pA/pF, n = 10, p = 0.1, 3 min after IL-1β) compared with its effect at a holding potential of −70 mV.
These data suggest that IL-1β increases sodium current by partially relieving resting slow inactivation of TTX-r sodium channels due to an alteration in its voltage dependence.
p38 MAPK activation underlies the effect of IL-1β on the excitability of nociceptor-like DRG neurons
IL-1β acts via p38 and p42/44 MAPK, JNK and NFκB signal transduction pathways to produce its diverse effects (O'Neill, 2002). IL-1β applied to adult DRG cultures (1–10 ng/ml for 15 min) induced a 3.6-fold increase in phospho-p38 MAP kinase in neurons (Fig. 6 A). To test whether the activation of p38 MAPK contributes to the IL-1β-mediated increases in excitability and sodium currents, we investigated whether a p38 inhibitor blocked these changes. SB203580 dose-dependently reduced the effects of IL-1β on action potential threshold (Fig. 6 C; supplemental Table 3, available at www.jneurosci.org as supplemental material), total and TTX-r-per sodium current amplitude (Fig. 6 F,G; supplemental Table 3, available at www.jneurosci.org as supplemental material), and the voltage dependence of slow inactivation (V 1/2 (SB203580, 10μM) = −54 ± 4 mV, k = 18 ± 4 mV, n = 5 vs V 1/2 (SB203580,10μM + IL-1β) = −57 ± 5 mV, k = 17 ± 4 mV, p = 0.09, n = 5). SB203580 dose-dependently reduced the IL-1β mediated hyperpolarizing shift of AP threshold and modulation of sodium currents, and completely reversed these effects at a concentration of 10 μm (Fig. 6).

IL-1β modulates activity of DRG neurons via p38 MAPK. A , Increase in phosphorylated p38 MAP kinase (pp38) in cultured DRG neurons after IL-1β (1 ng/ml, 10 min) demonstrated by immunocytochemistry and Western blot. B–E , Preincubation of DRG neurons for 40 min with the p38 inhibitor SB203580 (10 μm) prevented the IL-1β mediated increases in excitability. C , Dose dependent effect of SB203580 on IL-1β-mediated decrease of spike threshold. (□, 10 μm; ○, 100 nm; ▵, 10 nm). Arrow indicates time of application of IL-1β. Time point “-5” refers to data collected before application. Note no change in spike threshold after preincubation with 10 μm SB203580 and significant decrease of threshold after preincubation with 10 nm SB203580. D , SB203580 (10 μm) prevents IL-1β -mediated changes in numbers of spikes per stimulus (mean ± SEM) expressed as multiples of the rheobase for each recorded cell (n = 10). E , Ramp (333 pA/s) activated trains of action potentials showed no change in threshold after IL-1β in presence of SB203580 (10 μm), but still demonstrated reduced spike adaptation. F , The ratio of the mean peak total and TTX-r sodium current amplitude (IL-1β, 20 min/control) (n = 10) showed no change after IL-1β in cells preincubated with 10 μm SB203580 but increased significantly in presence of 10 nm SB203580. G , Quasi-steady-state current– voltage curves during a slow (35 mV/s) ramp demonstrating that SB203580 (10 μm) blocked the IL-1β -mediated change in both early (amplitudeVehicle 148 ± 34 pA vs 151 ± 42 pA, p = 0.23; current activation onsetVehicle −53 ± 8 mV vs −36 ± 9 mV, p = 0.21; and peakVehicle −42 ± 8 mV vs −45 ± 10 mV, p = 0.11, n = 10) and late components (amplitudeVehicle 112 ± 25 pA vs 105 ± 12 pA, p = 0.1; current activation onsetVehicle −23 ± 6 mV vs −24 ± 4 mV, p = 0.09 and peakVehicle −12 ± 5 mV vs −16 ± 5 mV, p = 0.26, n = 10) of resulting inward current. Recordings for B–E were made using extracellular solution 4, those for F , solution 1, and those for G , solution 3 (Table 1).
Although SB203580 blocked the effect of IL-1β on the early response to a depolarizing current (Fig. 6 D,E) it did not alter the reduction in spike adaptation (Fig. 6 E; supplemental Table 3, available at www.jneurosci.org as supplemental material), indicating that this particular IL-1β-mediated change is p38 independent, and may result from a reduction in potassium currents (Takeda et al., 2008).
To establish whether the action of IL-1β on C-fiber peripheral terminals in vivo involves p38 or other MAPK, we analyzed the levels of phospho-p38, phospho-JNK and phospho-ERK immunohistochemically in the plantar skin.
Intradermal injection of IL-1β (1 and 10 pg) rapidly (10 min) induced phospho-p38 in contrast to sham controls (Fig. 7 A). No significant IL-1β mediated effects on phospho-JNK or phospho-ERK could be detected (data not shown). In keeping with this, preinjection of 50 μg of SB203580 into the hindpaw 20 min before an intraplantar injection of IL-1β attenuated both the mechanical (Fig. 7 B, left) and thermal pain hypersensitivity it normally produces (Fig. 7 B, right).

Activation of p38 MAPK contributes to the IL-1β-mediated behavioral decrease in mechanical threshold and thermal responsiveness. A , Phosphorylation of p38 in peripherin-positive cutaneous sensory fibers after intraplantar IL-1β injection (1 ng). B , Pretreatment with the p38 inhibitor SB203580 (50 μg) attenuated the effects of IL-1β on mechanical threshold and noxious heat response latency (mean ± SEM); *p < 0.05.
Discussion
The high threshold of nociceptors and their normal selective sensitivity only to noxious stimuli is mediated by expression of transducer ion channels on their peripheral membrane, of which TRP channels are the most prominent. Exogenous chemical irritant stimuli that activate nociceptors include capsaicin and mustard oil acting on TRPV1 and TRPA1, whereas endogenous nociceptor chemical activators include ATP (via P2X2/3) and protons (TRPV1/ASICs) (Caterina et al., 1997; Burgard et al., 1999). Our data demonstrate that IL-1β is itself an endogenous activator that can initiate action potential discharge and act by augmenting voltage gated sodium currents rather than on transducer channels.
Inflammation, by changing the chemical milieu of the peripheral terminals of nociceptors, produces many changes in the transduction of stimuli by these sensory neurons. Many inflammatory mediators act on receptors that indirectly via intracellular kinases, activate transducer ion channels, as for bradykinin via B2 and its coupling with the TRPA1 channel (Bandell et al., 2004; Bautista et al., 2006). Other mediators are not, however, coupled to increased ion flux, but instead reduce the threshold or increase the density of transducer channels, heightening transduction sensitivity, as for the action of NGF on TRPV1 (Ji et al., 2002; Zhang et al., 2005), and of PGE2 also on TRPV1 (Moriyama et al., 2005).
Transduction channels are not the only substrate for post-translational changes downstream of nociceptor sensitizer-activated signaling; these also include voltage-gated sodium channels. PGE2 and epinephrine, increase sodium currents in DRG neurons and in this way, amplify the effects of transducer currents (England et al., 1996; Gold et al., 1996; Rush and Waxman, 2004; Baker, 2005).
Although cytokines are well known to promote pain hypersensitivity this has been considered to be largely secondary to the recruitment and activation of immune cells that synthesize and release the inflammatory mediators that then produce peripheral sensitization (Cunha et al., 2005), rather than to a direct action of the cytokines on neurons. In particular, IL-1β induces cyclooxygenase 2 to produce PGE2 (Maier et al., 1990), and also increases NGF synthesis (Safieh-Garabedian et al., 1995). Such changes take, however, some time for increased transcription, protein synthesis, mediator release and action, and yet inflammatory pain generally has a minimal time lag. Our data suggest that this is contributed to by the rapid, transcription-independent action of cytokines on nociceptors. In keeping with this, application of IL-1β in vitro increased a heat activated inward current (Obreja et al., 2002) and CGRP release from peripheral terminals (Opree and Kress, 2000), whereas single unit nociceptor recordings in vivo demonstrated a sensitization to external stimuli and transient spontaneous discharge shortly after intraplantar injection of IL-1β (Fukuoka et al., 1994).
Our study demonstrates that IL-1β activates nociceptors and also enhances their response to depolarizing current and that it does this by augmenting TTX-r sodium currents by removing their slow inactivation.
TNFα also increases TTX-r sodium currents in nociceptor DRG neurons via p38 (Jin and Gereau, 2006) but its mechanism of action has not been explored.
The carriers of TTX-r slow (Na(v) 1.8) (Akopian et al., 1996) and TTX-r-per (Na(v) 1.9) (Cummins et al., 1999; Fang et al., 2002) are expressed both in cell bodies (Akopian et al., 1996; Dib-Hajj et al., 1998; Amaya et al., 2006) and the peripheral terminals of nociceptors in the skin (Liu et al., 2001; Zimmermann et al., 2007). The effect of IL-1β on these currents can explain the increased nociceptor excitability we observe because TTX-r slow is responsible for the majority of the inward charge during the upstroke of AP in nociceptors (Renganathan et al., 2001) and for the ability of the neurons to fire repetitively (Renganathan et al., 2001). TTX-r-per is too slow to contribute to the depolarizing phase of AP, but operates at subthreshold membrane potentials to regulate threshold and excitability (Herzog et al., 2001). An increase in TTX-r-per in a simulated neuron model leads to a decrease in single AP threshold and repetitive firing during prolonged subthreshold depolarizations (Baker, 2005). Such changes were also observed in DRG neurons when the TTX-r-per sodium current was increased either by GTP (Baker et al., 2003) or PKC (Baker, 2005), and as we now report, also after IL-1β. Moreover, an enhancement of the Na(v) 1.9 current underlies inflammatory soup-mediated changes in neuronal excitability, such as an increase in the number of action potentials in response to injected current and a decrease in the latency to the first spike (Maingret et al., 2008), similar to those produced by IL-1β. The hyperpolarizing shift and increase in activation of TTX-r-per by IL-1β can explain the dramatic induction by IL-1β of spontaneous generation of action potentials at the resting potential.
Acutely applied PGE2 increases the excitability of DRG neurons by a PKA dependent increase in the composite TTX-r current (England et al., 1996; Gold et al., 1996), but not TTX-r-per (Zheng et al., 2007; Maingret et al., 2008). The effects of PGE2 on TTX-r are G-protein dependent (England et al., 1996; Gold et al., 1996; Baker et al., 2003), as are similar effects of 5-HT (Cardenas et al., 2001), whereas the effects of IL-1β are mediated by a membrane receptor tyrosine kinase and by p38 activation rather than PKC or PKA. Unlike PGE2's effect on the TTX-r current (Gold et al., 1996), the IL-1β mediated increase in total and composite TTX-r sodium current is not accompanied by changes in the voltage dependence of activation or fast inactivation. However, a hyperpolarizing shift in the activation curve of TTX-r-per, similar to that described in response to chronic application of PGE2, does occur (Rush and Waxman, 2004).
In addition to fast inactivation, TTX-r slow currents also exhibit prominent slow inactivation (Ogata and Tatebayashi, 1992; Blair and Bean, 2003; Choi et al., 2007). Here we show that the midpoint of slow inactivation was near −55 mV, and that in consequence ∼40% of sodium channels are inactive at the resting membrane potential. The IL-1β-mediated relief of slow inactivation, leads to an additional availability of sodium channels, a change sufficient to substantially increase the current evoked at the resting potential. Removal of inactivation by hyperpolarization at –100 mV has a similar effect to IL-1β, it increases the sodium current. In addition, because slow inactivation limits the responsiveness of small DRG neurons to maintained stimuli (Blair and Bean, 2003), a reduction in slow inactivation (Choi et al., 2007) may explain the dramatic decrease in accommodation we find in response to current ramps (Fig. 1 B) that mimic depolarization by transducer channels such as TRPV1). 5-HT modulates the voltage dependence of slow inactivation of TTX-s channels in cortical pyramidal neurons (Carr et al., 2003), but in the opposite direction, reducing sodium current by enhancing resting slow inactivation, and by a different mechanism, one involving G-proteins and PKA or PKC. These findings and our own point, therefore, suggest modulation of slow inactivation as powerful means to increase or decrease sodium currents. The action of IL-1β on slow inactivation of TTX-r currents is of special interest because of the recent description of an enhancement on Na(v) 1.8 slow inactivation by lacosamide, an anticonvulsant with antinociceptive properties (Sheets et al., 2008).
IL-1β exerts its cellular effects by interacting with the cell surface (IL-1R) type I and type II receptors (McMahan et al., 1991), with most effects mediated by IL-1RI (Liege et al., 2000). This receptor is widely expressed, including in the nervous system, and as we find, in small DRG neurons. We find moreover, that the regulation of neuronal excitability and sodium currents by IL-1β is largely mediated by phosphorylation and activation of p38 MAP kinase. Activation of p38 was detected in peripheral terminals almost immediately after IL-1β injection, and administration of a p38 inhibitor blocked the electrophysiological and acute pain enhancing effects of IL-1β. We cannot rule out the possibility, however, that the IL-1β-mediated effect is contributed to by stimulating release of signaling molecules from non-neuronal cells.
The induction of pp38 MAPK by IL-1β was not restricted to small DRG neurons (Fig. 6 A). This is in agreement with our immunohistochemical data showing IL-1RI expression predominantly but not exclusively in small diameter neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Recently modulation of the Na(v) 1.8 sodium channel by a direct p38 MAPK-mediated phosphorylation of serine 551 and 556 on the L1 loop was demonstrated in nociceptive DRG neurons (Hudmon et al., 2008). The p38-mediated increase in TTX-r slow current density was not accompanied by changes in activation or fast inactivation voltage dependence. The site of p38 mediated effects on Nav1.9 needs to be elucidated, as does whether the effect of TNFα on these channels (Jin and Gereau, 2006), also acts by a reduction in slow inactivation.
Although many reports describe pronociceptive effects of IL-1β when administered peripherally and centrally (Sommer and Kress, 2004; Wieseler-Frank et al., 2005), intrathecal administration of high doses (0.1 μg, 1 μg) increases nociceptive thresholds in naive and inflamed rats (Souter et al., 2000). We observed a similar dose-dependent response to peripheral injection; high doses (1 μg) were antinociceptive. This suggests that the signaling kinetics of the IL-1β receptor complex may depend on the concentration of the ligand, with a ceiling/desensitizing effect at very high doses. In trigeminal ganglion neurons a short (5 min) incubation of IL-1β (20 ng/ml) was found by (Liu et al., 2006) to reduce sodium currents; a result different from ours, although they did find that a long exposure (24 h) increased them. The discrepancy of the two sets of findings may be because the IL-1β-mediated changes in slow inactivation are prominent at −70 mV but not at −80 mV (where the partial fraction availability before IL-1β was 84 ± 3% vs 89 ± 5% after, unlike the 20% increase found at −70 mV). In our experiments we used a holding potential before activation of sodium channels of −70 mV (close to natural resting potential of −65 to −70 mV) whereas Liu et al. used one of −80 mV.
Nociceptors by responding directly to cytokines can directly “sense” the immune response in inflamed tissue; essentially they are, therefore, not only noxious stimulus detectors, but also inflammation sensors or “inflammaceptors”. Blocking IL-1β synthesis or action may not only be anti-inflammatory, but also analgesic for inflammatory pain.
Footnotes
-
This work was supported by the National Institutes of Health (C.J.W.). We thank Dr. Huijuan Hu for helpful discussion.
- Correspondence should be addressed to Tarek A. Samad, Pain Molecular Neurobiology Group, Neuroscience Discovery, Wyeth Research, 865 Ridge Road, Princeton, NJ 08852. samadt{at}wyeth.com





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}