
Abstract
Mints/X11s are neuronal adaptor proteins that bind to amyloid-β precursor protein (APP). Previous studies suggested that Mint/X11 proteins influence APP cleavage and affect production of pathogenic amyloid-β (Aβ) peptides in Alzheimer's disease; however, the biological significance of Mint/X11 binding to APP and their possible role in Aβ production remain unclear. Here, we crossed conditional and constitutive Mint1, Mint2, and Mint3 knock-out mice with transgenic mouse models of Alzheimer's disease overproducing human Aβ peptides. We show that deletion of all three individual Mint proteins delays the age-dependent production of amyloid plaque numbers and Aβ40 and Aβ42 levels with loss of Mint2 having the largest effect. Acute conditional deletion of all three Mints in cultured neurons suppresses the accumulation of APP C-terminal fragments and the secretion of ectodomain APP by decreasing β-cleavage but does not impair subsequent γ-cleavage. These results suggest that the three Mint/X11 proteins regulate Aβ production by a novel mechanism that may have implications for therapeutic approaches to altering APP cleavage in Alzheimer's disease.
- Mint
- X11
- APP
- β-amyloid
- Alzheimer's disease
- knock-out
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder leading to cognitive decline (Selkoe, 2001; Sisodia and St George-Hyslop, 2002). A neuropathological hallmark of AD is neuritic plaques containing deposits of 40–43 amino acid amyloid-β (Aβ) peptides that are derived from the β-amyloid precursor protein (APP). APP is a type I membrane glycoprotein that is physiologically processed by sequential cleaved site-specific proteases (Haass and De Strooper, 1999; Selkoe, 2001; Sisodia and St George-Hyslop, 2002). First, α- or β-secretase cleave APP into a large secreted extracellular fragment and a smaller membrane-associated C-terminal fragment (CTF). The APP-CTF is composed of a short extracellular stub, transmembrane region, and cytoplasmic tail, which subsequently is digested by γ-secretase within the transmembrane region (De Strooper et al., 1998; Struhl and Adachi, 2000; Yu et al., 2001). Cleavage of the APP-CTF by γ-secretase releases an intracellular cytoplasmic APP fragment that translocates to the nucleus (Cupers et al., 2001; Kimberly et al., 2001) and may regulate transcription (Cao and Südhof, 2001). In addition, cleavage of APP by γ-secretase generates small secreted peptides, including the pathogenic Aβ peptides that form the major constituents of β-amyloid plaques in AD (Glenner and Wong 1984).
The cytoplasmic domain of APP contains a conserved sorting signal (YENPTY motif) (Haass et al., 1994; Marquez-Sterling et al., 1997) that interacts with several proteins containing phosphotyrosine binding-domains (PTB-domain), including Mint/X11 adaptor proteins (Borg et al., 1996; McLoughlin and Miller, 1996; Zhang et al., 1997). Mint1 and Mint2 are neuron-specific, whereas Mint3 is ubiquitous (Okamoto and Südhof, 1997, 1998). They are composed of an isoform-specific N-terminal sequence, a central PTB-domain that binds to APP, and two C-terminal PDZ (postsynaptic density-95/Discs large/zona occludens-1) domains that bind to a number of proteins in vitro, including presenilins (Okamoto and Südhof 1997; Lau et al., 2000, Biederer et al., 2002).
Overexpression and/or knockdown of Mints/X11s suggested that Mints modulate APP processing and Aβ generation but led to conflicting conclusions (reviewed in supplemental Table 1, available at www.jneurosci.org as supplemental material). Mint overexpression increases APP steady-state levels and decreases Aβ secretion by inhibition of γ-secretase (Borg et al., 1998; Sastre et al., 1998; Mueller et al., 2000; Lee et al., 2003, 2004). RNA interference-mediated partial knockdown of Mint1 or Mint2 also led to a decrease in Aβ levels (Xie et al., 2005). Thus, it is unclear how Mint/X11s affect APP cleavage and Aβ production physiologically and whether their function may be involved in the pathogenesis of AD.
To test this, we have crossed conditional and constitutive Mint1, Mint2, and Mint3 knock-out mice (Ho et al., 2003, 2006) to two separate lines of transgenic mice that overproduce human Aβ that serve as models for AD. We found that deletion of each of the three Mint isoforms decreases amyloid plaque production and lowers Aβ40 and Aβ42 levels, with homozygous loss of Mint2 having the largest effect. This effect is attributable to a decrease in β-secretase but not γ-secretase cleavage of APP. Our results suggest a novel mechanism of action for Mints/X11s in regulating APP cleavage that could be exploited in future searches for new drug therapies.
Materials and Methods
Mouse breedings.
Mint knock-out mice (Ho et al., 2003, 2006) were crossed to two transgenic mouse lines that overproduce human Aβ (APPswe/PS1dE9; Jackson ImmunoResearch Laboratories, stock #004462) (Borchelt et al., 1997) (APPswe/Ind; Jackson ImmunoResearch Laboratories, stock #004661) (Mucke et al., 2000). To generate Mint knock-outs carrying the doubled-transgene APPswe/PS1dE9, Mint1−/−, Mint2+/− mice were mated with APPswe/PS1dE9 transgenic mice, and the offspring of these crosses were mated back to either Mint1+/−, Mint2+/− or Mint1−/−, Mint2+/− mice, which should result in mice homozygous for the Mint1 or Mint2 knock-out alleles carrying APPswe/PS1dE9 transgene. Mint3−/− and homozygous-floxed CASK mice (Atasoy et al., 2007) were also mated to APPswe/PS1dE9 transgenic mice to generate Mint3−/− or CASK hypomorphic mice carrying the transgene. Homozygous Mint1−/− were mated to APPswe/Ind transgenic mice to generate Mint1 knock-out carrying the APPswe/Ind transgene.
Immunohistochemistry and Aβ plaque quantitation.
Brains were cut sagitally in half for Aβ immunohistochemical staining and Aβ ELISA quantifications. For immunohistochemistry, brains were fixed overnight at 4°C in 4% fresh paraformaldehyde, cryoprotected in 30% sucrose in PBS, and frozen in OCT (optimal cutting temperature) compound. Sagittal cryostat sections were systematically collected in series, and every fourth section (40 μm apart) from a complete series, with a random starting section, was immunostained. Sections were permeabilized with 0.5% Triton X-100, blocked with 2% goat serum/0.1% Triton X-100, and incubated with Aβ (U6590) antibody overnight at 4°C. This was followed by incubation with horseradish peroxidase-coupled secondary antibodies and developed in 3,3′-diaminobenzidine tetrahydrochloride with nickel chloride as a metal enhancement and analyzed by standard light microscopy. Aβ immunostaining was analyzed blindly without the knowledge of the mouse genotype. Images were captured with an Olympus BX-51 research microscope using a SPOT CCD camera. Aβ pixels were determined using MetaMorph imaging software (Molecular Devices) in which we first apply the same threshold value (between 100 and 256) to all images and then calculate the pixel area of individual plaques and total number of the plaque within the cortex and hippocampus for each section. Statistical significance was determined by Student's t test.
Aβ peptide measurements by ELISA.
Cortical and hippocampal areas were homogenized in 2 ml 20 mm Tris pH 8.5, 10 mm EDTA pH 8.0, 1 mm PMSF with proteinase inhibitors. One milliliter of this starting lysate was set aside and used for Western blot analysis. The remaining starting lysate was centrifuged at 135,000 g for 1 h at 4°C. The supernatant “soluble” fraction was removed into a new tube, and the pellet was resuspended in 1 ml of 10 mm Tris pH 7.6, 150 mm NaCl, 2% Triton X-100, 2% NP-40, 1 mm PMSF and proteinase inhibitors and extract for 1 h at 4°C. This was followed by a second centrifugation at 100,000 g for 1 h at 4°C. The supernatant “membrane” fraction was removed into a new tube, and the remaining pellet was resuspended in 1 ml of fresh 50 mm Tris pH 8, 5 m guanidine HCl, 1 mm PMSF and proteinase inhibitors (insoluble fraction). Protein levels were quantified (Pierce Chemical), and ELISAs were performed according to the instructions provided by the manufacturer's protocol [ImmunoBiological Laboratories Kits for human Aβ42 (27711) and Aβ40 (27718)].
Neuronal cultures.
Dissociated high-density neocortical cultures were prepared from newborn mice (Kavalali et al., 1999). Recombinant lentiviruses were produced by transfecting human embryonic kidney 293T cells with VSVg, CMV Δ8.9 (plasmids encoding the elements essential for packing of viral particles), and pFUGW (a shuttle vector encoding the gene of interest and recombination arms for incorporating into mammalian genome) with FuGENE reagent as previously described (Ho et al., 2006). The pFUGW plasmids contained the enhanced green fluorescent protein (EGFP) with nuclear localization signal (NLS) carrying cre recombinase sequence (EGFP-NLS-CRE) with ubiquitin promoter or a pFUGW plasmid containing only EGFP-NLS (CreΔ). Neurons were infected for 48 h for expression, and media were exchanged back to normal growth media and sustained until 13–15 d in vitro (DIV) for biochemical analyses. Neurons were treated with 2 μm N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester (DAPT), a γ-secretase inhibitor IX (Calbiochem) at various time points, and lysates were collected in 1× sample buffer. For DAPT washout experiments, neuronal cultures at 12 DIV were treated with 2 μm DAPT for 24 h, after which the neuronal media was washout with PBS and replaced with neuronal growth media for the duration of the experiment in which lysates were collected at 2, 4, 6, 12, and 24 h after DAPT washout.
Miscellaneous.
SDS-PAGE and immunoblotting were performed using standard procedures, and most antibodies used were described previously (Ho et al., 2006). NuPAGE 4–20% gradient gels were used to detect the low molecular weight APP-CTF fragments (Invitrogen).
Antibodies.
Antibodies used were as follows: Aβ 1–16 (6E10) monoclonal antibody (SIG39320; Covance Research Products); anti-human sAPPα (2B3) mouse IgG monoclonal antibody (11088; ImmunoBiological Laboratories); anti-human sAPPβ wild-type rabbit IgG affinity purify antibody (18957; ImmunoBiological Laboratories); and mouse sAPP 22C11 (MAB348; Millipore).
Results
Deletion of Mints decreases Aβ plaque formation in transgenic mouse models of AD
To test whether Mint/X11 proteins control the production of β-amyloid by APP cleavage, we crossed the APP/presenilin double-transgenic mice with constitutive knock-out mice lacking Mint1, Mint2, or Mint3 (Ho et al., 2003, 2006). We analyzed littermate offspring containing the double transgene that either contained all three Mints or selectively lacked one of the three Mint isoforms. These studies purposely used sex-matched littermate pairs with a hybrid genetic background to avoid possible interference by homozygous background mutations in inbred strains and to control for genetic background.
We first measured the amyloid plaque load as a function of the Mint1, Mint2, or Mint3 deletion at 6, 9, and 12 months of age (Fig. 1; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Cortical and hippocampal sections of littermate mice containing or lacking the various Mint isoforms were stained with Aβ antibody, and plaque load was measured by image analysis using a nonbiased automatic procedure. In double-transgenic mice with wild-type Mints, plaque load increased ∼10-fold from 6 to 12 months of age (Fig. 1B). At 6 months, deletion of each of the three Mint isoforms decreased Aβ plaque load approximately twofold to fivefold in both the hippocampus and cortex. At 9 months of age, deletion of the neuron-specific isoforms Mint1 and Mint2 still significantly decreased plaque load by approximately twofold, whereas deletion of Mint3 did not. By 12 months, only the Mint2 deletion significantly decreased the Aβ plaque load and only in the hippocampus, whereas the Mint3 deletion even caused an increase. As a control, we crossed the double-transgenic APP/presenilin-1 mice with hypomorphic CASK-mutant mice (Atasoy et al., 2007), because CASK is a neuronal adaptor protein that binds to Mint1 but not to Mint2 or Mint3 (Butz et al., 1998). The CASK mutation had no effect on Aβ plaque production (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), confirming that the effect of the Mint deletions on Aβ plaque load is specific. These observations indicate that deletion of Mints delays development of Aβ plaques in the transgenic mice, with differential effects of Mint isoforms most likely attributable to their distinctive expression patterns in the brain. Specifically, Mint2 that has the biggest effect is also expressed most highly in neurons and enriched in excitatory neurons, whereas Mint1 is enriched in inhibitory neurons and Mint3 having the lowest abundance in the brain (Okamoto and Südhof 1997, 1998; Ho et al., 2003, 2006). Immunostaining for neuronal isoform Mint1 and Mint2 and APP revealed distinct localization in the CA3 region of the hippocampus. Mint1 mainly stains inhibitory interneurons, whereas Mint2 and APP localize mostly to the soma of excitatory pyramidal neurons in the CA3 region of the hippocampus (Fig. 2). Therefore, it is possible that Mint1 and Mint3 effects is itself not enough to overcome the continued plaque accumulation with age.

Deletion of Mint/X11s decreases plaque formation in double-transgenic mice expressing mutant APP and presenilin-1. A, Representative images of hippocampal sections from transgenic mice (tg+) that are either wild type for Mints (Mint1+/+2+/+3+/+; top row), or lack Mint1 (Mint1−/−2+/−3+/+; second row), Mint2 (Mint1+/−2−/−3+/+; third row), or Mint3 (Mint1+/+2+/+3−/−; bottom row). Sections were from mice at 6, 9, and 12 months of age as indicated, stained with antibodies to Aβ, and visualized by HRP-labeling. Scale bar, 300 μm. See supplemental Figure 1, available at www.jneurosci.org as supplemental material, for representative images of cortical sections. B, Quantitations of the Aβ-positive plaque load in transgenic mice in the hippocampus and cortex. Images shown in A were analyzed with the MetaMorph program to calculate the coverage of the total section area occupied by plaques (means ± SEMs; n = 6–10; *p < 0.05, **p < 0.01, ***p < 0.001). Note that in the analyses of constitutive Mint knock-out mice, all analyses were performed on littermate mice that contain the double transgene for APP and presenilin and deletions for Mint1, Mint2, or Mint3 as indicated.

Distinct localization of Mint1, Mint2, and APP in the CA3 region of the adult hippocampus. A–C, Immunostaining for Mint1 (A), Mint2 (B), and APP (C) displayed distinct localization in the CA3 region of the hippocampus. Mint1 (A) mainly stained inhibitory interneurons, whereas Mint2 (B) and APP (C) localized mostly to the soma of excitatory pyramidal neurons in the CA3 region of the hippocampus. Scale bar, 50 μm.
Deletion of Mints decreases Aβ40 and Aβ42 peptide levels in transgenic mouse models of AD
To examine whether the decrease in Aβ plaques caused by Mint deletions is attributable to an effect of Mints on Aβ production or Aβ deposition, we determined the levels of Aβ40 and Aβ42 peptides in the brains from double-transgenic mice containing or lacking the various Mint proteins (Fig. 3). We analyzed Aβ peptides in three fractions: the soluble fraction (i.e., Aβ peptides that have not yet aggregated), membranes (defined as material that is soluble in 2% Triton X-100/2% NP40), and detergent-insoluble material (Aβ peptides in plaques). The membranous fraction was analyzed as an intermediate between soluble and detergent-insoluble materials to cleanly separate these two fractions. In double-transgenic mice containing Mints, the level of Aβ peptides increased approximately threefold in the soluble and ∼10-fold in the detergent insoluble fraction from 6 to 12 months (Fig. 3). Deletion of each of the three Mints suppressed the levels of Aβ40 and Aβ42 at 6 months. At 9 months, deletion of individual Mints continued to decrease both Aβ40 and Aβ42 levels in the insoluble fraction by approximately twofold to threefold, but only the neuronal isoforms Mint1 and Mint2 (and not Mint3) were able to decrease Aβ peptides in the soluble and membrane fraction. By 12 months, only the Mint2 deletion was able to decrease soluble Aβ peptides, with insoluble Aβ peptides exhibiting a small decrease that was not significant. This large effect of Mint2 deletion on Aβ production is interesting because when we measured endogenous Aβ40 and Aβ42 levels that did not carry the transgene in each of the Mint knock-out mice, only deletion of Mint2 significantly reduced Aβ42 levels in the insoluble fraction at 6 and 13–15 months of age (supplemental Figs. 3, 4, available at www.jneurosci.org as supplemental material). Thus, deletion of each of the three Mints independently delays Aβ production and deposition in the mutant APP/presenilin double-transgenic mouse model for AD, leading to more than a threefold decrease in Aβ levels at 6 months of age, and more than a twofold decrease in Aβ levels at 9 months of age.

Effect of Mint1, Mint2, or Mint3 deletions on Aβ40 or Aβ42 levels in mouse brain at 6, 9, and 12 months of age. Brains from transgenic littermate mice expressing mutant human APP and presenilin-1 (tg+) either containing or lacking Mint1, Mint2, or Mint3 were harvested at 6, 9, or 12 months of age and separated into soluble (A), membranous (B), and detergent-insoluble fractions (C). Aβ40 and Aβ42 levels were then measured by ELISA for all three fractions. (A, soluble proteins: supernatant of detergent-free brain homogenates centrifuged at 135,000 g for 1 h; B, membranous proteins: proteins in the pellet from the initial centrifugation that can be solubilized with 1% Triton X-100; and C, detergent insoluble proteins: proteins that remain insoluble after addition of Triton X-100). Data shown are means ± SEMs (n = 6–10; *p < 0.05, **p < 0.01, ***p < 0.001). Note that in C, the scale of the y-axis is reduced 1000-fold.
The effect of Mint deletions is selective, because protein quantitations revealed that Mint deletions had no major effects on the overall composition of the brain (supplemental Table 2, available at www.jneurosci.org as supplemental material). We detected no changes in APP, Nicastrin, or BACE1 expression but found that a small decrease in ApoE levels by Mint deletions. This is a potentially interesting observation in light of recent results suggesting that binding of ApoE to its receptor triggers the endocytosis of APP, which in turn may lead to Aβ production by a mechanism involving Mint1 or Mint2 (He et al., 2007). We also detected a significant decrease in NMDA-receptor 2A levels in Mint1 and Mint2 knock-out mice carrying the transgene; however, none of the synaptic proteins we analyzed were altered (supplemental Table 2, available at www.jneurosci.org as supplemental material).
A potential concern about our studies is that the double-transgenic mouse line we used as a model of AD may have special features that are not generally applicable, such as problems caused by a particular transgenic integration site. To exclude this possibility, we analyzed in a second unrelated mouse model of AD, namely single transgenic mice expressing human APP with both the Swedish and Indiana mutation (APPswe/Ind) (Mucke et al., 2000). In this mouse line, deletion of Mint1 again decreased the plaque load significantly in the hippocampus at 9 months (Fig. 4A,B). These mice produce lower concentrations of Aβ peptides than the double transgenic mice and have a very low plaque load in cortex that were not altered by deletion of Mint1. Deletion of Mint1 in the APPswe/Ind transgenic mice also suppressed the levels of Aβ40 and Aβ42 significantly, both for soluble and detergent-insoluble Aβ peptides (Fig. 4C,D). Thus, deletion of Mints lowers Aβ production and deposition in two independent mouse models of AD.
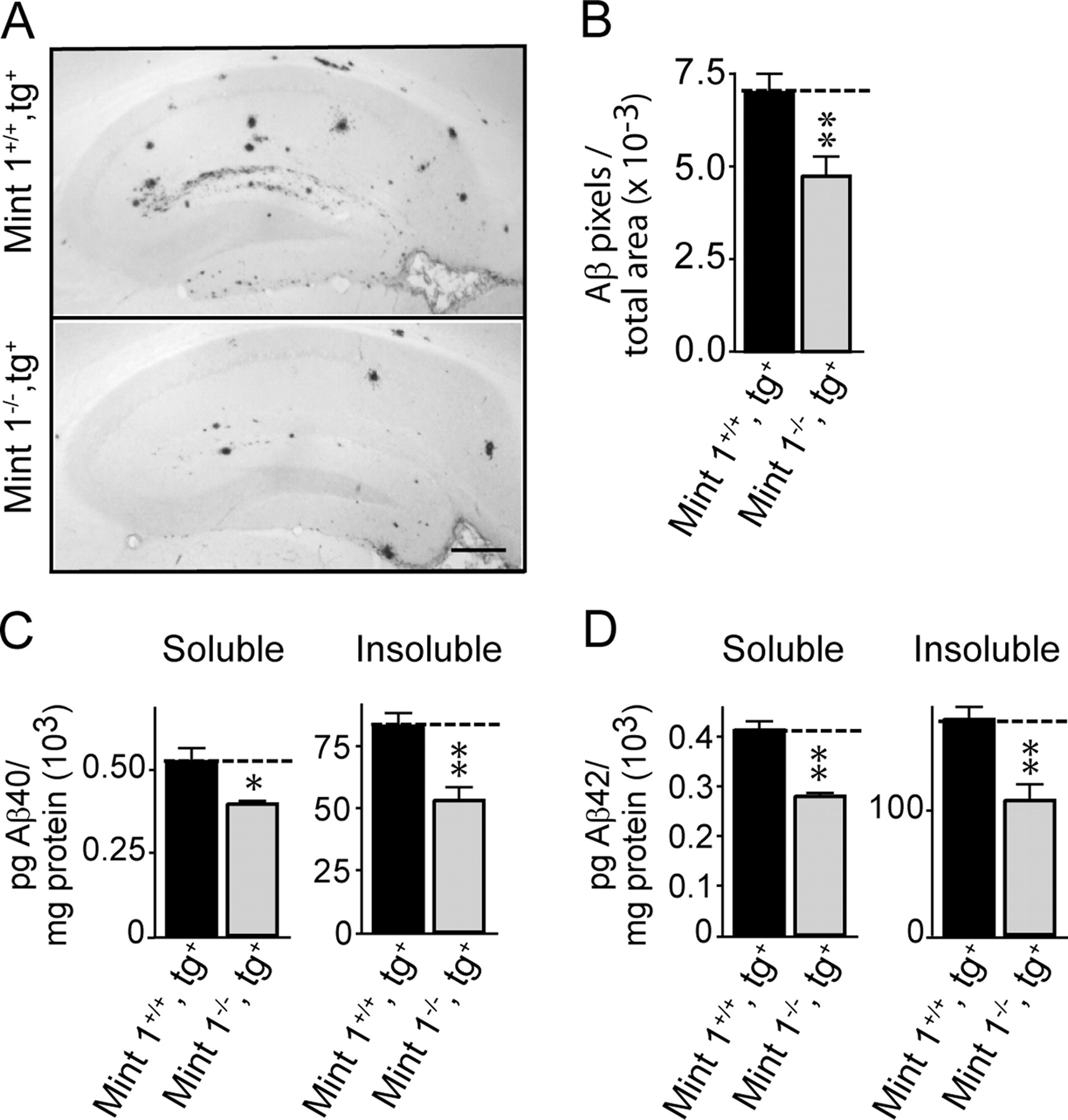
Effect of Mint1 deletion on plaque formation and Aβ production in transgenic mice expressing mutant human APP with the swedish and indiana mutations at 9 months of age. A, Representative images of hippocampal sections stained for Aβ (scale bar, 300 μm). B, Quantitations of plaque loads uncovered in such images in APP-transgenic mice containing or lacking Mint1. C, D, Peptide levels for Aβ40 (C) and Aβ42 (D) in APP-transgenic mice containing or lacking Mint1. All summary graphs depict means ± SEMs (n = 3 independent cultures performed in triplicates). *p < 0.05, **p < 0.01.
Acute deletion of Mint proteins decreases Aβ peptide levels
The decreased Aβ production in Mint-deficient APP-transgenic mice could be attributable to the following: decreased extracellular β-cleavage of APP, increased extracellular α-cleavage of APP, or impaired intramembranous γ-cleavage of the APP-CTF by β-cleavage. To differentiate between these possibilities, we crossed triple conditional Mint knock-out mice (Ho et al., 2006) with the double-transgenic APP/presenilin-1 expressing mice, and cultured neurons from mice that were homozygous for all three floxed Mint genes and additionally contained a single allele of the double transgene. We then infected the cultured neurons with lentivirus expressing mutant or active cre recombinase (CreΔ or Cre, respectively) (Ho et al., 2006) to obtain precisely matched pairs of cultured neurons that either contain or lack Mints.
Immunoblotting confirmed that infection with lentivirus expressing wild-type cre recombinase (Cre), but not mutant cre recombinase (CreΔ) completely abolished Mint expression (Fig. 5A). In these neurons, the expression of APP and its localization to the trans-Golgi network (Borg et al., 1996; Sastre et al., 1998; Biederer et al., 2002) were unchanged, suggesting that Mints are not required for the normal maintenance of APP levels or its normal localization (Fig. 5B).
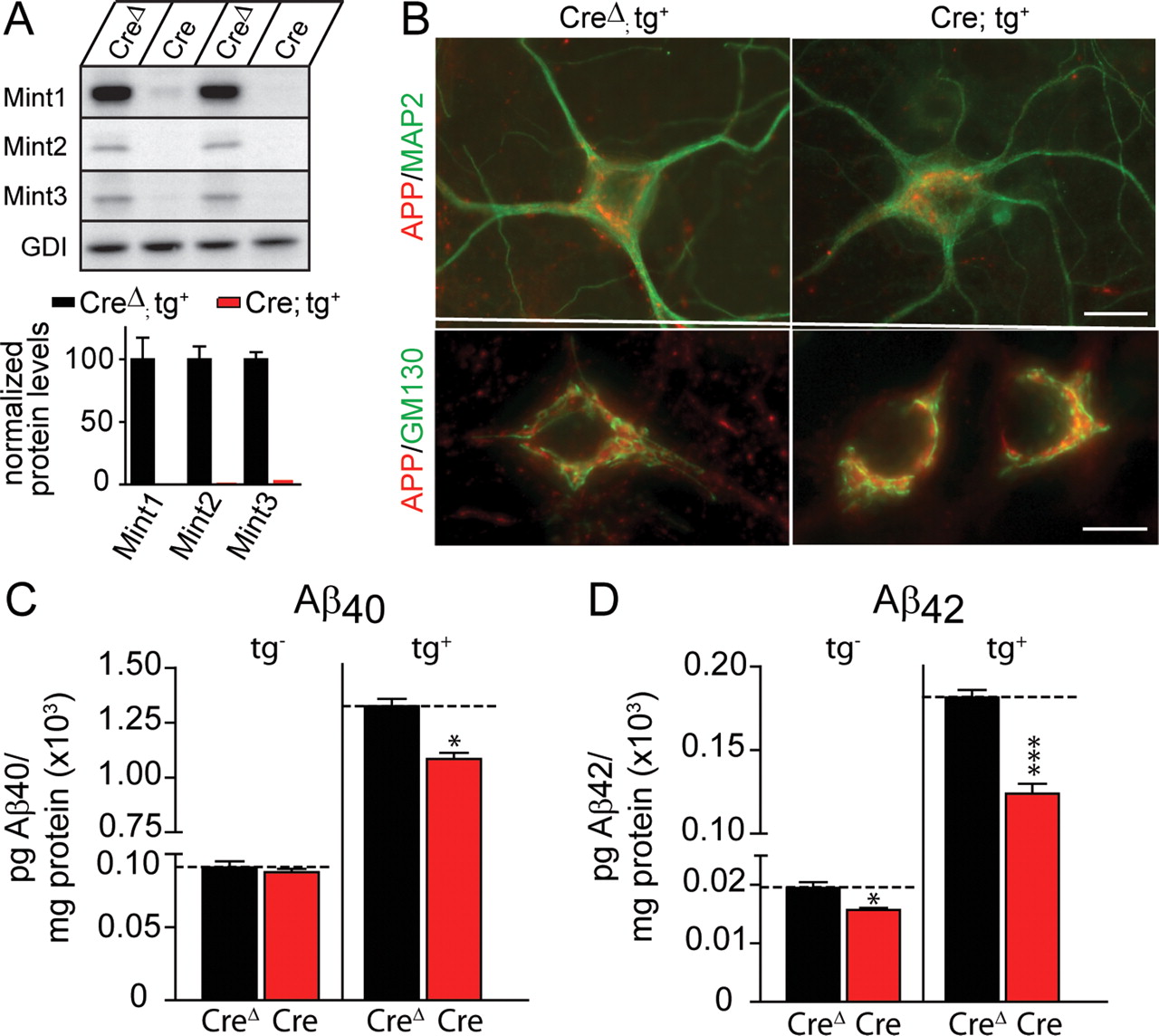
Conditional deletion of all three Mint proteins in neuronal cultures decreases Aβ levels. Neurons were cultured from Mint triple-floxed newborn pups carrying one allele of double transgene (tg+) for mutant human APP and presenilin-1 and infected with either lentiviral mutant (CreΔ) or wild-type cre recombinase (Cre). A, Representative immunoblots (top) and quantitations (bottom) for Mint1, Mint2, and Mint3 proteins in cultured neurons. Neuronal cultures infected with mutant CreΔ virus showed normal expression of all three Mint proteins, whereas Cre efficiently deleted all three Mint protein expression. B, Double immunofluorescence analysis of APP and MAP2 (top), or APP and Golgi marker GM130 (bottom) in cultured neurons infected with CreΔ or Cre displayed normal expression and localization (scale bars: top, 15 μm; bottom, 10 μm). C, D, Conditioned medium from Mint-deficient neurons infected with Cre that express transgenic mutant human APP and presenilin-1 (tg+) showed a significant decrease in Aβ40 and Aβ42 levels compared with control neurons infected with CreΔ measured by ELISA. A small significant decrease in Aβ42 levels but not Aβ40 was observed in Mint-deficient neurons lacking the transgene (tg−). Data shown are means ± SEMs (*p < 0.05, ***p < 0.001).
We next measured Aβ peptide levels to determine whether deleting Mint proteins in cultured neurons decreased Aβ levels similar to the intact mice. Indeed, we found that both Aβ40 and Aβ42 levels were significantly decreased in the conditioned medium of Mint-deficient neurons that contained the single allele of the double transgene (Fig. 5C,D). When we analyzed endogenous Aβ levels in neurons lacking the double transgene, we observed that Aβ42 levels were also significantly lowered by deletion of Mints, whereas the Aβ40 levels, which are extremely low in mice, were not measurably changed (Fig. 5C,D).
Acute deletion of Mints alters APP cleavage
We next examined whether deletions of Mints alter the extracellular or the intramembranous cleavage of APP. To address this, we measured the levels of APP-CTFs in cultured neurons containing or lacking Mints, before or after addition of the γ-secretase inhibitor DAPT. An impairment of β-cleavage should decrease the accumulation of APP-CTFs after γ-secretase inhibition by DAPT because less APP-CTFs are produced. In contrast, an activation of α-cleavage or an impairment of γ-cleavage should cause either no change or an increase in the accumulation of APP-CTFs after γ-secretase inhibition.
Using antibodies to the C terminus of APP, three APP-CTF species can be detected by immunoblot analysis: an 83 amino acid fragment produced by α-cleavage (C83), and 89 and 99 amino acid fragments produced by β-cleavage (C89 and C99) (Haass and De Strooper, 1999). However, the C83 and C89 bands are difficult to resolve, allowing us only to separately quantify the C99 and the combined C83 and C89 APP-CTFs in DAPT-treated cultured neurons (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). We measured the accumulation of these APP-CTFs as a function of time after addition of the γ-secretase inhibitory DAPT (Fig. 6). Without DAPT treatment, no significant Mint-dependent change in the levels of any APP-CTF was detected in neuronal cultures even by immunoprecipitations followed by quantitative immunoblotting, and it would have been difficult to detect a small change (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). After addition of DAPT, C99 levels increased linearly for 12 h in wild-type (CreΔ) and Mint-deficient (Cre) neurons, after which C99 levels saturated, whereas C83+C89 levels continued to rise (Fig. 6A,B). Strikingly, deletion of Mints significantly decreased the accumulation of C99 and of the combined C83+C89 APP-CTFs (Fig. 6A,B). Because APP-CTFs are highly phosphorylated in neurons, we treated immunoprecipitates of APP-CTFs with λ protein phosphatase and blot with 6E10 antibody to identify C99 species. After DAPT treatment, deletion of Mints significantly decreased the accumulation of both phosphorylated and unphosphorylated forms of C99 (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The decrease in APP-CTF production produced by deletion of Mints was specific because we observed no Mint-dependent change in APP levels as a function of DAPT treatment (Fig. 6C). The small decrease in APP levels that we observed at 3 and 6 h after DAPT treatment might be attributable to a decrease in APP half-life while increasing α and β processing and/or accumulation of APP. The decrease in APP-CTF production in Mint-deficient neurons suggests that less of the N-terminal ectodomain of APP (sAPP) should be secreted. Indeed, sAPP levels, as measured by immunoblotting with three different antibodies (monoclonal antibody 6E10 that recognizes soluble sAPP, and polyclonal anti-sAPPα and sAPPβ antibodies), were decreased in conditioned medium from Mint-deficient neurons (Fig. 6D). Consistent with these results, we also examined C99 and sAPP levels in the mouse brain homogenates of brain-specific Mint1 and 2 knock-out mice. Immunoprecipitation with a specific APP C-terminus antibody (U955) and immunoblotting with 6E10 antibody that recognizes the C99 fragment, only Mint2 knock-outs had a significant decrease in C99 levels (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Correspondingly, we also detected a small decrease in sAPP levels in transgenic mice lacking either Mint1 or Mint2 (data not shown).

Conditional deletion of all three Mint proteins in neuronal cultures decreases the accumulation of APP-CTFs induced with the γ-secretase inhibitor DAPT. Quantitative analysis of APP-CTF and full-length APP levels in neurons containing or lacking Mints as a function of time after addition of γ-secretase inhibitory DAPT. A, Top, Representative immunoblots for APP-CTFs with GDI, a ubiquitously expressed protein used as internal loading control from control neurons infected with CreΔ and Mint-deficient neurons infected with Cre after DAPT treatment for 0, 3, 6, 12, and 24 h. Bottom, Summary graph of APP-C99 levels as relative values normalized to GDI. Mint-deficient neurons (Cre) carrying the transgene (tg+) showed a significant decrease in accumulation of APP-C99 levels induced with DAPT compared with control neurons (CreΔ). B, Summary graph showing total APP-C83 and APP-C89 levels of CreΔ and Cre after DAPT treatment. C, Representative immunoblots and quantitative analysis of full-length APP levels from cultured neurons expressing transgenic mutant human APP and presenilin (tg+) containing (CreΔ) or lacking Mint1, Mint2, and Mint3 (Cre). Bottom, Plot showed no changes in full-length APP levels. D, Quantitative analysis of secreted sAPP levels from conditioned media of cultured neurons expressing transgenic mutant human APP and presenilin (tg+) containing (CreΔ) or lacking all three Mint proteins (Cre) and probed with anti-6E10, anti-sAPPα, and anti-sAPPβ antibodies and normalized against total protein and plotted as a percentage of wild-type control (CreΔ). All summary graphs depict means ± SEMs (n = 3 independent cultures performed in triplicates). *p < 0.05, **p < 0.01, ***p < 0.001.
The decrease in the accumulation of APP-CTFs after DAPT treatment, with a corresponding decrease in sAPP secretion, demonstrates that deletion of Mints impairs extracellular β- and possibly α-cleavage of APP. However, these experiments do not exclude the possibility that deletion of Mints simultaneously impair γ-cleavage. To test this possibility, we measured the kinetics of accumulated APP-CTFs following DAPT treatment that are cleaved by γ-secretase after DAPT was washed out (Fig. 7). When plotted as absolute values [the ratio of C99 to GDP dissociation inhibitor (GDI) or C83+C89 to GDI as internal control], the deletion of Mints dramatically decreased all APP-CTF levels as expected (Fig. 7A,B). However, when we normalized for the peak CTF levels to allow for comparison in kinetics of decay, we found that deletion of Mints had no significant effect on the cleavage of APP-CTFs by γ-secretase (Fig. 7C,D). Moreover, no significant effect on APP levels was observed (Fig. 7E). Thus, Mint proteins are unlikely to affect γ-secretase activity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Deletion of Mints does not alter γ-secretase cleavage of APP-CTFs. A, Representative immunoblot of cultured neurons pretreated with 2 μm DAPT for 24 h to cause the accumulation of APP-CTFs. DAPT was then washed out with PBS and replaced with neuronal growth media for the duration of the experiment in which lysates were collected at 2, 4, 6, 12, and 24 h after DAPT washout. Plot shows the quantitation of APP C99 relative values normalized for GDI. B, Summary graph showing total APP-C83 and APP-C89 levels of CreΔ and Cre after DAPT washout. C, D, APP-CTF levels normalized to the peak value to compare the decay kinetics as a function of time after DAPT washout. E, Representative immunoblots and quantitative analysis of full-length APP levels from same experiment of cultured neurons expressing transgenic mutant human APP and presenilin (tg+) containing (CreΔ) or lacking Mint1, Mint2, and Mint3 (Cre) after DAPT washout. Bottom, Plot showed no changes in full-length APP levels. All summary graphs depict means ± SEMs (n = 3 independent cultures performed in triplicates). *p < 0.05, **p < 0.01.
Discussion
In the present study, we have made three principal observations about the in vivo function of Mint/X11 proteins in APP cleavage that may be important for our thinking of the biology of Mint/X11 proteins and about the pathogenesis of AD: (1) deletion of Mint proteins decreases plaque formation at 6–9 months of age with Mint2 deletion the most potent suppressor of Aβ production, most likely attributable to the relative expression levels of these proteins in the brain (Figs. 1⇑⇑–4). (2) Deletion of Mints decreases the extracellular cleavage of APP by β-secretase and possibly α-secretase in neurons but does not appear to alter intramembranous cleavage of the APP-CTF by γ-secretase (Figs. 6, 7). The observed effects are primarily mediated by β-secretase, because we measured in our experiments changes in Aβ peptides that are produced by β-secretase, and because C99 is affected more strongly than the combined C83 and C89 APP-CTFs which are produced by α- and β-cleavage, respectively. (3) Deletion of Mints does not significantly alter γ-secretase activity because it does not dramatically increase APP-CTF levels in cultured neurons or adult Mint knock-out mice (Fig. 6; supplemental Figs. 6, 7, available at www.jneurosci.org as supplemental material), and it does not change the kinetics of APP-CTF cleavage (Fig. 7).
Our results differ from earlier conclusions that suggested that Mints regulate γ-secretase (supplemental Table 1, available at www.jneurosci.org as supplemental material) and demonstrates a role for Mint proteins as intracellular adaptor proteins in regulating extracellular cleavage reaction. However, it is important to note that because β-cleavage of APP is predominantly intracellular in endosomes that perhaps Mint proteins may indeed affect intraendosomal pathway in APP processing. The following conditions of our experiments ensure the specificity of our results. First, different from earlier studies, we used both acute and constitutive deletions of Mints/X11s. Second, we used two different mouse models of AD to investigate the effects of Mint deletions. Third, we studied both intact brains and cultured neurons. Fourth, we measured a variety of parameters using a panoply of different antibodies. Last, we tested three different Mint isoforms in independent lines of mice, excluding genetic position effects.
Different from our results, previous studies suggested that Mints control γ-cleavage of APP-CTFs. Specifically, overexpression studies of Mints/X11suggested that overexpressed Mints inhibit γ-cleavage of APP-CTFs (supplemental Table 1, available at www.jneurosci.org as supplemental material), whereas RNAi knockdowns indicated that decreases in Mint1 or Mint2 levels also inhibit γ-cleavage of APP-CTFs and thereby decrease Aβ production (Xie et al., 2005). It is difficult to understand how overexpression and knockdowns of a protein has the same effects. Overexpressed proteins often assume functions incidental to those of the endogenous proteins, whereas RNAi occasionally exhibits off-target effects that need to be control for by rescue experiments. Furthermore, because γ-secretase has a major developmental role via Notch cleavage, any change in γ-secretase by Mint/X11 deletions should have led to a developmental phenotype that was not observed in Mint-deficient mice (Ho et al., 2003, 2006). Finally, two Mint1 and Mint2 knock-out studies (Sano et al., 2006; Saito et al., 2008) found an increase in endogenous mouse APP-CTFs and Aβ with a decrease in secreted APPs, changes that are difficult to reconcile but possibly attributable to strain differences and genetic background between our knock-out mice. Our data currently points that Mint isoforms may indeed perform distinct functions in APP processing in brain attributable to their differential expression, but not because of intrinsic differences in their functions. Mint1 is expressed mostly in inhibitory neurons, whereas Mint2 is expressed higher in excitatory neurons preferentially in the pyramidal neurons of the hippocampus, and deletion of Mint1 and Mint2 have been shown to alter inhibitory and excitatory synaptic transmission, respectively (Ho et al., 2003, 2006). It is possible that deletion of Mint proteins affects synaptic transmission thereby modulating the formation and secretion of amyloid peptides because neural activity has been shown to control APP processing (Kamenetz et al., 2003).
What is the mechanism by which intracellular Mints modulate extracellular APP cleavage? The phenotype of the Mint knock-outs described here is most consistent with an effect of Mints on APP trafficking, although the steady-state distribution of APP and cell surface APP levels were not changed by deletion of Mints (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). A role for Mints in protein trafficking is supported by two interactions that have been described previously. First, their binding to Munc18 proteins that are involved in plasma membrane fusion reactions, an interaction that is consistent with our previous Mint knock-out analyses (Ho et al., 2003, 2006), and, second, their binding to Arfs (ADP-ribosylation factors) that are particularly important for Golgi membrane trafficking (Hill et al., 2003). It also agrees with the colocalization of Mints with APP in the trans-Golgi network of neurons (Borg et al., 1996; Sastre et al., 1998; Biederer et al., 2002). In neurons, the majority of APP is axonally transported to the synaptic terminal, and during endocytosis of cell surface, APP directs it toward an amyloidogenic processing pathway (Nordstedt et al., 1993; Ikin et al., 1996; Marquez-Sterling et al., 1997). However, the lack of APP enrichment in presynaptic terminals indicates that APP is not stably retained in the terminal. Therefore, it is conceivable that Mint proteins could alter APP trafficking either by altering APP secretory pathway or endocytotic trafficking of reinternalized APP thereby affecting APP processing, but further studies will be necessary to investigate this possibility.
The striking effects of Mint deletions on Aβ production and Aβ plaque formation suggest that even small changes in the expression of Mints, when present chronically, could have dramatic consequences for the production of Aβ, the deposition of Aβ plaques, and the development of AD. Thus, increases in Mint expression, even if small, may be a risk factor for AD, and a possible therapeutic strategy to decrease Aβ production could be to interfere with the Mint/APP interaction. If a pharmacological avenue to selectively block the Mint/APP interaction could be found, such a strategy would be attractive because of the relatively mild effects of deletions of either APP or individual Mints, and such a selective block would presumably not impair the other functions of Mints and APP.
Footnotes
- Received June 2, 2008.
- Revision received October 15, 2008.
- Accepted November 18, 2008.
-
This work was supported by National Institutes of Health Grants R01-MH069585 (to T.C.S.) and K01-AG027311 (to A.H.). We are grateful to I. Kornblum, A. Roth, and E. Borowicz for excellent technical assistance and to L. Fan and J. Mitchell for animal care. We thank Dr. J. Shen (Center for Neurological Disease, Brigham Women's Hospital at Harvard Medical School) for her helpful advice; Dr. J. Herz for generous gifts of antibodies; and Drs. H. Li, J. Burre, A. Boucard, and M. Khvochtchev for their assistance with this work.
- Correspondence should be addressed to either of the following at their present addresses: Angela Ho, Department of Biology, Boston University, 5 Cummington Street, Boston, MA 02215, Email: aho1{at}bu.edu; or Thomas C. Südhof, Institutes of Medicine, Department of Molecular and Cellular Physiology, and Howard Hughes Medical Institute, Stanford University, 1050 Arastradero Road, Palo Alto, CA 94304-5543, E-mail: Email: tcs1{at}stanford.edu
- Copyright © 2008 Society for Neuroscience 0270-6474/08/2814392-09$15.00/0