

Abstract
Experience-dependent changes in the structure of dendritic spines may contribute to learning and memory. Encoded by three genes, the Shank family of postsynaptic scaffold proteins are abundant and enriched in the postsynaptic density (PSD) of central excitatory synapses. When expressed in cultured hippocampal neurons, Shank promotes the maturation and enlargement of dendritic spines. Recently, Shank3 has been genetically implicated in human autism, suggesting an important role for Shank proteins in normal cognitive development. Here, we report the phenotype of Shank1 knock-out mice. Shank1 mutants showed altered PSD protein composition; reduced size of dendritic spines; smaller, thinner PSDs; and weaker basal synaptic transmission. Standard measures of synaptic plasticity were normal. Behaviorally, they had increased anxiety-related behavior and impaired contextual fear memory. Remarkably, Shank1-deficient mice displayed enhanced performance in a spatial learning task; however, their long-term memory retention in this task was impaired. These results affirm the importance of Shank1 for synapse structure and function in vivo, and they highlight a differential role for Shank1 in specific cognitive processes, a feature that may be relevant to human autism spectrum disorders.
Introduction
Activity-induced changes in synaptic structure and efficacy have been proposed to underlie learning and memory. Dendritic spines, specialized protrusions that form the postsynaptic compartment of most excitatory synapses, can undergo bidirectional morphological changes in response to neuronal activity (Matsuzaki et al., 2004; Nagerl et al., 2004; Okamoto et al., 2004; Zhou et al., 2004; Tada and Sheng, 2006). Interestingly, the size and shape of individual dendritic spines seem to correlate with their capacity for structural change (Kasai et al., 2003; Matsuzaki et al., 2004). It has been proposed that small, thin spines are specialized for plasticity, whereas large, stable spines serve as “memory” structures (Kasai et al., 2003); however, there is no direct evidence to support this idea.
At the interface between the postsynaptic membrane and the spine cytoplasm, the postsynaptic density (PSD) consists of a complex network of proteins that link glutamate receptors to the actin cytoskeleton and postsynaptic signaling pathways (Kennedy, 2000; Sheng and Kim, 2002; Sheng and Hoogenraad, 2007). Encoded by three genes (Lim et al., 1999; Boeckers et al., 2002; Bockers et al., 2004), the Shank family of scaffold proteins (also known as ProSAP, SSTRIP, cortBP, Synamon, and Spank) are abundant in the PSD (Cheng et al., 2006). When overexpressed in cultured hippocampal neurons, Shank proteins strongly promote the enlargement of dendritic spines, particularly the spine heads (Sala et al., 2001).
Through multiple protein interaction domains, Shank serves as a scaffold for a variety of postsynaptic molecules, including the guanylate kinase-associated protein (GKAP) family of PSD proteins (Boeckers et al., 1999; Naisbitt et al., 1999), Homer (Tu et al., 1999), and several actin regulatory proteins (Du et al., 1998; Soltau et al., 2002; Park et al., 2003; Qualmann et al., 2004). Lying at the cytoplasmic face of the PSD (Valtschanoff and Weinberg, 2001) and interfacing between glutamate receptor complexes and actin regulatory proteins, Shank appears to be well situated to play a role in spine morphogenesis and its regulation by synaptic activity (Sheng and Kim, 2000). Interestingly, mutations in the human Shank3 gene have been linked to autism spectrum disorders (ASDs) (Durand et al., 2007), suggesting that Shank family proteins may play an important role in human cognitive development.
To investigate the function of Shank in vivo, we disrupted in mice the Shank1 gene, which is expressed exclusively in the brain. Shank1−/− mice showed altered protein composition of the PSD and smaller dendritic spines and synapses, which correlated with a weakening of basal synaptic transmission. These results demonstrate that Shank1 is important in vivo for regulating dendritic spine morphology and synaptic strength. Behaviorally, the Shank1−/− mutant mice were defective in hippocampal-dependent contextual fear memory, despite normal hippocampal long-term potentiation (LTP), long-term depression (LTD), and late-phase LTP (L-LTP). Surprisingly, however, they demonstrated enhancement in spatial learning in the eight-arm radial maze, but this was associated with impaired long-term retention of that information. We suggest that Shank1 promotes the maturation of smaller, more plastic spines into larger, more stable spines, a cellular process required for normal cognitive development.
Materials and Methods
Generation of Shank1 knock-out mice.
An ∼110 kb bacterial artificial chromosome (BAC) clone containing the complete genomic sequence of mouse Shank1 was isolated from a high-density colony array of mouse C57BL/6 genomic BAC clones (Genome Systems, St. Louis, MO). To construct the Shank1 targeting vector, a 14 kb EcoR1-BamH1 Shank1 genomic fragment was cloned into pBluescript II SK vector. A 2 kb BstXI-HindIII fragment containing exons 14 and 15 encoding almost the entire PDZ domain was then replaced by the PGK-neo cassette in the same transcriptional orientation as Shank1. A thymidine kinase cassette was added at the end of the long arm allowing for double selection. The targeting vector was linearized with NotI and electroporated into J1 embryonic stem (ES) cells, which were subsequently selected in geneticin (G418) and 1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil (FIAU) containing medium as described previously (Li et al., 1992). Genomic DNA isolated from G418- and FIAU-resistant colonies was digested with BamHI or EcoRV and analyzed by Southern blotting. Two independent clones (362R and 388R) from 179 drug-resistant colonies were isolated. Chimeric mice were produced by injecting targeted ES cell clones into C57BL/6 blastocysts, and heterozygous offspring were backcrossed into C57BL/6 and 129SvJae strains (gift from R. Jaenisch, Massachusetts Institute of Technology, Cambridge, MA). The animals used for experiments in this study were in a 129SvJae/C57BL/6 hybrid genetic background. For PCR genotyping, primer pairs amplifying a portion of the deleted region (wild-type locus) or the neo cassette (mutant locus) were used.
Antibodies and biochemistry.
The rabbit polyclonal Shank1-specific antibody (1356) and the “pan-Shank” antibody (3856) have been described previously (Lim et al., 1999). Other antibodies used in this study include rabbit antibodies against GKAP (Kim et al., 1997), Homer (gift from E. Kim, Korea Advanced Institute of Science and Technology, Daejeon, Korea), glutamate receptor-interacting protein (GRIP) (Wyszynski et al., 1999), glutamate receptor 1 (GluR1) (Oncogene Research, San Diego, CA), GluR2/3 (Millipore, Bedford, MA), NR2B (Sheng et al., 1994), mGluR5 (gift from R. Huganir, Johns Hopkins University, Baltimore, MD), β-PIX (β-p21-activated kinase-interacting exchange factor) (Millipore), and α-calcium calmodulin-dependent protein kinase II (α-CaMKII) (Sigma, St. Louis, MO); and monoclonal antibodies against PSD-95 (K28/43; gift from J. Trimmer, University of California at Davis, Davis, CA), NR1, insulin receptor substrate p53 (gift from S. H. Ong and T. Pawson, University of Toronto, Toronto, Ontario, Canada), cortactin (4F11; gift from T. Parsons, University of Virginia, Charlottesville, VA), and Bassoon (Stressgen, Ann Arbor, MI). The Triton-extracted PSD fraction (PSDI) was purified from the forebrains of individual 2- to 3-month-old mice, as described previously (Cho et al., 1992).
Neuron culture and immunocytochemistry.
Primary hippocampal cultures were prepared from the brains of individual mouse embryos at embryonic day 18 (E18) to E19 as described previously (Brewer et al., 1993) with some modifications. Each culture was derived from a single embryo to ensure a genotypically pure neuronal population. Hippocampi from each embryo were dissected and digested with trypsin, which was inactivated by addition of fetal calf serum. Tissue was pelleted by brief centrifugation and then dissociated in Neurobasal/B27 medium. Cells were plated onto coverslips coated with poly-d-lysine (30 μg/ml) and laminin (2 μg/ml) at a density of 65,000 cells/well. Genomic DNA was isolated from embryo tails and genotyped by PCR. For immunocytochemistry, neurons were fixed after 18–19 d in vitro (DIV) briefly with 1% formaldehyde and 4% sucrose, followed by methanol at −20°C. Primary and secondary antibodies were applied in GDB buffer (30 mm phosphate buffer, pH 7.4, containing 0.2% gelatin, 0.5% Triton X-100, and 0.45 m NaCl).
Confocal images were obtained using a 63× objective (numerical aperture, 1.4) with sequential acquisition settings of 1024 × 1024 pixels. Each image was a z-series projection of ∼7–15 images, each averaged three to four times and taken at 0.4–0.7 μm depth intervals. Morphometric analysis and quantification were performed using MetaMorph software (Universal Imaging Corporation, West Chester, PA) by investigators who were blind to genotype and experimental manipulation. Colocalization was measured by color-separating PSD-95 and pan-Shank channels, manually setting a threshold level for each channel (identical for each neuron), and then determining the overlapping puncta by manual inspection.
Morphological analysis of spines.
Brains from adult male mice were fixed with 4% paraformaldehyde by transcardial perfusion. Vibratome sections of 50 μm were prepared and stained with cresyl violet. To examine dendritic spine number and morphology, 200 μm coronal hippocampal sections were subjected to “Diolistic” labeling, as described previously (Gan et al., 2000; Grutzendler et al., 2003). Tungsten particles (1.7 μm; Bio-Rad, Hercules, CA) coated with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate crystals (DiI; Invitrogen, Eugene, OR) were propelled into fixed tissue with biolistic Helios gene gun (Bio-Rad) through a membrane filter with a 3 μm pore size (Falcon 3092; BD Biosciences, Franklin Lakes, NJ). Sections were postfixed in 4% paraformaldehyde overnight and then mounted for confocal microscopy. A Zeiss (Thornwood, NY) 100× objective was used to obtain an image series of sections (1024 × 1024 pixels, 0.09 μm/pixel) at 0.5 μm intervals, with close attention given to achieving similar fluorescence intensity across different regions and imaging sessions. All imaging and analysis was performed in blinded manner. For quantification, 30–50 μm dendritic segments were identified, and protrusions were measured as described previously (Sala et al., 2001).
Electron microscopy.
Mice were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused through the heart with heparinized saline followed by 100 ml of a mixture of 2% paraformaldehyde and 2% glutaraldehyde in phosphate buffer (0.1 m, pH 7.4). Brains were postfixed for 2 h in the same fixative, sectioned on a Vibratome at 40 μm, and processed for osmium-free embedment (Phend et al., 1995). Thin sections (80 nm) from CA1 hippocampus were cut and collected on 300 mesh uncoated copper grids and counterstained with uranyl acetate and Sato's lead. Grids were examined on a Philips Tecnai 12 electron microscope at 80 kV accelerating voltage. For quantitative study, digital images at 40,000× original magnification were acquired. All measurements were made independently by two investigators “blind” to the genetic makeup of the animal. For each grid studied, only the first 20 synapses that had clearly visible synaptic structures (presynaptic membrane, synaptic cleft, postsynaptic membrane, and PSD) were considered. In perforated synapses, the entire length of the synapse, including all fragments with PSD and all perforations, was measured as a single value.
Electrophysiology.
Transverse hippocampal slices (400 μm thickness) were prepared from 3- to 5-week-old mice in ice-cold dissection buffer (in mm: 238 sucrose, 2.5 KCl, 1 CaCl2, 5 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, gassed with 5% CO2/95% O2, pH 7.4). Slices were incubated in an interface incubation chamber containing extracellular artificial CSF (aCSF; in mm: 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, gassed with 5% CO2/95% O2, pH 7.4) and allowed to recover for 30 min at 30°C and then maintained at room temperature (24–26°C) for at least 2 h. Slices were then transferred to a submerged recording chamber and continuously perfused with aCSF. The recording pipettes (1.5–2 MΩ, 3 m NaCl) for field recording were placed in the stratum radiatum. The tungsten bipolar electrode (Frederick Haer Company, Bowdoinham, ME) was placed in the stratum radiatum, and the Schaffer collateral/commissural fibers were stimulated at 0.1 Hz. For LTP or LTD experiments, the stimulus strength was adjusted to evoke an EPSP of slope value between 0.12 and 0.15 or 0.15 and 0.19 mV/ms, respectively. The patch recording pipettes (2–5 MΩ) for whole-cell recording were filled with internal solution containing the following (in mm): 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 adenosine triphosphate disodium salt, 0.4 guanosine triphosphate trisodium salt, 10 sodium phosphocreatine, and 0.6 EGTA, pH 7.25, with CsOH. Picrotoxin (0.15 mm; Sigma) was dissolved in aCSF to block GABAA receptor-mediated synaptic transmission for whole-cell patch-clamp recordings. AMPA receptor-mediated EPSCs were recorded at −70 mV, and NMDA receptor-mediated EPSCs were recorded at +40 mV with the same stimulus strength in the presence of 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline-7-sulfonamide (0.01 mm; Tocris Cookson, Ballwin, MO). Paired-pulse facilitation (PPF) of AMPA receptor-mediated EPSCs was measured at −70 mV by delivering two afferent stimulations (50 ms interstimulus interval). All experiments were performed at 28°C, using a temperature controller (TC-324B; Warner Instruments, Hamden, CT). Results are reported as mean ± SEM. Statistical significance was evaluated by Student's t test. Statistical significance was set at p < 0.05. Miniature EPSCs (mEPSCs) were recorded in the presence of 0.001 mm tetrodotoxin (Calbiochem, La Jolla, CA) and were analyzed using the Mini Analysis software (Synaptosoft, Decatur, GA). Three hundred mEPSCs were sampled from each experiment; events >5 pA were analyzed. All experiments and the analysis of data were performed in a blind manner. Recordings were performed using a MultiClamp 700B amplifier and Digidata 1320B, and data were acquired and analyzed using Clampex 9.2 and Clampfit 9.2 (Molecular Devices, Union City, CA).
Behavioral analysis.
Mice were backcrossed at least five to six generations into the C57BL/6 genetic background. Wild-type and Shank1−/− F1 offspring from intercrosses between these animals and 129/SvJae heterozygotes were then used for behavioral experiments to minimize the potential effects of neighboring loci. All mice were male littermates, 3–5 months of age at the start of behavioral testing. The generation and maintenance of mice and all experimental procedures were performed in compliance with the National Institutes of Health, and all experiments were conducted and analyzed by investigators who were “blind” to the genotype. Statistical analyses [two-way ANOVA, Kolmogorov–Smirnov (K–S) t test] were performed using GraphPad Prism and StatsDirect software (GraphPad Software, San Diego, CA). Open-field testing, accelerating Rotarod testing, and the light–dark transition test were performed as described previously (Miyakawa et al., 2001).
Contextual and cued fear conditioning.
Mice were trained in a standard fear conditioning apparatus (Med Associates, Georgia, VT). They were allowed to explore freely for 3 min. A 30 s 2800 Hz pure tone (conditioned stimulus) was then delivered, coterminating with a 1 s, 0.75 mA footshock (unconditioned stimulus). Two additional conditioned stimulus–unconditioned stimulus pairs were presented with 1 min interstimulus intervals. Context testing was conducted after retention delays of 1 and 24 h in the same chamber. Cued testing with altered context was performed 48 h after conditioning. White opaque plastic was placed onto the floor and arched inside the chamber, and vanilla extract odor was added to further change the context. After a 3 min baseline period, a tone identical to the original conditioning was played for 2 min. Freezing was scored and analyzed automatically using FreezeFrame (Actimetrics, Wilmette, IL), with thresholds set to give agreement with blinded human observation.
Eight-arm radial maze.
The eight-arm radial maze test was conducted as described previously (Miyakawa et al., 2001), with minor modifications. One to 2 weeks before training, animals were food restricted until body weight was reduced to 80–85%. The maze was surrounded by prominent distal cues. During acquisition training, two of eight arms were baited with one hidden food pellet each, separated by 135° (either arms 1 of 6 or arms 2 of 5). The trial was terminated once both pellets were found and consumed or after a maximum of 20 min had elapsed. Mice were confined to the center platform for 5 s after each arm choice by automatically controlled guillotine doors. A “reference” memory error was scored each time a mouse entered one of the six unbaited arms. A “working” memory error was scored when a mouse revisited an arm that it had already entered during the same training trial. The number of reference or working memory errors was scored for each trial, and the average number of errors per trial was determined for each mouse over a block of four trials (block 1, trials 1–4; block 2, trials 5–8; etc.). For the original training protocol, two separate cohorts of mice were studied (cohort 1, n = 7 for each genotype; cohort 2, +/+, n = 8; −/−, n = 7). Similar results were obtained with each cohort, and data were pooled for subsequent analysis. In the standard protocol, animals were trained with one trial per day for 14 consecutive days and then two trials per day (trials 15–40) and four trials per day (trials 41–84). After completion of acquisition training, the mice were returned to their home cage for 4 weeks without exposure to the maze; mice were again food restricted to the same target body weight before retraining. After 28 d of “rest,” the mice were retested with the same bait configuration, receiving four trials on two consecutive days. For reversal training, positions of the baits were switched by 180° (mice previously baited with arm 1 of 6 were changed to arms 2 of 5 and vice versa). Reversal training was performed initially with two trials per day (trials 1–8) and then increased to four trials per day (trials 9–36). For the intensive training protocol, two additional cohorts of mice (cohort 1, n = 9 for each genotype; cohort 2, +/+, n = 8; −/−, n = 9) received training on five successive days, followed by two off days. They received one trial per day for four training days, increased to two trials per day (trials 5–20) and then four trials per day (trials 21–60). The first cohort was retested in the original bait configuration after 28 d. Data acquisition and control of guillotine doors were accomplished with NIH Image RM software.
Results
Generation of Shank1 mutant mice
To generate Shank1 knock-out mice, we constructed a gene-targeting vector to disrupt exons 14 and 15 of the Shank1 gene by homologous recombination (Fig. 1A,B). These exons encode a highly conserved region of Shank1, including the PDZ domain that is required for synaptic targeting of the protein (Sala et al., 2001). Two independent ES cell lines were identified by Southern blotting and used to generate chimeras that transmitted the mutation into the germ line (Fig. 1C). Intercrosses of Shank1 heterozygous mice yielded offspring at the expected Mendelian ratio (representative litter shown in Fig. 1D). Shank1−/− mice were grossly indistinguishable from wild-type littermates in their home cage and showed similar survival.

Generation and characterization of Shank1 mutant mice. A, Domain structure of Shank1, showing ankyrin repeats (Ank), SH3 and PDZ domains, proline-rich region (Pro), and sterile α motif (SAM) domain. B, Schematic diagram of the Shank1 gene locus, the targeting vector, and the mutant allele after homologous recombination. The neomycin resistance cassette (NEO) replaces the exons coding the PDZ domain (white box) and their intervening introns. BamHI and EcoRV probes used for Southern analysis and PCR primers are shown. Restriction sites: B, BamHI; R, EcoRI; RV, EcoRV. C, Southern blot analysis of wild-type (+/+) and two independent clones of targeted ES cells (362 and 388). D, PCR genotype analysis of a representative litter from heterozygote intercross. The upper 517 bp band corresponds to amplified product of wild-type allele, and 282 bp band corresponds to the PCR product from the neo cassette. E, Immunoblot of forebrain membrane fractions from wild-type (+/+), heterozygous (+/−), and homozygous Shank1−/− mice, probed with Shank1-specific antibody 1356 (left) or pan-Shank antibody 3856 (right). F, Cresyl violet staining of coronal sections of hippocampus from wild-type and Shank1−/− mice.
To confirm loss of Shank1 protein expression, we used a Shank1-specific peptide antibody (1356) raised against amino acids 425–440 of Shank1 (Lim et al., 1999), which lie N-terminal to the targeted deletion. Immunoblotting of wild-type mouse forebrain extracts with antibody 1356 showed a major band of ∼240 kDa and several smaller bands that presumably represent alternative splicing and/or degradation products; all these bands decreased in the heterozygote and were eliminated in the homozygous brain (Fig. 1E, left). No bands were seen even in the low molecular weight region of the gel (data not shown). Immunoblotting with pan-Shank antibody 3856, which recognizes Shank2 and Shank3 as well as Shank1 (Lim et al., 1999), confirmed the loss of the 240 kDa and lower bands corresponding to Shank1 polypeptides. However, an additional set of proteins recognized by 3856 (molecular weight, ∼160–200 kDa) were unchanged in Shank1 knock-outs; these presumably correspond to Shank2 and Shank3 gene products (Fig. 1E, right). These data indicate that the gene targeting resulted in a Shank1 null mutant, with no compensatory increase in Shank2 and Shank3.
We detected no gross abnormalities in the size or histological structure of the brain (including cortex, hippocampus, and cerebellum), based on Nissl-stained sections (Fig. 1F) (data not shown).
Altered PSD protein composition in Shank1−/− brain
Via its multiple protein–protein interactions, the Shank scaffold has been proposed to play an important role in assembling the PSD (Sheng and Kim, 2000). We therefore examined the effect of Shank1 disruption on the protein composition of PSD fractions purified from the forebrains of wild-type and Shank1−/− mice. We used PSD fractions extracted once with Triton X-100 (PSDI), because the twice-extracted (PSDII) or Sarkosyl-extracted (PSDIII) fractions did not yield sufficient protein from individual mice for additional analysis. One-dimensional SDS-PAGE (Fig. 2A) and two-dimensional gel electrophoresis (data not shown) of purified PSDs revealed no obvious change in the overall protein patterns. By immunoblotting, the Shank1-specific 1356 antibody confirmed the absence of Shank1 protein in the PSD fraction of Shank1 knock-out brains (Fig. 2B, top left). The pan-Shank 3856 antibody showed ∼40% reduction in total Shank immunoreactivity in PSDs from Shank1−/− mice (Fig. 2B,C). Interestingly, the ∼240 kDa Shank1 protein is more prominent in the PSD preparation compared with crude membrane extracts [compare “Shank (total)” in Figs. 1E, 2B], suggesting that Shank1 may be more highly enriched in the PSD than Shank2/3.
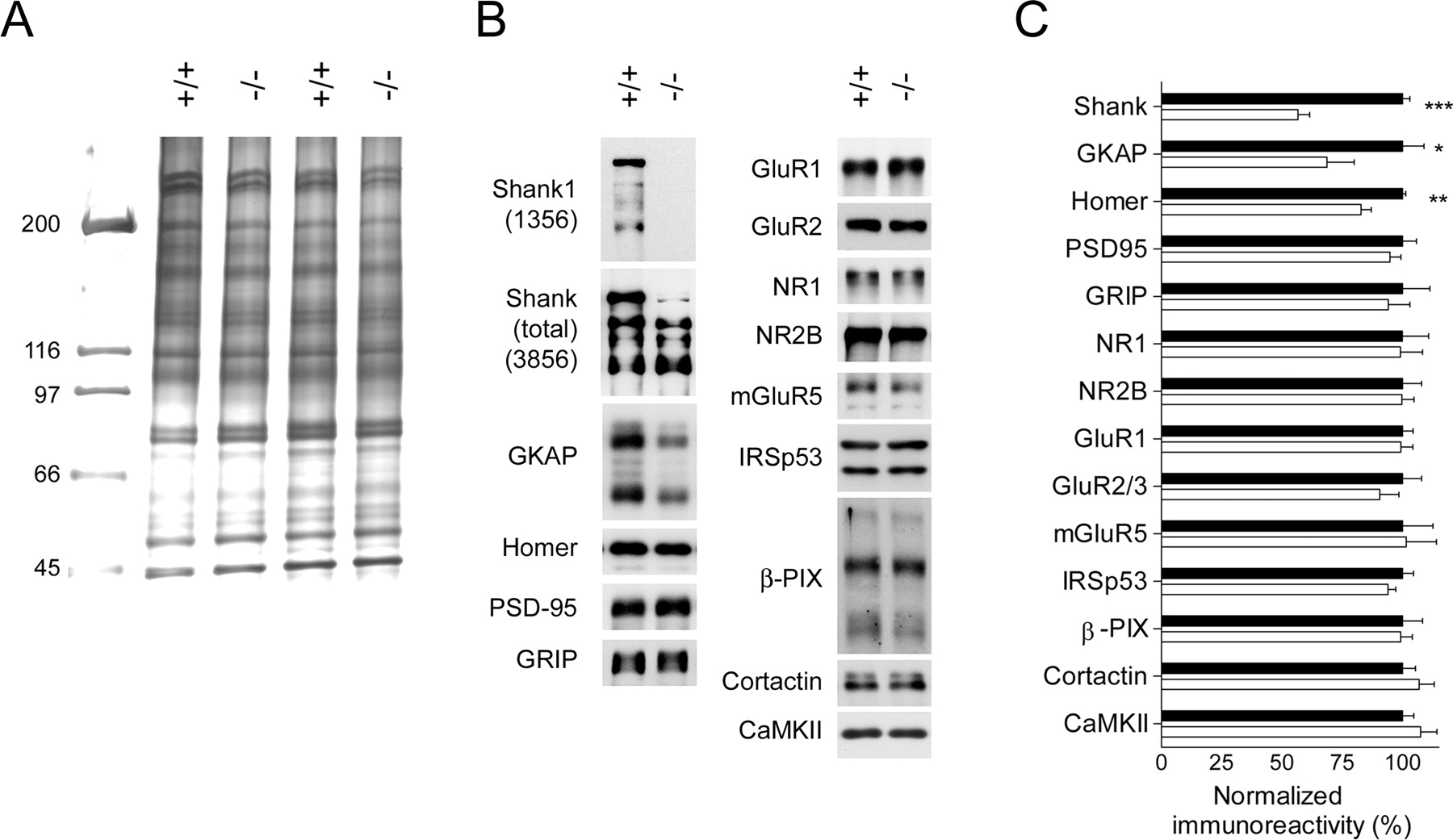
Altered composition of PSD fractions in Shank1 mutant mice. Analysis of Triton-extracted PSD fractions (PSDI) purified from forebrain of adult wild-type (+/+) and Shank1 knock-out (−/−) mice. A, SDS-PAGE and silver staining of PSD proteins revealed no major differences. B, Immunoblot analysis of PSD fractions from individual wild-type and Shank1−/− mice for the indicated proteins. C, Quantitation of various proteins in PSD fractions based on results as shown in B, normalized to wild-type (100%; black histograms). Histograms show mean ± SEM (>6 mice from each genotype). There is a significant difference in total Shank (+/+, n = 6; −/−, n = 8; ***p < 0.0001), GKAP (+/+, −/−, n = 12 each; *p < 0.05), and Homer (+/+, n = 12; −/−, n = 14; **p < 0.002).
A variety of glutamate receptors, scaffold proteins, and signaling molecules were measured by quantitative immunoblotting (Fig. 2B,C). In Shank1-mutant PSDs, there was a significant reduction of GKAP (∼30%). In addition, the level of Homer1b/c showed a modest but significant decrease (∼20%). Thus, the levels of two scaffold/adaptor proteins that bind directly to Shank were reduced in Shank1-deficient PSDs. In the same preparations, we detected no significant difference in the abundance of many other proteins that are known to be associated with synapses or the PSD, including NMDA, AMPA, and metabotropic glutamate receptors PSD-95 and GRIP (Fig. 2B,C). Also unchanged were the PSD levels of β-PIX and cortactin, actin regulatory proteins that can interact directly with Shank but that are not highly enriched in the PSD (Naisbitt et al., 1999; Park et al., 2003).
We then performed immunocytochemistry to examine the distribution of synaptic proteins in dissociated hippocampal neurons cultured from individual wild-type or Shank1−/− mouse embryos. As expected, staining with the Shank1-specific 1356 antibody was abolished in neurons from mutant animals (Fig. 3A). As reported previously (Naisbitt et al., 1999), staining with pan-Shank antibody 3856 showed Shank to be localized to dendritic clusters that colocalized with the excitatory synapse marker PSD-95 (Fig. 3A). In Shank1−/− neurons, the linear density of Shank puncta (labeled with pan-Shank antibody 3856) along the dendrite was reduced ∼50% (Fig. 3C). Similar to wild-type neurons (in which 90.3 ± 1.6% of Shank puncta colocalized with PSD-95 clusters), the vast majority of the 3856-immunoreactive Shank puncta in Shank1−/− neurons (97.1 ± 1.2%; n = 10 neurons per genotype) also colocalized with PSD-95. These data indicate that the remaining Shank staining (presumably resulting from Shank2 and Shank3) still localizes at excitatory synapses. In line with the biochemical results, there was also a significant decrease in the density of GKAP puncta in Shank1-deficient neurons (Fig. 3B,C). Although we detected no significant change in density of Homer puncta, the immunostaining pattern of Homer was more diffuse in Shank1-deficient neurons (Fig. 3B), supporting the idea that Shank is involved in recruiting or stabilizing Homer at synapses (Sala et al., 2001). There was no difference between cultured wild-type and Shank1 knock-out neurons in the cluster density or staining pattern of PSD-95 or the presynaptic active zone protein Bassoon. Overall, the biochemical and immunostaining data provide consistent evidence that Shank1 is important for synaptic accumulation of GKAP and Homer, supporting previous conclusions based on overexpression and dominant-negative studies of Shank in cultured neurons (Sala et al., 2001).
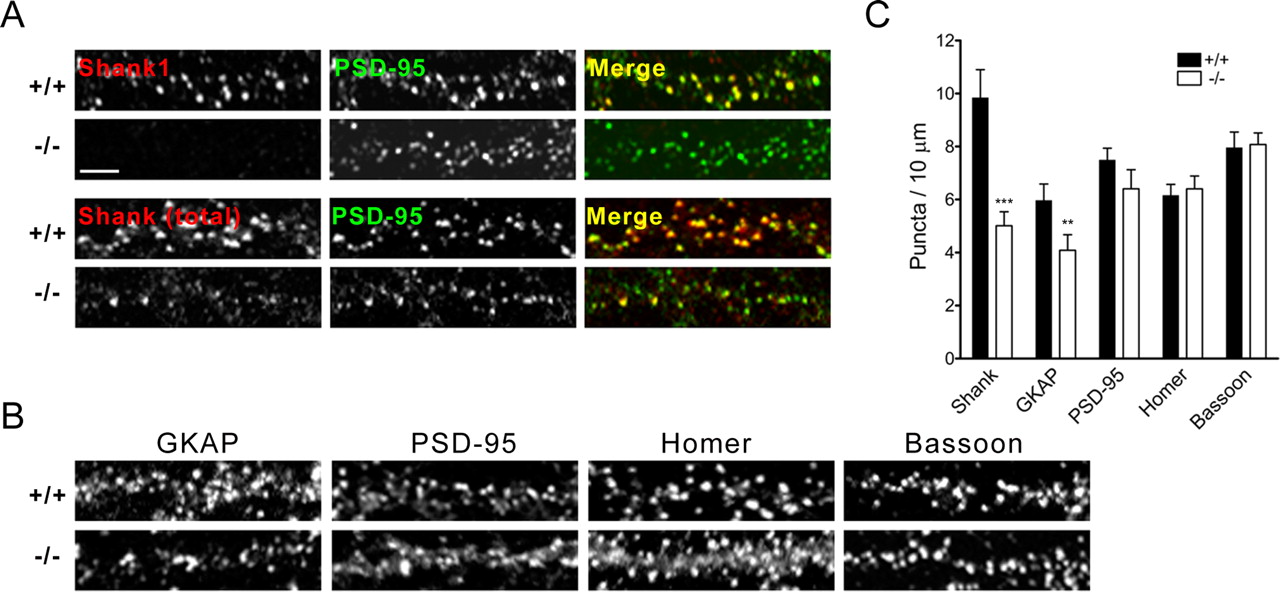
Altered immunostaining of PSD proteins in Shank1-deficient neurons. A, B, Hippocampal neurons in dissociated culture (18–19 DIV) from wild-type (+/+) or Shank1−/− mice were immunostained for the indicated proteins. A, Representative dendrites double labeled for PSD-95 (green) and with Shank1-specific antibody (1356) or pan-Shank antibody (3856) (red). Merged image is shown in color on the right. Scale bar, 5 μm. B, Dendrites immunostained for GKAP, PSD-95, Homer, or presynaptic marker Bassoon. C, Quantification of puncta density per 10 μm dendrite length for the indicated proteins (mean ± SEM). Shank1−/− dendrites show a significant reduction in pan-Shank (n = 10 cells each; ***p < 0.001) and GKAP puncta density (+/+, n = 21 cells; −/−, n = 16 cells; **p < 0.01).
Altered synapse morphology in Shank1 mutant brain
To examine the in vivo role of Shank1 in spine morphology, we performed blinded quantitative analysis of the number and size of dendritic spines in adult wild-type and Shank1−/− mice (Fig. 4A). We focused on apical dendrites of CA1 pyramidal neurons of hippocampus, where synapse morphology and plasticity have been studied extensively. Mean spine density showed a slight decrease in Shank1 knock-out mice (Fig. 4B) (p < 0.05; t test). Cumulative frequency plots of spine length (Fig. 4C) and spine head width (Fig. 4D) revealed a highly significant shift toward smaller spine size (length, p < 0.001, t test, and p < 0.0005, K–S; width, p < 0.001, t test, and p < 0.001, K–S). Although the absolute differences are small, possibly related to the presence of the remaining Shank isoforms, these data affirm that Shank1 is important for spine growth or maintenance in vivo.

Smaller dendritic spines and thinner PSDs in Shank1 mutant mice. A, DiI-labeled dendrites from CA1 pyramidal neurons of adult wild-type (+/+) and Shank1−/− mice (three representative segments shown). Scale bar, 5 μm. B, Quantification of dendritic spine density in wild-type versus Shank1−/− neurons. Data are presented as scattergrams (with mean ± SEM superimposed), each point corresponds to the mean spine density for a single neuron (+/+, n = 33 neurons from 4 animals; −/−, n = 27 neurons from 4 animals). Shank1−/− mice show a slight reduction in spine density (p < 0.05; t test). A total of 9210 wild-type spines and 7797 mutant spines were measured. C, D, Cumulative frequency plots for spine length (C) and spine head width (D) from wild-type (black line) and Shank1−/− mice (gray line). Scattergrams, with mean ± SEM, are shown at the right. Shank1−/− mice showed a small but highly significant shift toward smaller spines (length, p < 0.0005, K–S; width, p < 0.001, K–S). E, Representative electron micrographs of hippocampal CA1 striatum radiatum synapses from wild-type and Shank1−/− mice. The PSD is visible as an electron-dense layer adjacent to the postsynaptic membrane. Note thinner PSD in Shank1−/− synapses. Scale bar, 100 nm. F, G, Cumulative frequency distribution of PSD length (F) and thickness (G) from wild-type (black line) and Shank1−/− mice (gray line). Sixty randomly selected synapses from each of four wild-type and six Shank1−/− mice were measured by blind observers.
We next examined the ultrastructure of synapses, analyzing thin-section electron microscopic (EM) micrographs from wild-type and Shank1−/− mouse brains (Fig. 4E). In hippocampal CA1 region, the mean PSD thickness of Shank1−/− synapses was significantly reduced relative to wild type, and cumulative frequency graphs revealed a roughly parallel shift across the range of PSD thickness (Fig. 4G) [+/+, PSD thickness, 48.4 ± 1.6 nm (mean ± SEM); −/−, 45.8 ± 1.4 nm; p < 0.004, K–S]. The mean length of PSDs decreased in Shank1 mutants but did not reach statistical significance (+/+, 310.5 ± 3.5 nm; −/−, 299.4 ± 8.9 nm; p = 0.295, K–S). However, the cumulative frequency distribution showed that Shank1 mutant synapses suffered primarily a loss of the largest PSDs relative to wild type (Fig. 4F). Indeed, PSD length among the largest quartile of CA1 synapses measured per animal was significantly decreased in Shank1 mutants (+/+, 441.8 ± 12.9 nm; −/−, 397.9 ± 15.4 nm; p < 0.003, K–S). These data suggest that although Shank1 is not required for synapse formation, it may be critical for the development and/or maintenance of the largest subset of PSDs and synapses in particular.
Reduced basal synaptic transmission and normal synaptic plasticity in Shank1 mutant mice
What is the effect of Shank1 deficiency on synaptic function? We investigated excitatory synaptic transmission at the Schaffer collateral/commissural-CA1 synapse in acute hippocampal slices from wild-type and Shank1−/− mice (3–5 weeks of age). First, we examined synaptic transmission in Shank1−/− mice by measuring AMPA receptor-mediated field EPSPs in the stratum radiatum using extracellular recording techniques (Fig. 5A). The input–output curve for Shank1−/− mice was significantly shifted downward compared with wild type, especially at high stimulus intensities, demonstrating that Shank1 deficiency reduces basal synaptic transmission. PPF of AMPA receptor-mediated EPSCs, measured by whole-cell patch clamping, was similar in wild-type and Shank1−/− slices (+/+, 1.84 ± 0.13, n = 12 cells/6 mice; −/−, 1.74 ± 0.11, n = 9 cells/5 mice; p = 0.588). These results imply that the change in input–output relationship is unlikely to be caused by a change in presynaptic release probability. The ratio of AMPAR and NMDAR EPSCs (AMPA/NMDA ratio) was not significantly different between wild-type mice and Shank1−/− mice (+/+, 1.77 ± 0.35, n = 9 cells/7 mice; −/−, 1.91 ± 0.35, n = 9 cells/5 mice; p = 0.785), suggesting that the Shank1 deficiency does not affect the proportion of synaptic AMPA and NMDA receptors.

Decreased synaptic strength in Shank1 mutant mice. A, Left, Sample traces (average of 10 consecutive responses) represent the responses evoked with seven different stimulus intensities from wild-type (+/+) or Shank1−/− hippocampal slices. The same sample traces are shown at two different scales. Stimulus artifacts were truncated. Right, Summary graph of the input–output relationships of field EPSPs of wild-type mice (+/+; n = 15 slices from 9 mice) and Shank1−/− mice (−/−; n = 12 slices from 7 mice). Symbols indicate the mean ± SEM. The input–output relationship of Shank1−/− is significantly weaker than that of wild-type mice (*p < 0.05; Student's t test). B, Left, Top and Middle, Two consecutive sample mEPSC traces from wild-type and Shank1−/− mice. Left, Bottom, Averaged mEPSC from wild-type mice (left) and Shank1−/− mice (center) (average of 300 traces) and superimposed traces (sup; right). Note that the time course of the events is the same. Right, Summary graphs of the amplitude and frequency of mEPSCs in wild-type (n = 12 cells/6 mice) and Shank1−/− (n = 13 cells/6 mice) animals. There was no significant difference in mEPSC amplitude (NS, not significant). The frequency of mEPSCs in Shank1−/− mutants is significantly less than that of wild-type mice (*p < 0.02; Student's t test).
Given no change in PPF, the observed difference in synaptic response may be explained by a reduction in the number of functional synapses, in the number of glutamate receptors per synapse, or a combination of the two. To address this question, we examined the amplitudes and frequencies of the AMPAR-mediated mEPSCs (Fig. 5B). There was no significant difference in the average mEPSC amplitude between wild-type and Shank1−/− mice. However, the frequency of the mEPSC from Shank1−/− mutants was significantly reduced, suggesting that decreased basal transmission is primarily caused by a reduction in the number of functional synapses. We found no difference in the total length of CA1 apical dendrites in Shank1 knock-out versus wild-type mice (data not shown), implying that the weaker synaptic transmission is not a result of loss of dendrites and total synapses per neuron.
We next examined the effect of Shank1 deficiency on synaptic plasticity at Schaffer collateral/CA1 synapses. LTP was induced in acute hippocampal slices (3- to 5-week-old mice) using tetanic stimulation (100 Hz, 1 s). Similar magnitudes of LTP were obtained in wild-type (1.55 ± 0.06 of baseline at 60 min after tetanus) and mutant (1.58 ± 0.10; p = 0.800) slices (Fig. 6A). We also did not detect a difference in LTP in older mice (7- to 9-week-old mice) (data not shown). LTD evoked by low-frequency stimulation (1 Hz, 15 min) was also indistinguishable between wild-type (0.81 ± 0.06 of baseline at 75 min) and Shank1−/− (0.86 ± 0.02; p = 0.464) mice (Fig. 6B).

Synaptic plasticity is unchanged in Shank1 mutant mice. A, Top, Sample traces of field EPSPs of wild-type (+/+) and Shank1−/− mice (−/−) recorded at the times indicated in summary graph. Below, Summary graph of the averaged time course of LTP (+/+, n = 12 slices/10 mice; −/−, n = 12 slices/12 mice). Initial EPSP slopes were measured, and the values were normalized to the averaged slope value measured during the baseline period (−30 to 0 min). Tetanic stimulation (100 Hz, 1 s) was applied at 0 min. B, Sample traces and summary graph of the averaged time course of LTD (+/+, n = 9 slices of 9 mice; −/−, 7 slices of 7 mice). Low-frequency stimulation (1 Hz, 15 min) was applied at 0 min. C, Sample traces and summary graph of the averaged time course of late-phase LTP (+/+, n = 9 slices of 9 mice; −/−, n = 8 slices of 8 mice). Four trains of tetanic stimulation (100 Hz, 1 s) were applied from 0 min at 5 min intervals. There was no statistically significant difference between wild-type and Shank1−/− mice.
We further tested a stronger stimulus protocol (four tetanic trains separated by 5 min intervals), which has been used widely to induce L-LTP (Kelleher et al., 2004). This protocol elicited a long-lasting potentiation that was similar in wild-type and Shank1−/− slices (+/+, 1.29 ± 0.14 of baseline at 180 min; −/−, 1.30 ± 0.08; p = 0.916) (Fig. 6C). Thus, although loss of Shank1 decreases basal synaptic strength, it does not appear to affect standard forms of electrophysiologic plasticity lasting up to a few hours.
Increased anxiety-like behaviors in Shank1 mutant mice
We investigated the behavior of adult Shank1−/− mice in a variety of assays. Although they showed no obvious differences in the home cage, the Shank1−/− mice were significantly less active in a novel open-field environment than their wild-type littermates, as measured by horizontal activity, total distance traveled, and movement time (Fig. 7A,B). The relative reduction in distance traveled and time spent moving (movement time) was similar in the mutant mice; thus, average velocity of movement (calculated as total distance/movement time) was only slightly decreased in mutant mice (+/+, 8.47 ± 0.17 cm/s; −/−, 7.55 ± 0.28 cm/s; p < 0.01). Additionally, mutant mice spent significantly less time in the center zone, a measure of anxiety-like behavior (+/+, 148.6 ± 25.6 s; −/−, 56.1 ± 12.2 s; p = 0.002). The Shank1 mutants demonstrated a mild deficit in motor performance, as revealed by a reduced latency to fall in the accelerating Rotarod test (Fig. 7C). Nevertheless, given the small difference in open-field average movement velocity, it is unlikely that motor impairment alone accounts for the large decrease in open-field activity. In the light/dark exploration test, another anxiety-related task, Shank1−/− mutants similarly had fewer transitions between compartments and showed longer latencies to enter the light side (Fig. 7D).

Hypoactivity, rotarod performance, and increased anxiety-related behaviors in Shank1 mutant mice. A, Horizontal activity in open-field test is shown as number of beam breaks (mean ± SEM). Black line, Wild type (n = 24); gray line, Shank1−/− (n = 24). Shank1−/− mice show significantly reduced activity (genotype effect, F(1,46) = 21.60; p < 0.0001, repeated measures two-way ANOVA). B, Total distance traveled and movement time during 30 min open-field session is significantly reduced in Shank mutants (****p < 0.0001; t test). C, Mice (n = 15 per genotype) were tested for their ability to stay on the accelerating Rotarod (measured as latency in seconds to falling off). Three trials were performed on two consecutive days, with each trial terminated after 300 s. Repeated measures ANOVA revealed a significant genotype effect (F(1,28) = 21.19; p < 0.0001). D, Shank1−/− mutants make significantly fewer light–dark transitions and have longer latencies to enter the light compartment in the light–dark transition assay (***p = 0.001; *p = 0.05; n = 7 per genotype).
We also observed that Shank1−/− homozygous mice were poor breeders, giving birth only rarely. Moreover, homozygous mutant females did not nurture their pups, and their litters generally died before weaning. Thus, all our studies were performed on offspring of heterozygote intercrosses.
Impaired fear conditioning in Shank1 mutant mice
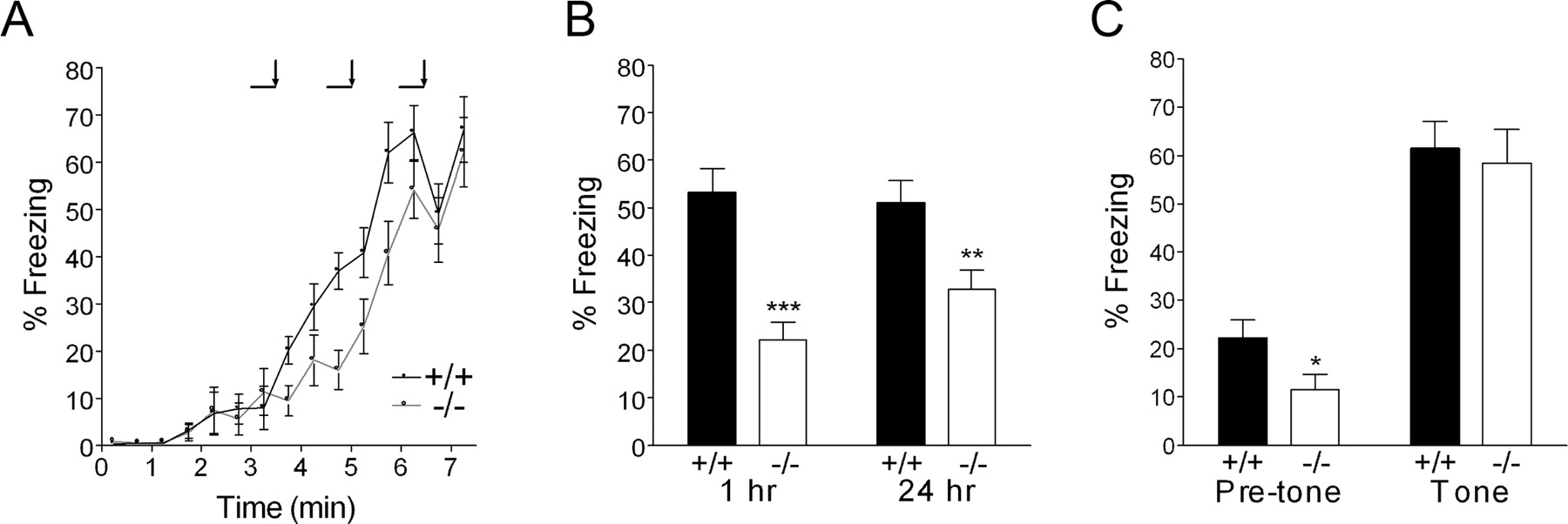
To examine the role of Shank1 in hippocampus-dependent learning and memory, we turned to a contextual fear conditioning task, in which long-term memory can be established with a single conditioning trial. During the conditioning period, wild-type and Shank1−/− mice were similarly active before the tone-shock pairs and showed similar levels of freezing after footshocks (Fig. 8A). However, the Shank1−/− mutants showed significantly less freezing during context testing conducted at 1 h (Fig. 8B) (+/+, 53.1 ± 5.0%; −/−, 22.2 ± 3.7%; p < 0.0001) and at 24 h after conditioning (+/+, 51.0 ± 4.6%; −/−, 32.8 ± 4.1%; p = 0.006). Given that Shank1 mutants show a decreased freezing response, the data are unlikely to be confounded by the overall hypoactivity of the Shank1 mutants, which could potentially mimic freezing. In contrast, when the conditioned stimulus (tone) was presented in an altered context 48 h after conditioning (cued testing), the Shank1−/− mutants froze to a similar extent as wild type (Fig. 8C) (+/+, 61.4 ± 5.8%; −/−, 58.4 ± 7.1%; p = 0.757). Thus, Shank1 is required selectively for contextual fear memory, a process believed to depend on intact hippocampus and amygdala function.

Impaired contextual fear memory in Shank1 mutant mice. A, Wild-type mice (black line; n = 20) and Shank1−/− mice (gray line; n = 18) showed similar freezing responses during the conditioning phase. The horizontal bar denotes exposure to conditioned stimulus (tone), and the vertical arrow notes timing of unconditioned stimulus (footshock). B, Decreased freezing in Shank1−/− mice at 1 and 24 h after fear conditioning when exposed to the same context (**p < 0.01; ***p < 0.0001). C, Cued fear memory was tested in an altered context 48 h after conditioning. The mean percentage of time spent freezing before tone and during presentation of the tone is shown. There is no significant difference in freezing in response to the conditioned stimulus.
Enhanced spatial learning in Shank1−/− mice
We then used the eight-arm radial maze task to assess spatial memory (Olton and Papas, 1979). This kind of learning accrues over days and weeks and also requires the hippocampus. With successive daily trials, the mice learn the locations of the two arms baited with food, allowing simultaneous evaluation of reference memory (errors counted by entries into arms never containing the reinforcing bait) and working memory (errors counted by entries into arms previously visited during the same trial). Thus, in this task, animals must learn and remember the position of baited arms between trials while rapidly establishing memory of previously visited arms within a trial. The eight-arm radial maze task is less sensitive than the Morris water maze to differences in motor performance, because learning is scored based on the number of errors made rather than the time required to complete the task.
During the acquisition phase, both wild-type and Shank1−/− mice improved their performance with repetitive training (Fig. 9) (see Materials and Methods for details of training). Remarkably, the Shank1 mutants showed a steeper learning curve and reached a better performance level with fewer reference memory errors than the wild-type animals (Fig. 9A) (genotype effect, F(1,27) = 10.98; p < 0.003, repeated measures two-way ANOVA). Even after prolonged training in this protocol, the wild-type mice did not catch up to the mutants' level of performance. Moreover, the Shank1−/− mice made fewer working memory errors than wild type; indeed, the knock-out animals made virtually no mistakes of “revisiting” toward the end of the training period (Fig. 9B) (genotype effect, F(1,27) = 10.41; p < 0.004).

Enhanced acquisition and impaired retention of spatial memory by Shank1 mutants in the eight-arm radial maze task. Two of eight arms were baited to test simultaneously reference and working memory. A, Total number of reference memory errors during acquisition training (+/+, black line, n = 15; −/−, gray line, n = 14). Mice received a total of 84 trials; data are presented in blocks of four trials. B, Total number of working memory errors (revisiting errors) across training. Shank1−/− mice make significantly fewer reference memory errors (genotype effect, F(1,27) = 10.98; p < 0.003, repeated measures two-way ANOVA) and working memory errors (genotype effect, F(1,27) = 10.41; p < 0.004) than wild-type mice. C, Shank1−/− mice correctly select a baited arm with their first arm selection more frequently than wild type (genotype effect, F(1,27) = 49.72; p < 0.0001). Data are shown in blocks of eight trials. D, Latency (time in seconds) to complete the trials in the eight-arm radial maze task is presented as means of four trials. E, Impaired spatial memory retention in Shank1−/− mice. Mice were retested in the radial maze task 28 d after completion of initial training (white bars); data are the mean of four trials. “End-training” (black bars) shows the performance during the last four-trial block of the acquisition phase shown in A. There is no significant difference in the performance of wild-type mice, but mutants perform significantly worse after 28 d without exposure to the maze. *p < 0.02; NS, p > 0.05, t test. F, Reversal training shows enhanced behavioral plasticity in Shank1−/− mutants. Position of baits was changed, and mice were retrained to learn new positions of baits. There was no difference in initial reversal trials (trials 1–16, genotype effect, F(1,27) = 0.30; p = 0.59), but Shank1−/− mice made fewer reference memory errors compared with wild type over last 20 trials (genotype effect, F(1,27) = 7.81; p < 0.01). G, Graph comparing standard (black) and intensive training protocols (gray) in the eight-arm radial maze task. For the intensive training, wild-type and Shank1−/− mice received a total of 60 trials over 22 training days. H, Despite intensive training, wild-type mice (n = 17) still make more reference memory errors than Shank1−/− mice (n = 18) (genotype effect, F(1,33) = 10.86; p < 0.003, repeated measures two-way ANOVA). Data are presented in blocks of four trials. I, Defect in memory retention in Shank1−/− mutants after intensive training (+/+, −/−, n = 9 mice each). When tested 28 d after completion of intensive training, mutant mice make more reference memory errors compared with last four-trial acquisition block, whereas wild-type performance shows no significant deterioration. “End-training” represents last four-trial block shown in H. **p < 0.005; NS, p > 0.05, t test.
As another measure of reference memory performance, we also quantified the percentage of trials in which a baited arm was chosen correctly with the first arm selection. Both genotypes selected a baited arm ∼25% of the time in initial trials (as expected from random selection of 2 of 8 baited arms), but the Shank1−/− mice improved more quickly and to a higher level (Fig. 9C) (genotype effect, F(1,27) = 49.72; p < 0.0001). Together, these data indicate that Shank1-deficient mice learn faster and more effectively during repetitive training in the eight-arm radial maze.
Obviously, the enhanced spatial learning of Shank1−/− mice in the radial maze task cannot be accounted for by their relative hypoactivity, which might be expected to prolong the duration of the task. In fact, although the Shank1−/− mice initially took a longer time to complete the task (trials 1–8, genotype effect, F(1,27) = 7.22; p < 0.02, repeated measures two-way ANOVA), the same animals were later able to find the baits in a shorter time than wild-type (trials 21–84, genotype effect, F(1,27) = 4.23; p < 0.05) (Fig. 9D).
The surprising results above indicate that Shank1-deficient animals have enhanced spatial learning, seemingly counterintuitive for a gene that encodes a major PSD scaffold protein. Therefore, we examined the long-term stability of spatial memory. To assess memory retention, we retested the mice in the same radial maze task after a period of 28 d in their home cage, during which they had no exposure to the maze. The wild-type animals showed no deterioration in their performance of the radial maze (measured by reference memory errors) after the 4 week “rest” (Fig. 9E). In contrast, the Shank1−/− mice performed significantly worse after the 28 d rest, regressing to the wild-type level of reference memory performance. Thus, Shank1 mutants are unable to retain long term the learning enhancement that they gained over the wild-type mice during training.
We further tested the “re-learning” ability of Shank1−/− mice by reversal training, in which the radial maze is rebaited in two different arms (Fig. 9F). Although wild-type and Shank1 mutants performed similarly in initial reversal trials (reversal trials 1–16, F(1,27) = 0.30; p > 0.5), the knock-out mice learned the new locations more quickly and made significantly fewer reference memory errors with repetitive training in the newly baited maze (reversal trials 17–36, genotype effect, F(1,27) = 7.81; p < 0.01).
Differences in training protocol can affect learning and memory (Kogan et al., 1997). We therefore subjected a different cohort of mice to the same radial maze task but applied a more intensive training program in which the number of trials per day was accelerated (Fig. 9G). Under this training regimen, the Shank1−/− mutants performed similarly to the mutant cohort trained under the original protocol, but the wild-type mice improved, making fewer reference memory errors with the more intensive protocol. Nevertheless, despite this improvement, the wild-type mice did not catch up to the performance of Shank1 mutants (Fig. 9H) (genotype effect, F(1,33) = 10.86; p < 0.003). After training, the mice were again tested for long-term retention. Twenty-eight days after the end of intensive training, wild-type mice retained their memory, completing the task with no significant change in reference memory errors. In contrast, Shank1−/− animals made markedly more reference memory errors after the 4 week break from training (Fig. 9I). In summary, Shank1 mutant mice show enhanced acquisition of spatial memory in the radial maze task but impaired long-term retention of this memory after training has finished.
Discussion
Regulation of PSD composition by Shank
In this study, we focused on the in vivo function of Shank1, a PSD scaffold of the Shank family, the expression of which is restricted to the brain. Previous biochemical studies showed that Shank binds to the adaptor proteins GKAP and Homer and hence to the NMDA receptor complex and metabotropic glutamate receptors (Naisbitt et al., 1999; Tu et al., 1999). In PSDs purified from Shank1−/− brain, we found depletion of GKAP and Homer, direct binding partners of Shank that are highly enriched at the synapse. The degree of loss of GKAP and Homer from the PSD (∼20–30%) can be viewed as relatively substantial, considering that disruption of the Shank1 gene reduced total Shank in the PSD by only ∼40% and that the remaining Shank2 and Shank3 also binds these proteins.
By immunoelectron microscopy, the PSD has a laminar organization, with Shank and GKAP lying in the “deeper” (cytoplasmic) side of the PSD (Naisbitt et al., 1999; Valtschanoff and Weinberg, 2001). The thinning of the PSD in Shank1 mutant mice could be explained, at least in part, by the loss of Shank1 and associated proteins from the cytoplasmic side of the PSD. Together, these findings corroborate the biochemical function of Shank as a PSD organizing scaffold in vivo.
Regulation of spine size and synapse function by Shank
At the cellular level, Shank has been proposed to promote spine growth and maturation, particularly enlargement of the spine head (Sala et al., 2001). Our finding that spines of CA1 pyramidal neurons are smaller in Shank1−/− animals confirms that Shank plays a role in spine morphogenesis in vivo. Shank1 mutants exhibited a selective loss of the largest PSDs in the hippocampal CA1 region. These results suggest that Shank1 is not required for spine formation but promotes the growth or stability of larger spines.
Dendritic spine size is positively correlated with synaptic strength, size of the PSD and presynaptic active zone, and the number of docked vesicles per active zone (Harris and Stevens, 1989; Matsuzaki et al., 2001). Here, we find that deletion of Shank1 weakens basal synaptic transmission. Most likely, the decrease in synaptic strength is a postsynaptic effect, because there is no change in paired-pulse facilitation. The reduced frequency but unaltered amplitude of mEPSC in the knock-out suggests primarily a loss of functional synapses in the absence of Shank1. The decrease in synaptic strength seems unlikely to be caused by a fall in dendritic spine number, because there is only a subtle reduction in spine density and no change in total dendrite length. A possible explanation is that some of the smaller spines in Shank1 knock-out neurons lack active functional synapses (that reach the detection threshold of this miniature analysis). Thus, the Shank1 postsynaptic scaffold is an important determinant in vivo of synaptic strength as well as morphology.
The electrophysiological phenotype that we observe is reminiscent of the effects of loss of function of the postsynaptic scaffold proteins PSD-95 and PSD-93, which also result in decreased mEPSC frequency but unaltered amplitude (Beique et al., 2006; Elias et al., 2006). However, in contrast to our findings in the Shank1−/− mutant, PSD-95−/− animals show enhanced LTP and deficits in spatial learning (Migaud et al., 1998; Beique et al., 2006).
Shank1 deletion genetically dissociates hippocampus-dependent memory mechanisms
To date, the vast majority of mutations reported to affect learning and memory in mice result in an impairment of performance. The few cases in which loss-of-function mutations led to improved spatial learning and memory include mutations that increase neuronal excitability (Collinson et al., 2002; Murphy et al., 2004; Nolan et al., 2004) or that affect synaptic calcium dynamics (Futatsugi et al., 1999; Jeon et al., 2003) (see below). In these cases, changes in long-term memory retention were not fully addressed. Despite the weakened basal synaptic transmission, loss of Shank1 resulted in improved acquisition of a spatial learning task, both in initial training and in reversal learning. However, they showed impairment in long-term retention of spatial memory (tested 4 weeks after training). Thus, Shank1 genetically dissociates the process of learning and memory that operates cumulatively during repetitive training over successive days from the process of long-term memory storage lasting several weeks or more. To our knowledge, this is the first example of a genetic manipulation that enhances formation of spatial memory during training while impairing retention of that memory after training.
Deletion of Shank1 also genetically dissociates spatial memory from contextual fear memory, both of which involve the hippocampus. Evidence from mutant mouse studies suggests that different molecular mechanisms may underlie specific forms of hippocampal-dependent memory. For example, mutations in CaMKII and the upstream kinase CaMKKβ result in a selective impairment of spatial memory, with no effect on contextual fear memory (Bach et al., 1995; Peters et al., 2003). In contrast, Shank1 is required for normal contextual fear conditioning but not spatial learning. It is notable that the Shank1 mutant shows significant impairment even at 1 h after fear conditioning, distinguishing it from other genetically altered mice that have specific deficits in the consolidation of fear memory (Hayashi et al., 2004). Thus, Shank1 appears to be important for both short- and long-term contextual fear memory while apparently inhibiting acquisition of spatial learning. This result is consistent with the notion that different mechanisms subserve single-trial versus gradual repetitive learning.
Spine morphology and the regulation of synaptic plasticity
Balancing new learning/plasticity with memory retention/stability is a dilemma faced by all network models for learning (Abraham and Robins, 2005). At a structural level, dendritic spines with different morphologies might contribute to these opposing functions. Dendritic spines with large heads (mushroom-shaped) exhibit more AMPA receptors and stronger excitatory postsynaptic responses (Matsuzaki et al., 2001) and are stable in vivo over months (Grutzendler et al., 2002; Trachtenberg et al., 2002; Holtmaat et al., 2005). In contrast, small thin spines, although having weaker synapses, are more susceptible to potentiation (Matsuzaki et al., 2004) and are structurally more dynamic and transient (Kasai et al., 2003; Holtmaat et al., 2005). These correlations have led to the proposal that the small dynamic spines are preferentially involved in learning, whereas larger stable spines mediate long-term memory storage (Kasai et al., 2003). If so, expression of proteins that promote growth of mushroom spines may alter the balance between spine plasticity and stability. Our data support this idea as the genetic deletion of Shank1, which results in decreased spine and synapse size, dissociates these processes, favoring new learning over long-term retention in a spatial memory task.
Shank protein expression is low at birth and increases during the first few weeks of postnatal development, reaching peak levels at 3–4 weeks of age (Lim et al., 1999), correlating with morphological maturation and reduced motility of dendritic spines (Majewska and Sur, 2003). It is tempting to speculate that accumulation of Shank and its associated proteins might contribute to the spine stabilization and the decline in behavioral plasticity that accompanies maturation of the nervous system.
Despite the overall trend toward decreased plasticity with age, the capacity for experience-dependent synaptic modification continues into adulthood, albeit at a lower level. Local protein synthesis and degradation has been proposed to be a mechanism for synapse-specific changes. Interestingly, Shank1 and Shank3 mRNAs are targeted to dendrites, raising the possibility of local translation of Shank proteins regulated by synaptic activity (Zitzer et al., 1999; Bockers et al., 2004). Furthermore, Shank and GKAP, the two families of scaffold proteins most significantly affected in the Shank1 mutant animals, are among the most prominent targets in the PSD for activity-dependent degradation by the ubiquitin-proteasome system (Ehlers, 2003). The regulated turnover of Shank in dendrites therefore provides a mechanism for local malleability of Shank levels, which may contribute to synapse-specific spine plasticity even in the mature brain.
Shank and autism spectrum disorders
No human mutations have yet been reported for Shank1, the closest relative of Shank3; however, the linkage of Shank3 mutations to a familial form of autism (Durand et al., 2007) clearly implicates Shank proteins in human cognitive development. The impairment of contextual fear learning, the normal cued fear conditioning, and the enhancement of spatial learning in the Shank1 knock-out mouse are reminiscent of the “mixed” cognitive phenotype often seen in human autism. Shank1 mutant mice also showed increased anxiety-related behaviors, which is typical of autistic patients. Most remarkably, the Shank1 mutants behaviorally resemble a knock-in mouse with an “autistic” mutation in neuroligin-3 (Tabuchi et al., 2007), in that both show enhanced spatial learning. We speculate that the improved spatial learning might be a useful endophenotype in animal models for some forms of ASD.
There is accumulating genetic evidence linking synapse development with ASDs, and dendritic spine abnormalities have been reported in some ASDs (Kaufmann and Moser, 2000). Our study of the Shank1 mutant mouse adds more support to the idea that aberrant development and function of synaptic connections contribute to pathogenesis of ASDs.
Footnotes
-
A.Y.H. is a recipient of National Institutes of Health K08 Award NS41411. C.S. is supported by the Giovanni Armenise–Harvard Foundation Career Development Program and by the European Community (LSHM-CT-2004–511995, SYNSCAFF). M.S. is an investigator of the Howard Hughes Medical Institute. We are grateful to I. Brito for assistance with mouse care, K. Phend and J. Ding for help with perfusion fixation and EM analysis, L. Leiter and V. Nelson for assistance with animal behavior, and A. Govindarajan for helpful discussions.
- Correspondence should be addressed to Morgan Sheng, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 46-4303, Cambridge, MA 02139. msheng{at}mit.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}