

Abstract
Uptake through the dopamine transporter (DAT) represents the primary mechanism used to terminate dopaminergic transmission in brain. Although it is well known that dopamine (DA) taken up by the transporter is used to replenish synaptic vesicle stores for subsequent release, the molecular details of this mechanism are not completely understood. Here, we identified the synaptic vesicle protein synaptogyrin-3 as a DAT interacting protein using the split ubiquitin system. This interaction was confirmed through coimmunoprecipitation experiments using heterologous cell lines and mouse brain. DAT and synaptogyrin-3 colocalized at presynaptic terminals from mouse striatum. Using fluorescence resonance energy transfer microscopy, we show that both proteins interact in live neurons. Pull-down assays with GST (glutathione S-transferase) proteins revealed that the cytoplasmic N termini of both DAT and synaptogyrin-3 are sufficient for this interaction. Furthermore, the N terminus of DAT is capable of binding purified synaptic vesicles from brain tissue. Functional assays revealed that synaptogyrin-3 expression correlated with DAT activity in PC12 and MN9D cells, but not in the non-neuronal HEK-293 cells. These changes were not attributed to changes in transporter cell surface levels or to direct effect of the protein–protein interaction. Instead, the synaptogyrin-3 effect on DAT activity was abolished in the presence of the vesicular monoamine transporter-2 (VMAT2) inhibitor reserpine, suggesting a dependence on the vesicular DA storage system. Finally, we provide evidence for a biochemical complex involving DAT, synaptogyrin-3, and VMAT2. Collectively, our data identify a novel interaction between DAT and synaptogyrin-3 and suggest a physical and functional link between DAT and the vesicular DA system.
Introduction
In the CNS, the neurotransmitter dopamine (DA) mediates a wide array of physiological functions including regulation of locomotor activity, cognitive processes, neuroendocrine secretion, and the control of motivated behaviors including emotion, affect, and reward mechanisms (Carlsson, 1987; Greengard, 2001). Consequently, dysfunctions in the DA system are believed to contribute to the development of several neurological and psychiatric conditions such as Parkinson's disease, dystonia, depression, schizophrenia, attention deficit/hyperactivity disorder (ADHD), Tourette's syndrome, and drug addiction (Carlsson, 1987; Roth and Elsworth, 1995; Koob and Le Moal, 1997; Greengard, 2001; Adinoff, 2004).
DA, released into the synaptic cleft, is transported back to the presynaptic terminal via the plasma membrane DA transporter (DAT). Thus, reuptake through DAT is the most effective way to limit the lifetime of DA signaling in the brain (Amara and Kuhar, 1993; Giros and Caron, 1993; Torres et al., 2003b; Cragg and Rice, 2004). DAT is the molecular target for therapeutic agents used in the treatment of mental disorders such as ADHD and depression as well as for cocaine and amphetamine, which are highly addictive and are major substances of abuse worldwide (Barker and Blakely, 1995; Amara and Sonders, 1998; Robbins and Everitt, 1999). Deletion of the DAT gene in mice leads to significant neurochemical changes characterized by a dramatic reduction of intracellular DA stores (95% decrease of DA levels compared with wild-type mice) (Giros et al., 1996; Jones et al., 1998). Indeed, depletion of DA stores in DAT knock-out mice is observed despite the fact that the DA synthesis rate was elevated by twofold. Reduction of monoamine levels in brain has also been observed after chronic treatment with monoamine transporter inhibitors in mice (Avni et al., 1975; Rattray, 1991; Wilson et al., 1996; Wu et al., 1997; Baumann et al., 1998; Volkow et al., 1999; Wang et al., 1999; Mateo et al., 2005). Thus, a complex series of neuroadaptive changes occur to try to maintain DA homeostasis in the absence of DAT. These findings illustrate the importance of DAT in the maintenance of normal DA levels and suggest that most of the DA taken up by the transporter is repackaged in synaptic vesicles for subsequent release; however, the molecular details for this mechanism are not completely understood. To begin to address these elusive mechanisms, we have embarked on a systematic search for proteins that interact with DAT and are examining their physiological significance.
Over recent years, an increasing number of DAT interacting proteins have been identified (for review, see Torres, 2006). These interactions suggest that the synaptic distribution, targeting, compartmentalization, trafficking, and functional properties of DAT can be regulated via protein–protein interactions. Here, using the mating-based split ubiquitin yeast two-hybrid system, we described the synaptic vesicle protein synaptogyrin-3 as a DAT interacting protein. Our findings show that synaptogyrin-3 and DAT interact in yeast, heterologous cells, mouse brain tissue, and live neurons in culture. Our results also demonstrate that the cytoplasmic N termini of both DAT and synaptogyrin-3 are sufficient for this interaction. Furthermore, the N terminus of DAT binds synaptic vesicles from a purified brain preparation. Overexpression of synaptogyrin-3 in neuronal HEK-293 cells results in an increase in DAT activity without changes in transporter levels at the plasma membrane. Instead, the effect of synaptogyrin-3 was abolished in the presence of the vesicular monoamine transporter-2 (VMAT2) inhibitor reserpine, suggesting that the ability of synaptogyrin-3 to regulate DAT activity depends on the vesicular DA storage system. Finally, we provide evidence for a biochemical complex involving DAT, synaptogyrin-3, and VMAT2. The physiological implications of this novel interaction are discussed.
Materials and Methods
Reagents.
The antibodies against synaptogyrin-1 and synaptogyrin-3 have been described previously (Belizaire et al., 2004). Antibodies against synaptotagmin, synaptophysin, clathrin, and Rab5 were obtained from BD Biosciences Transduction Laboratories. The synaptobrevin, SV2, and PSD-93 antibodies were obtained from Synaptic Systems. DAT H80, DAT C20, and VMAT2 C20 antibodies were from Santa Cruz Biotechnology. Millipore supplied the DAT AB5802, DAT MAB369, Na+/K+ ATPase C464.6, and VMAT 2 AB1767 antibodies, as well as the nonspecific IgGs from goat PP40 and rabbit PP64. The myc (9E10) and SNAP-25 antibodies were purchased from Sigma-Aldrich. The monoclonal transferrin receptor antibody was supplied by Zymed Laboratories. Secondary antibodies conjugated with HRP were from Jackson ImmunoResearch. Sulfo-NHS-SS-biotin, ultralink avidin beads, and dithiobis(succininidyl propionate) (DSP) were from Pierce. All other reagents were obtained from Sigma-Aldrich unless stated otherwise.
Mating-based split ubiquitin yeast two-hybrid system.
The mouse DAT cDNA was subcloned into the pCCW-Ste vector from Dualsystems Biotech, verified by automated sequencing, and expressed in the reporter yeast strain DSY-5. Yeast cells expressing mDAT were cotransformed with a mouse cDNA brain library expressed in the vector pNubG-x (Dualsystems Biotech). Selection of positive interactions was performed in single dropout media lacking tryptophan, leucine, and histidine (Clontech). Isolation of plasmid DNAs was performed as described previously (Torres et al., 2001) and verified by automated sequencing.
Cell culture and transfections.
HEK-293 and PC12 cells were obtained from the American Type Culture Collection. HEK-293 cells were cultured in MEM supplemented with 10% fetal bovine serum (FBS), 1 mm glutamine, and 50 μg/ml each penicillin and streptomycin at 37°C in a humidified, 5% CO2 incubator and transfected using the CaPO4 precipitation method with 5 μg of total DNA. PC12 cells were cultured in DMEM supplemented with 5% FBS, 5% horse serum, 1 mm glutamine, and 50 μg/ml each penicillin and streptomycin at 37°C in a humidified, 10% CO2 incubator and transfected using Lipofectamine 2000 (Invitrogen). MN9D cells were provided by Dr. Alfred Heller (University of Chicago, Chicago, IL) and maintained in DMEM high glucose, supplemented with 10% FBS, at 37°C in a humidified, 5% CO2 incubator. The human DAT cDNA was cloned into pcDNA3.1(+) using KpnI and XbaI sites and used to transfect MN9D cells with Lipofectamine 2000 combined with a CombiTag Magneto transfection reagent (OZ Biosciences). DAT-expressing single clones (MN9D-DAT cells) were selected with G418, verified by DAT immunoblot and immunofluorescence, and maintained in DMEM containing pyridoxal and 0.5 mg/ml G418. MN9D-DAT cells were transiently transfected using Lipofectamine 2000. For fluorescence resonance energy transfer (FRET) experiments, rat hippocampal neurons were prepared as described previously (Goetze et al., 2003). Briefly, hippocampi were dissected from neonatal Sprague Dawley rats, which had been killed by decapitation in accordance with the university's Institutional Animal Care and Use regulations. The tissue was cut into small pieces, incubated in 1 mg/ml papain for 30 min at 36°C, and dissociated by trituration in NMEM-B27 medium (MEM supplemented with 1.1 g of sodium pyruvate, 2 mm l-glutamine, 220 mg of NaHCO3, 600 mg of d-glucose, 2500 IU of penicillin, 2.5 mg of streptomycin, and 2 ml of B27 supplement per 100 ml). Approximately 300,000 cells were seeded into 35 mm culture chambers containing poly-d-lysine-coated glass coverslips. The medium was exchanged the day after the preparation and then once per week. Transfections of hippocampal neurons were performed with Lipofectamine 2000.
Generation of siRNA stable clones.
The pSilencer 4.1-CMV Neo vector was obtained from Ambion and used to insert small interfering RNAs (siRNAs) against synaptogyrin-3. siRNAs were constructed by inserting pairs of annealed DNA oligonucleotides into pSilencer 4.1-CMV Neo between BamHI and HindIII restriction sites according to the manufacturer's instructions. The synaptogyrin-3 siRNA target sequence was 5′-TCAGTGGCAACGTACAGCA-3′ corresponding to nucleotides 570–588 of rat synaptogyrin-3. The sequence 5′-ACTACCGTTGTTATAGGTG-3′ was used as a scramble control. To isolate siRNA-containing clones, PC12 cells were transfected with pSilencer 4.1-CMV-SYG3 siRNA along with pEF6 His A vector, which contains a blasticidin resistance gene. Twenty-four hours after transfections, 5 μg/ml blasticidin and 500 μg/ml neomycin were added to the medium. After selection, stable clones were maintained in 500 μg/ml neomycin. Western blot analysis for synaptogyrin-3 was performed as described previously.
Synaptosomal and synaptic vesicle preparation.
Striata from C57B1/6J mice were homogenized with 10 strokes in a glass Teflon homogenizer in 10 vol of 0.32 m sucrose, 10 mm HEPES-NaOH, pH 7.4, supplemented with protease inhibitors (1 μg/ml each pepstatin, leupeptin, and aprotinin, 40 μg/ml bestatin, and 2 μg/ml E-64). The homogenate was centrifuged at 1000 × g for 10 min at 4°C, and the resulting supernatant was centrifuged at 15,000 × g for 15 min at 4°C to yield a crude synaptosomal pellet. To isolate an enriched-synaptic vesicle preparation, the pellet containing the crude synaptosomes was lysed hypoosmotically through the addition of water containing protease inhibitors. The lysed synaptosomes were centrifuged at 23,000 × g for 20 min at 4°C, and the resulting pellet (synaptic plasma membrane) was discarded. The final supernatant was centrifuged for 2 h at 200,000 × g at 4°C to yield the synaptic vesicle-enriched pellet. This final pellet (termed P4) was resuspended in PBS and used in subsequent experiments. To obtain a pure synaptic vesicle pool, we isolated vesicles based on the protocol described by Morciano et al. (2005) with some modifications. Whole brain from C57B1/6J mice were homogenized with 20 strokes in a glass Teflon homogenizer in 10 vol of 0.32 m sucrose, 5 mm Tris/HCl, pH 7.4, supplemented with protease inhibitors. The homogenate was centrifuged at 1000 × g for 10 min at 4°C. The resulting pellet was discarded, and the entire supernatant was layered on top of a discontinuous Percoll gradient, prepared using three different layers of v/v Percoll solutions (3, 10, 23%) diluted in the original homogenization buffer. The Percoll gradient was centrifuged at 31,400 × g for 20 min at 4°C and the turbid fraction was collected containing isolated synaptosomes. Four volumes of the original homogenization buffer were added to the synaptosomes, and the sample was centrifuged again at 20,000 × g for 1 h at 4°C. The resulting pellet was hypoosmotically lysed in 6.5 ml of 5 mm Tris/HCl, pH 7.4, by pipetting the sample up-down-up 20 times and passing the sample through both a 22 and 27½ gauge needle. This suspension was centrifuged at 188,000 × g for 2 h at 4°C. The resulting pellet was resuspended in 0.5 ml of sucrose buffer (200 mm sucrose, 0.1 mm MgCl2, 0.5 mm EGTA, 10 mm HEPES, pH 7.4) and layered onto a discontinuous sucrose gradient ranging from 0.3 to 1.2 m sucrose (both prepared in 10 mm HEPES, 0.5 mm EGTA, pH 7.4). The sucrose gradient was centrifuged at 85,000 × g for 2 h at 4°C, and 500 μl fractions were collected top to bottom and used in subsequent experiments.
Coprecipitation with glutathione
S-transferase fusion proteins. cDNA fragments coding for intracellular domains of DAT, norepinephrine transporter (NET), or synaptogyrin-3 were amplified by PCR and subcloned into the pGEX4T-1 vector. Constructs containing glutathione S-transferase (GST) fusion proteins were sequenced, expressed in bacteria, and purified as described previously (Carneiro et al., 2002). Briefly, recombinant constructs were transformed in BL21 Escherichia coli and treated with 0.1 mm IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h at 37°C. The cells were harvested by centrifugation at 4300 × g for 10 min and resuspended in buffer A (50 mm EDTA in PBS) containing protease inhibitors. The cells were then sonicated and lysed by adding 1% Triton X-100 to the mixture for 1 h at 4°C followed by centrifugation at 9500 × g for 10 min. The resulting supernatants were incubated with glutathione-Sepharose 4B beads (GE Healthcare), washed three times with buffer A, and maintained at 4°C. To pull down proteins from brain lysates, fresh mouse brain or striatal tissue was homogenized in 10 vol of buffer B (20 mm HEPES, pH 7.6, 125 mm NaCl, 10% glycerol, 1 mm EDTA, 1 mm EGTA) containing protease inhibitors. After homogenization, 1% Triton X-100 or PBS was added and the mixture was incubated for 1 h at 4°C. The homogenate was then centrifuged at 16,000 × g for 10 min at 4°C, and the supernatant was incubated with 50 μg of glutathione beads containing GST fusion proteins. The mixtures were incubated overnight at 4°C, washed three times with buffer B and once with PBS, resuspended in 25 μl of SDS-PAGE sample buffer, and maintained at 4°C. For GST pull-down experiments using the P4 fraction, samples were cross-linked using 1 mm DSP for 30 min at room temperature (RT) after overnight incubation with 50 μg of the GST fusion proteins. The cross-linking reaction was inactivated by incubation with 50 mm Tris for 10 min at RT. The sample was lysed at 4°C with rotation using buffer B with or without 1% Triton X-100 and washed twice with buffer B, and once with PBS. Finally, GST pull-down assays using the purified synaptic vesicle pool used buffer B containing 1.2 mm CaCl2 and 1.2 mm MgSO4 and followed the same protocol used for pull downs in the P4 fraction. In all cases, the final sample was resuspended in 25 μl of SDS-PAGE sample buffer, analyzed by SDS-PAGE, and immunoblotted with the indicated antibodies.
Immunoprecipitations and Western blot analysis.
Striatal synaptosomes or transfected HEK-293 or MN9D cells were washed with ice-cold PBS and lysed in buffer B containing 1% Triton X-100 and protease inhibitors at 4°C for 1 h. Samples were centrifuged for 10 min at 14,000 × g to remove cellular debris. In some cases using striatal synaptosome preparations, interacting proteins were cross-linked with DSP as described previously. The protein concentration of the solubilized material was determined using the BCA reagent from Pierce. Immunoprecipitations were performed using 2–4 mg of total protein. Samples were incubated overnight with the indicated antibody at 4°C, followed by the addition of 30 μl of a mixture of protein A and protein G Sepharose beads (GE Healthcare). Immunoprecipitated proteins were recovered by centrifugation at 14,000 × g for 2 min, washed three times with buffer B, and resuspended in protein sample buffer. Samples were incubated at 37°C for 30 min, separated by SDS-PAGE on 10% Tris-HCl polyacrylamide gels, and transferred to nitrocellulose membranes using the Bio-Rad system. Whole-brain lysates were used in all experiments as positive control loading 50 μg of protein per lane. Nitrocellulose membranes were first blocked for 1 h in TBS buffer (50 mm Tris-HCl, 150 mm NaCl, 0.2% Tween 20) containing 5% dry milk, and then incubated with the indicated primary antibody for 1 h in blocking buffer, washed three times for 10 min each, and incubated with an HRP-conjugated secondary antibody. After all antibody incubations, membranes were washed three times with TBS buffer and protein bands were visualized using the West Pico system (Pierce).
Transport assays.
The conditions to examine DAT-mediated uptake in cultured cells have been described previously (Torres et al., 2003a). Briefly, 72–96 h after transfections, medium was removed, and DAT-mediated uptake was measured after incubation of cells for 5 min with 250 μl of uptake buffer (5 mm Tris base, 7.5 mm HEPES, 120 mm NaCl, 5.4 mm KCl, 1.2 mm CaCl2, 1.2 mm MgSO4, 1 mm ascorbic acid, and 5 mm glucose, pH 7.4). In some cases, reserpine (1 μm for PC12, 100 μm for HEK-293 cells) was added to the buffer for 10 min before the uptake reaction. For PC12 cells, 20 nm the substrate [3H]DA (3,4-[7-3H]dihydroxyphenylethylamine) (34.8 Ci/mmol; PerkinElmer) and increasing concentrations of cold DA ranging from 100 nm to 100 μm were used. In MN9D cells, 20 nm the substrate [3H]MPP+ (1-methyl-4-phenylpryridinium) (31.6 Ci/mmol; PerkinElmer) and increasing concentrations of cold MPP+ ranging from 100 nm to 100 μm were used. After rinsing with 1 ml of NaCl-free uptake buffer, cells were solubilized in 0.5 ml of 1% SDS and the radioactivity incorporated into the cells was measured by liquid scintillation counting. Nonspecific uptake was determined in the presence of 300 μm DA or MPP+. The protein concentration was measured using the BCA protein assay kit (Pierce). Data are presented as the mean ± SE.
Cell surface biotinylation.
Transfected PC12 and MN9D cells were washed three times with PBS and then incubated with gentle agitation for 30 min at 4°C with 1 ml of 1.5 mg/ml sulfo-NHS-SS-biotin prepared in 150 mm NaCl, 2 mm CaCl2, 10 mm triethanolamine, pH 7.8. This reaction was quenched by incubating the cells for an additional 10 min with 50 mm glycine in PBS. Cells were then washed with PBS and incubated in radioimmune precipitation assay buffer (RIPA) (10 mm Tris, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, and 1% sodium deoxycholate, pH 7.4) at 4°C for 1 h. Each sample was divided into two aliquots. One aliquot was used for isolation of biotinylated proteins with ultralink-immobilized avidin beads (Pierce). The second aliquot was used to determine total DAT levels. Samples were analyzed by SDS-PAGE and Western blotting with the MAB369 rat anti-DAT antibody and an HRP-conjugated secondary antibody.
FRET.
FRET (Schmid and Sitte, 2003) was measured with an epifluorescence microscope (Carl Zeiss TM210) using the “three-filter method” according to the method described by Xia and Liu (2001). Neonatal rat hippocampal neurons were prepared as described previously and seeded onto poly-d-lysine-coated glass coverslips (24 mm diameter). The next day, cells were transiently transfected using Lipofectamine 2000. Medium was replaced with Krebs'-HBS buffer (in mm: 10 HEPES, 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2), and images were taken using a 63× oil objective and a LUDL filter wheel that allows for rapid exchange of filters (<100 ms). The system was equipped with the following fluorescence filters: cyan fluorescent protein (CFP) filter (ICFP) (excitation, 436 nm; dichroic mirror, 455 nm; emission, 480 nm), yellow fluorescent protein (YFP) filter (IYFP) (excitation, 500 nm; dichroic mirror, 515 nm; emission, 535 nm), and FRET filter (IFRET) (excitation, 436 nm; dichroic mirror, 455 nm; emission, 535 nm). The acquisition of the images was done with MetaMorph (Meta Imaging; Molecular Devices; version 4.6.). Background fluorescence was subtracted from all images and fluorescence intensity was measured in different regions of interest. To calculate normalized FRET signals (nFRET), we used the following equation:
 where a and b represent the bleed-through values for YFP and CFP, respectively. Corrected FRET (FRETc) images were obtained according to Sorkin et al. (2000). Briefly, after background subtraction from all three images, CFP and YFP images were multiplied with their corresponding bleed-through value. The following equation was used for the calculation of FRETc images: FRETc = FRET − (b × CFP) − (a × YFP). Positive control experiments included either expression of the tandem CFP-YFP or coexpression of CFP-DAT and YFP-DAT. However, negative control experiments included coexpression of CFP and YFP or the coexpression of CFP-DAT and YFP-synaptophysin.
where a and b represent the bleed-through values for YFP and CFP, respectively. Corrected FRET (FRETc) images were obtained according to Sorkin et al. (2000). Briefly, after background subtraction from all three images, CFP and YFP images were multiplied with their corresponding bleed-through value. The following equation was used for the calculation of FRETc images: FRETc = FRET − (b × CFP) − (a × YFP). Positive control experiments included either expression of the tandem CFP-YFP or coexpression of CFP-DAT and YFP-DAT. However, negative control experiments included coexpression of CFP and YFP or the coexpression of CFP-DAT and YFP-synaptophysin.
Immunocytochemistry.
Male C57BL/6 mice were anesthetized with equithesin (50 mg/kg pentobarbital and 300 mg/kg chloral hydrate) and perfused through the heart with 10 ml of 0.1 m phosphate buffer (PB) followed by 20 ml of 4% paraformaldehyde in 0.1 m PB. All procedures were in strict accordance with guidelines of the Institutional Animal Care and Use Committee at the University of Pittsburgh. Brains were removed and stored in the same fixative for 2 h and then transferred to 30% sucrose in 0.1 m PB for 48 h. After this 2 d interval, brains were cut in the coronal plane at 40 μm as a one-in-six series. Striatal sections were rinsed in 10 mm PBS and nonspecific secondary IgG binding minimized by blocking in 10% normal goat serum with 0.3% Triton X-100 for 1 h at RT. After a brief rinse, sections were incubated overnight in primary antibodies at 4°C as follows: 1:400 rat anti-DAT (Millipore MAB369) and 1:600 rabbit anti-synaptogyrin-3 (Belizaire et al., 2004), in 1% normal goat serum and 0.3% Triton X-100, followed by three 10 min washes in 10 mm PBS and a 1 h incubation with secondary antibodies (goat anti-rat Alexa Fluor 555 and goat anti-rabbit Alexa Fluor 488; Invitrogen), each at 1:500, diluted in 0.3% Triton and 2% normal goat serum. Sections were then washed, mounted, placed on coverslips with Fluoromount G (Southern Biotechnology Associates), and viewed with a confocal microscope (Leica TCS-SL).
Data analysis.
Results are presented as mean ± SE. Significant differences between means were determined by Student's t test, with p < 0.05 considered statistically significant.
Results
Identification of the synaptic vesicle protein synaptogyrin-3 as a DAT interacting protein
To identify proteins that interact with the full-length DAT, we screened a mouse brain library using the mating-based split ubiquitin system in yeast (Fig. 1A) (Johnsson and Varshavsky, 1994; Stagljar et al., 1998). The cDNA encoding the full-length mouse DAT was cloned in-frame with the C-terminal one-half of ubiquitin and the LexA-VP16 transcription factor in the pCCW-Ste vector. This construct was then used to screen an adult mouse brain library of clones fused to the mutated form of ubiquitin in the pNubI13G vector (Dualsystems Biotech). Specifically, yeast cells expressing the DAT fusion protein were mated with cells pretransformed with an adult mouse brain library of ∼3 million clones. The screening resulted in 13 clones that induced activation of reporter genes in the presence of the mouse DAT. Two independent clones encoded the full-length sequence of synaptogyrin-3. These clones were in-frame with the N-terminal one-half of ubiquitin in pNubI13G, and interactions were confirmed in yeast mating assays (Fig. 1B). Synaptogyrin-3 is an integral membrane protein present in brain synaptic vesicles (Belizaire et al., 2004; Takamori et al., 2006). This protein contains four transmembrane domains with cytoplasmic N and C termini (Fig. 1C).

Identification of synaptogyrin-3 as a DAT interacting protein. A, Schematic representation of the yeast mating-based split ubiquitin system used in this study. *Cub, C-terminal one-half of ubiquitin (carrying the I13G mutation); Nub, N-terminal one-half of ubiquitin; PLV, protein LexA-VP16. A positive interaction between DAT and a protein from the cDNA library, when coexpressed in cells, reconstitutes a functional ubiquitin protein that releases the transcription factor PLV. This factor then translocates to the nucleus and induces the expression of the His3 and LacZ reporter genes. B, Retransformation of clones 4.1 and 9.2 (encoding synaptogyrin-3) for interaction with DAT. Incorporation of plasmids into yeast cells is verified by growth in selection medium lacking leucine and tryptophan (-L-W) (left plate); whereas specific interactions are detected by growth in selection medium lacking leucine, tryptophan, and histidine (-L-W-H) (right plate). C, Schematic representation of the synaptic vesicle protein synaptogyrin-3 depicting four transmembrane domains with cytoplasmic N and C termini.
Coimmunoprecipitation between DAT and synaptogyrin-3
To confirm this interaction in a mammalian cell context, we expressed recombinant full-length DAT and synaptogyrin-3 cDNAs in HEK-293 cells. The cells were lysed in the presence of detergent, and immunoprecipitation with a polyclonal synaptogyrin-3 antibody resulted in the coprecipitation of DAT only in cells transfected with both constructs (Fig. 2A, left panel). As expected, Western blotting for DAT after immunoprecipitation with the anti-DAT antibody revealed the presence of the transporter protein. Additionally, the anti-DAT antibody was able to coprecipitate synaptogyrin-3 from cell lysates coexpressing both proteins, but not from lysates of DAT-only or synaptogyrin-3-only transfected cells (Fig. 2A, right panel). Next, we examined the physiological relevance of the DAT–synaptogyrin-3 interaction by performing coimmunoprecipitation experiments in synaptosomal preparations from mouse striatum. As shown in Figure 2B, immunoprecipitation with three distinct commercially available anti-DAT antibodies resulted in synaptogyrin-3 coprecipitation from striatal synaptosomes. Cerebellum tissue was used as a negative control because it does not express detectable levels of DAT. As expected, synaptogyrin-3 did not coprecipitate from synaptosomes prepared from cerebellum (Fig. 2B), demonstrating the specificity of the DAT antibodies. To further confirm the specificity of this interaction, we also performed coimmunoprecipitation experiments in synaptosomes prepared in the presence of detergent. These synaptosomal lysates were incubated with the DAT antibody or alternatively with either nonspecific rabbit or goat antibodies. Indeed, our results showed that DAT and synaptogyrin-3 only coprecipitated with the DAT, but not with either nonspecific antibody (Fig. 2C). Finally, the specificity of the synaptogyrin-3 antibody was examined in HEK-293 cells expressing synaptogyrin-1, myc-tagged synaptogyrin-2, or synaptogyrin-3. No cross-reactivity was detected between our polyclonal synaptogyrin-3 antibody and synaptogyrin-1 or synaptogyrin-2 proteins (Fig. 2D). Similarly, immunoprecipitation of DAT from the striatum did not result in the coprecipitation of the synaptic vesicle proteins synaptophysin or synaptobrevin. We did observe a small, but considerably weaker, interaction between DAT and synaptogyrin-1 compared with that of DAT with synaptogyrin-3 (Fig. 2E). These results demonstrate a physical interaction between DAT and synaptogyrin-3.

Coimmunoprecipitation of DAT and synaptogyrin-3. A, HEK-293 cells were transfected with the indicated cDNAs. Immunoprecipitations (IP) were performed with the synaptogyrin-3 (left panels) or the DAT antibody (right panels). Control lysates are also shown. B, Immunoprecipitation of DAT using antibodies directed against different DAT epitopes results in the coprecipitation of synaptogyrin-3 from mouse striatum brain lysates. Western blot analysis of DAT and synaptogyrin-3 in control lysates from cerebellum tissue are also shown (bottom 2 panels). C, Control experiments showing immunoprecipitation with nonspecific rabbit or goat IgGs do not precipitate DAT or synaptogyrin-3. Lysate controls are also shown. D, The polyclonal synaptogyrin-3 antibody only recognizes synaptogyrin-3 but not synaptogyrin-1 (SYG1) or myc-tagged synaptogyrin-2 (SYG2-Myc) in transfected HEK-293 cells. E, Immunoprecipitation with the DAT antibody does not result in coprecipitation of synaptogyrin-1 (SYG1), synaptophysin (SYPH), or synaptobrevin (SYBV) from striatum or cerebellum lysates.
Colocalization of DAT and synaptogyrin-3 in striatum
Since the striata receives dense projections from midbrain DA neurons and previous reports found terminals within the striatum to be immunoreactive for synaptogyrin-3 (Belizaire et al. 2004), we next decided to examine whether DAT and synaptogyrin-3 colocalize in striatum. DAT immunolabeling was confirmed to be present in the substantia nigra and ventral tegmental DA neurons (data not shown), in fibers of the medial forebrain bundle ascending into the striatum, and within dense fiber networks and varicosities in the dorsal and ventral striatum (Fig. 3, top right panel). As expected, DAT-positive fibers were far fewer in the cortex and absent in the corpus callosum (cc) (Fig. 3, top right panel). However, synaptogyrin-3 immunolabeling was present in punctate varicosities across the dorsal and ventral striatum as well as in the cortex (Fig. 3, top left panel). As expected, synaptogyrin-3 labeling was absent from myelinated fiber bundles such as the internal capsule (ic), cc, and anterior commissure (data not shown). This presence and absence of antibody staining within these expected regions supports the specificity of our primary antibodies. Finally, specificity of secondary antibodies was also verified on each immunohistochemical run by loss of signal when either of the primary antibodies was omitted from the reaction (data not shown). In the dorsolateral mouse striatum, we observed extensive colocalization of synaptogyrin-3 and DAT signals within immunopositive fibers (Fig. 3, bottom right panel). Analysis using the colocalization function of MetaMorph software estimated that ∼40% of synaptogyrin-3-labeled structures were also positive for DAT signal (38.3 ± 6.3%; n = 3 animals; images in random fields in dorsolateral striatum). The estimates are to be viewed as approximations only, because they are based on confocal images after immunohistochemistry, a procedure whose efficacy varies with antibody penetration, quality of perfusion, and strength of the antibody–protein interaction, and among numerous other variables. Nevertheless, the observation that only a fraction of synaptogyrin-3 labeled puncta expressed DAT signal is entirely consistent with the widespread distribution of this synaptic protein within many striatal inputs versus the more restricted DAT expression limited to dopaminergic striatal fibers (Belizaire et al., 2004). Additional analysis on the same images revealed that nearly 80% of DAT-labeled structures stained positive for synaptogyrin-3 (76.5 ± 6.5%). Again, the lack of complete colocalization of DAT signal within the winding dopaminergic fibers with signal positive for synaptogyrin-3 at putative presynaptic terminals is not surprising and likely to reflect the presence of DAT within fibers outside of the presynaptic structure. These data indicate that some fraction of synaptogyrin-3 and DAT coexist and colocalize at presynaptic terminals in DA neurons.

Immunocytochemical analysis of DAT and synaptogyrin-3 in striatum. Slices were immunostained with a monoclonal rat anti-DAT (red; labeled with anti-rat Alexa Fluor 555) and a polyclonal rabbit anti-synaptogyrin-3 (green; labeled with anti-rabbit Alexa Fluor 488) antibodies. Low-magnification views of the dorsal striatum (Str), overlying cortex (Ctx), and the arch of the corpus callosum (cc) extending between the two macroscopic structures are shown in the top panels. Scale bar: top panels, 250 μm. High-power confocal images suggest the colocalization of the DAT and synaptogyrin-3 signals with punctate varicosities, appearing yellow in the merged view on the bottom right. Scale bar: bottom panels, 10 μm. Five examples of colocalization are indicated with white arrows. Areas of no signal in the internal capsule (labeled “ic”) are evident in all five images. Note the absence of both red and green fluorophores in these myelinated fiber bundles of the striatal internal capsule and of the cc support the specificity of the primary and secondary antibodies in use.
FRET analysis of DAT and synaptogyrin-3 interaction on live neurons
As an additional independent approach, we used FRET microscopy to confirm whether the biochemically observed interaction between DAT and synaptogyrin-3 also occurs in live cells. Since the advent of the jellyfish green fluorescent protein and its spectral variants, FRET microscopy has become an invaluable tool to discern protein–protein interactions in live cells. This approach relies on the close proximity between the proteins of interest (Foerster distance for CFP-YFP, <50 Å) and an appropriate relative orientation that allows an excited donor to transfer its energy to a second, longer-wavelength acceptor in a nonradiative manner. Neonatal hippocampal neurons from rats were cultured and transiently cotransfected with an amino CFP-tagged DAT and an amino YFP-tagged synaptogyrin-3. Visualization of physical protein interaction in intact cells was achieved by the method of Sorkin et al. (2000), which generates corrected FRET images (FRETc) as described in Materials and Methods. Control experiments included transfection with CFP-YFP in tandem (Schmid et al., 2001), cotransfection of CFP with YFP, CFP-DAT with YFP-DAT, and CFP-DAT with YFP-synaptophysin. We observed robust FRET signals when CFP-DAT and YFP-synaptogyrin-3 were coexpressed in neurons. FRET, according to Xia and Liu (2001), normalizes on the expression levels of the fluorescent proteins in the region of interest (ROI); this is correlated in a linear manner over a wide range of expression levels (CFP/YFP ratios, 1:6 and 6:1). Therefore, we included only neurons that displayed a maximum ratio in fluorescent expression ratios of 1:2 or 2:1. Our ROI was defined as areas on the cell membrane or at synaptic processes (Fig. 4A). Quantitative measurements of these ROIs were performed on 68 regions from 31 different transfected neurons on 5 experimental days revealing a mean FRET value of 0.213 ± 0.023 (Fig. 4B). As a positive control, we coexpressed CFP-DAT and YFP-DAT because previous studies have demonstrated the oligomerization of DAT (Sorkina et al., 2003; Torres et al., 2003a). As expected, we detected a robust FRET signal (mean value of 0.222 ± 0.042) between CFP-DAT and YFP-DAT (Fig. 4A,B). To assess the specificity of the DAT–synaptogyrin-3 interaction measured by FRET analysis, we coexpressed neurons with CFP-DAT and YFP-synaptophysin since our previous results indicate that these two proteins do not interact in coimmunoprecipitation experiments (Fig. 2D); FRET signals (mean value of 0.091 ± 0.033) (Fig. 4B) were observed in a range not significantly different from the negative control CFP plus YFP, confirming our biochemical findings. Together with the coimmunoprecipitation and colocalization experiments, these findings indicate that synaptogyrin-3 and DAT coexist and interact in brain.

Interaction between DAT and synaptogyrin-3 as visualized by FRET microscopy. Hippocampal neurons from neonatal rats were cotransfected with CFP-DAT and YFP-synaptogyrin-3 (YFP-SYG3). A, High magnification of a neuron transfected with CFP-DAT and YFP-synaptogyrin-3 (top left panel, CFP channel; top right panel, YFP channel; bottom left panels, FRET channel; bottom right panels, corrected FRET channel). The arrowheads indicate neuronal processes in which FRET was detected. FRETc images are displayed using pseudocolors with the spectrum inset representing fluorescence intensity. B, The FRET was analyzed as described in Materials and Methods. Control experiments included cells transfected with a CFP-YFP tandem construct, cells cotransfected with CFP and YFP, cells coexpressing CFP-DAT and YFP-DAT, and cells coexpressing CFP-DAT and YFP-synaptophysin (YFP-SYPH). The intensity of FRET was measured for the different combination of CFP/YFP pairs and expressed as normalized FRET values as described in Materials and Methods. Error bars indicate SEM.
The cytoplasmic N termini of DAT and synaptogyrin-3 are sufficient for the DAT–synaptogyrin-3 interaction
To delineate the DAT domains involved in the interaction with synaptogyrin-3, GST fusion proteins containing the intracellular N terminus (DAT-N, residues 1–60), the first intracellular loop (DAT-L1, residues 119–139), and the C terminus (DAT-C, residues 582–620) of the human DAT were generated. GST fusion proteins were produced in bacteria, purified with glutathione-agarose beads, and incubated with mouse brain lysates overnight. As shown in Figure 5A, only DAT-N, but not DAT-L1, DAT-C, or GST alone bound synaptogyrin-3 from mouse brain lysates. In addition, DAT-N did not exhibit detectable binding to the related synaptic vesicle proteins synaptogyrin-1 or synaptophysin (Fig. 5B). We also tested whether the N terminus of the related NET might interact with synaptogyrin-3. A GST fusion protein containing the first 66 residues of NET (NET-N) did not exhibit substantial interaction with synaptogyrin-3 when compared with the interaction with DAT-N (Fig. 5C). Similarly, to delineate the domains of synaptogyrin-3 involved in the interaction with DAT, we generated GST fusion proteins containing the cytoplasmic N terminus (SYG3-N; residues 1–26), loop (SYG3-L; residues 89–108), and C terminus (SYG3-C; residues 169–229) of synaptogyrin-3. As shown in Figure 5D, only the N terminus of synaptogyrin-3 was able to interact with DAT. All these pull-down assays were repeated under denaturing conditions and for shorter incubation periods (1 h), and our results remained consistent (data not shown). Together, these findings suggest that the cytoplasmic N termini of both DAT and synaptogyrin-3 are sufficient for this specific interaction.

The N termini of both DAT and synaptogyrin-3 are sufficient for the DAT/synaptogyrin-3 interaction. A, B, Aliquots containing GST only, DAT-N, DAT-L1, or DAT-C fusion proteins (50 μg each) were incubated with whole-brain lysates, and the precipitated proteins were analyzed by Western blot using antibodies against synaptogyrin-3 (SYG3), synaptogyrin-1 (SYG1), or synaptophysin (SYPH). C, GST alone, DAT-N, or NET-N was incubated with whole-brain lysates, and the pull downs were analyzed by Western blot using the anti-synaptogyrin-3 antibody. The DAT-N, but not NET-N, interacts with synaptogyrin-3. D, Aliquots containing SYG3-N, SYG3-L, or SYG3-C GST-fusion proteins (50 μg) were incubated with striatum lysates and the precipitated proteins were analyzed by Western blot using the anti-DAT antibody. The SYG3-N interacts with DAT.
The N terminus of DAT is capable of binding synaptic vesicles
Having demonstrated an interaction between the N terminus of DAT and solubilized synaptogyrin-3 from mouse brain lysate, we hypothesized that synaptogyrin-3 might facilitate an interaction between DAT and synaptic vesicles. Thus, we sought to investigate whether, indeed, DAT can bind synaptic vesicles. To do this, we obtained an enriched synaptic vesicle preparation (called P4 fraction) from mouse brain. Western blot analysis of P4 confirmed the presence of specific synaptic vesicle proteins (Fig. 6A). After incubation with GST alone or DAT-N, complexes were cross-linked with DSP, lysed with buffer B containing 1% Triton X-100, and precipitated by centrifugation. Western blot analysis revealed that DAT-N was able to interact with synaptogyrin-3 from the solubilized synaptic vesicle preparation (Fig. 6B). No detectable interaction was observed between DAT-N or GST alone and other synaptic vesicle proteins including synaptogyrin-1, synaptophysin, synaptotagmin, or synaptobrevin (Fig. 6B). Next, we incubated this preparation with either GST alone or DAT-N in the absence of detergent to preserve the integrity of the organelles. Analysis of the samples precipitated with DAT-N revealed the presence of synaptogyrin-3, synaptotagmin, and SV2, suggesting that the N terminus of DAT could actually bind synaptic vesicles from this P4 preparation. We did not detect the plasma membrane marker Na+/K+ ATPase in the DAT-N precipitate (Fig. 6C).

The N terminus of DAT interacts with synaptic vesicles. A, An enriched synaptic vesicle preparation (P4) was obtained from mouse brain and analyzed by Western blot for synaptic vesicle markers. The final supernatant from the preparation of P4 (S4 fraction) is also shown. B, Western blot analysis of P4 samples (with detergent) precipitated with GST alone or DAT-N. C, Western blot analysis of P4 samples (without detergent) precipitated with GST alone or DAT-N. D, Synaptic vesicles separate in two distinct populations when separated through sucrose gradients. Western blot analysis was performed with the indicated antibodies: synaptogyrin-3 (SYG3), synaptotagmin (SYTM), synaptobrevin (SYBV), synaptogyrin-1 (SYG1), synaptophysin (SYPH), VMAT2, postsynaptic density protein 93 kDa (PSD93), transferrin receptor (TfR). Fractions 7–11 from the sucrose gradient were pooled and incubated with GST alone or DAT-N in the presence (E) or absence (F) of detergent. Mixtures were treated with 0.5 mg/ml DSP for 10 min and solubilized in PBS. Bound proteins were precipitated with glutathione agarose beads binding either GST alone or DAT-N, separated by SDS-PAGE, and immunoblotted with the indicated antibodies.
Having demonstrated the feasibility of an interaction between DAT-N and vesicles from an enriched preparation, we performed similar experiments using purified synaptic vesicles from Percoll and sucrose gradients. Western blot analysis of several synaptic vesicle markers revealed that vesicles were distributed between two populations. The synaptic vesicle markers synaptogyrin-3, VMAT2, synaptophysin, SV2, synaptogryin-1, and synaptobrevin were distributed throughout the entire gradient; however, only fractions 7–11 were devoid of cytosolic, plasma membrane, endosomal, presynaptic plasma membrane, and postsynaptic markers (Fig. 6D). The remainder of the fractions was contaminated with proteins from additional subcellular compartments. Specifically, fractions 1–5 contained clathrin-enriched fractions, whereas fractions 13–25 also contained plasma membrane (Na+/K+ ATPase), endosomal (transferrin receptor, Rab 5), presynaptic (SNAP25), and postsynaptic (PSD93) markers (Fig. 6D). Our results are consistent with those obtained by Morciano et al. (2005) under similar conditions. Using only fractions 7–11, containing a purified synaptic vesicle pool, we performed subsequent pull-down experiments with GST alone or DAT-N. In the presence of detergent, DAT-N was able to bind synaptogyrin-3, but not other synaptic vesicle proteins including SV2 and synaptophysin (Fig. 6E). In the absence of detergent, Western blot analysis revealed that synaptogyrin-3 as well as other synaptic vesicle proteins including SV2, VMAT2, synaptotagmin, and synaptophysin coprecipitated with DAT-N (Fig. 6F). There was no detectable interaction between GST alone and any of the tested synaptic vesicle proteins. Thus, our results using this purified pool of vesicles confirmed our previous results using P4 and indicate that the N terminus of DAT is capable of interacting with synaptic vesicles.
Functional interaction between DAT and synaptogyrin-3
To test the functional consequences of the interaction between DAT and synaptogyrin-3, we used MN9D cells, a dopaminergic neuronal cell line that has been extensively characterized as a model of midbrain dopaminergic neurons (Choi et al., 1992; Hermanson et al., 2003; Chen et al., 2005; Rick et al., 2006). To facilitate the biochemical and functional characterization of DAT, we generated a stable cell line overexpressing the human DAT (MN9D-DAT). MN9D cells contain an abundance of catechol-O-methyltransferase (COMT), and since DA is a substrate of COMT, we opted to use MPP+, which binds saturably and with high affinity to DAT (Kitayama et al., 1993), as the substrate in these uptake assays. We observed an increase in DAT activity in MN9D-DAT cells overexpressing synaptogyrin-3 (Km = 13.29 ± 2.48 μm; Vmax = 2.37 ± 0.09 pmol · min−1 · mg protein−1; n = 3) compared with MN9D-DAT mock-transfected cells (Km = 11.03 ± 2.29 μm; Vmax = 1.39 ± 0.17 pmol · min−1 · mg protein−1; n = 3) (Fig. 7A). Similar results were obtained regardless of whether DAT was transiently or stably expressed (data not shown). In conjunction with these uptake assays, we performed biotinylation experiments using sulfo-NHS-SS-biotin to examine whether synaptogyrin-3 overexpression resulted in changes to the levels of cell surface DAT. Sulfo-NHS-SS-biotin, which binds to lysine and arginine residues in proteins, is cell impermeant and can be used to label cell surface proteins. Although the levels of synaptogyrin-3 increased approximately twofold (Fig. 7A, inset), the total and cell surface levels of DAT were not altered by synaptogyrin-3 overexpression (Fig. 7B). Thus, these results indicate that the increase in DAT uptake activity by synaptogyrin-3 is not a consequence of an increase in cell surface levels of DAT.

Synaptogyrin-3 overexpression increases DAT activity in MN9D-DAT cells. A, MN9D-DAT cells transfected with synaptogyrin-3 vector (▴) show an increased DA uptake activity (Vmax = 2.37 ± 0.09 pmol · min−1 · mg protein−1; n = 3) compared with MN9D-DAT mock-transfected cells (·) (Vmax = 1.39 ± 0.17 pmol · min−1 · mg protein−1; n = 3). Overexpression of synaptogyrin-3 results in a twofold increase of protein levels (inset). B, Total (top) and cell membrane (bottom) expression were analyzed by Western blot for each transfection and normalized to α-tubulin (TUB) levels. Densitometric analysis was performed for each experiment and plotted as a fraction of control. Results are presented as the mean ± SE (*p < 0.05).
Additionally, we used PC12 cells as a second cell model system. Nixosetine (300 nm) was used in these experiments to block the endogenous NET. Again, we observed an increase in DAT activity in PC12 cells transfected with synaptogyrin-3 and DAT compared with PC12 cells transfected only with DAT (Fig. 8A). Here, Western blot experiments showed levels of synaptogyrin-3 to be increased approximately fourfold (Fig. 8A), whereas the total and cell surface levels of DAT were not altered by synaptogyrin-3 overexpression (Fig. 8B). In addition, synaptogyrin-3 overexpression failed to alter alanin uptake, ruling out nonspecific effects on membrane ion gradients (data not shown). Finally, to assess the effect of eliminating synaptogyrin-3 on DAT function, we used siRNA technology to reduce the levels of synaptogyrin-3 from PC12 cells. In our hands, overexpressing an siRNA construct targeting residues 570–588 of synaptogyrin-3 results in a reduction of ∼40% of protein levels compared with control cells overexpressing a scramble siRNA sequence. The expression levels of several synaptic vesicle proteins including synatogyrin-1, amphiphysin, and synaptophysin were not affected. In addition, DAT levels also remained unaffected (Fig. 8C). In cells overexpressing the synaptogyrin-3 siRNA construct, we observed a decrease in DAT activity compared with scramble control cells (Fig. 8D). Biotinylation assays showed unaltered cell surface levels of the transporter (Fig. 8E). Thus, these results indicate that changes in synaptogyrin-3 expression levels correlate with changes in DAT activity and, furthermore, that these changes are not the result of changes in the cell surface levels of the transporter.

Changes in synaptogyrin-3 expression levels correlate with changes in DAT activity in PC12 cells. A, PC12 cells were transiently transfected with DAT (white) or cotransfected with both synaptogyrin-3 and DAT (black), and uptake activity was performed with [3H]DA 96 h after transfections. The data represent the means ± SE of four independent experiments. Overexpression of synaptogyrin-3 was analyzed by Western blot for each transfection, and protein levels were normalized to α-tubulin (TUB) levels. B, Overexpression of synaptogyrin-3 has no effect on the cell surface (top) or total (bottom) levels of DAT. Representative blot images of cells expressing DAT and cells coexpressing DAT and synaptogyrin-3 are shown. Densitometric analysis was performed for each experiment and plotted as a fraction of control. Results are presented as the mean ± SE. C, Densitometric analysis of synaptogyrin-3 protein levels from cells transfected with a synaptogyrin-3 siRNA (black bar) or a scramble siRNA control (white bar). The expression levels of proteins involved in regulated exocytosis or endocytosis were unaltered by synaptogyrin-3 siRNA. D, DAT activity in synaptogyrin-3 knockdown cells. Uptake assays in PC12 cells expressing a synaptogyrin-3 siRNA (▴) or scramble siRNA (·) were performed in the presence of the NET inhibitor, nixosetine, with [3H]DA 96 h after transfections. The data represent means ± SE of four independent experiments. E, Levels of surface DAT remain unaltered in synaptogyrin-3 siRNA cells. AMPH, Amphiphysin; SYPH, synaptophysin; SYG1, synaptogyrin-1. *p < 0.05.
To further examine the specificity of the effect of synaptogyrin-3 on DAT activity, we next explored whether it could also alter NET activity. In the absence of the NET inhibitor nixosetine, PC12 cells exhibit low levels of uptake that is attributed to NET-mediated transport (Brüss et al., 1997). Synaptogyrin-3 overexpression in WT PC12 cells did not alter this basal uptake activity (Fig. 9A). Next, we transfected cells with NET alone or together with synaptogyrin-3 and measured uptake activity. Although overexpression of NET increases basal uptake approximately fivefold, it is not further affected by synaptogyrin-3 overexpression (Fig. 9A). Consistent with our previous data showing that the amino NET does not interact with synaptogyrin-3, immunoprecipitation of NET did not result in the coprecipitation of synaptogyrin-3 in PC12 cells (Fig. 9B). The failure of synaptogyrin-3 to physically or functionally interact with NET supports the specificity and importance of the interaction between synaptogyrin-3 and DAT.

Synaptogyrin-3 overexpression does not increase NET activity. A, Uptake assays in nontransfected PC12 cells (■), cells transfected with synaptogyrin-3 (▾), cells transfected with NET (·), and cells transfected with NET and synaptogyrin-3 cDNAs (▴) were performed in the absence of the NET inhibitor nixosetine 96 h after transfections. The data represent the means ± SE of four independent experiments. B, PC12 cells were transfected with either DAT alone, NET alone, or NET and synaptogyrin-3. Immunoprecipitation experiments were performed on these lysates using either antibodies directed against DAT (left) or NET (right). Western blot analysis showed successful transfection of DAT and NET (bottom) but show that synaptogyrin-3 only coprecipitates with DAT and not NET (top).
Synaptogyrin-3 does not affect DAT activity in HEK-293 cells
We next considered the alternative explanation that the effect of synaptogyrin-3 on DAT function was attributable to a direct effect of the DAT/synaptogyrin-3 interaction. Since our previous experiments used MN9D and PC12 cells as a neuron-like model system, it was not possible to discriminate a direct effect of synaptogyrin-3 on DAT versus a potential contribution of the vesicular system. Thus, we decided to perform similar experiments in HEK-293 cells, a non-neuronal cell system devoid of a DA vesicular pool. These cells were successfully transfected with either DAT alone or in combination with synaptogyrin-3 (Fig. 10A). DAT activity was not different in cells transfected with DAT alone compared with cells coexpressing DAT and synaptogyrin-3 (Fig. 10B) despite the fact that these two proteins coimmunoprecipitate in this cell line (Fig. 2A). This result indicates that synaptogyin-3 does not change DAT activity through a direct effect on the transporter protein through its protein–protein interaction.

Synaptogyrin-3 does not affect DAT activity in HEK-293. A, Western blot analysis confirmed that HEK-293 cells were transiently transfected with either DAT alone or in combination of synaptogyrin-3. B, Uptake assays in cells expressing only DAT (black) or DAT and synaptogyrin-3 (red) were performed with [3H]DA 96 h after transfections. The kinetic values for DAT activity were Km = 3.61 ± 0.67 μm and Vmax = 3.63 × 106 ± 2.02 × 105 pmol · min−1 · mg protein−1 in cells expressing DAT and synaptogyrin-3 versus Km = 3.21 ± 1.13 μm and Vmax = 3.40 × 106 ± 3.49 × 105 pmol · min−1 · mg protein−1 in cells expressing only DAT. The data represent the means ± SE of three independent experiments (*p < 0.05).
The VMAT2 inhibitor reserpine decreases DAT activity and prevents the effect of synaptogyrin-3
As a third explanation, we explored the possibility that the vesicular DA storage system influences the ability of synaptogyrin-3 to regulate DAT activity. Thus, we performed DA uptake experiments in the presence of the VMAT inhibitor reserpine. Acute reserpine preincubation (1 μm; 10 min) of PC12 cells transfected with DAT alone resulted in ∼50% inhibition of DAT activity (Fig. 11A,C). The reduction in DAT activity by reserpine was observed even at concentrations as low as 100 nm (data not shown). These results are consistent with those previously reported by Metzger et al. (2002). [3H]DA uptake in HEK-293 cells transfected with the transporter was not altered by reserpine at concentrations as high as 100 μm (Fig. 11B) indicating that the effect of reserpine on DAT activity was not attributable to a direct inhibition of the transporter protein. Consistent with our previous data, DAT activity was increased in the presence of synaptogyrin-3 overexpression (Fig. 11C). Interestingly, synaptogyrin-3 overexpression failed to increase DAT activity in the presence of reserpine in PC12 cells (Fig. 11C). These results suggest that synaptogyrin-3 may facilitate a functional link between DAT and the DA vesicular storage system.
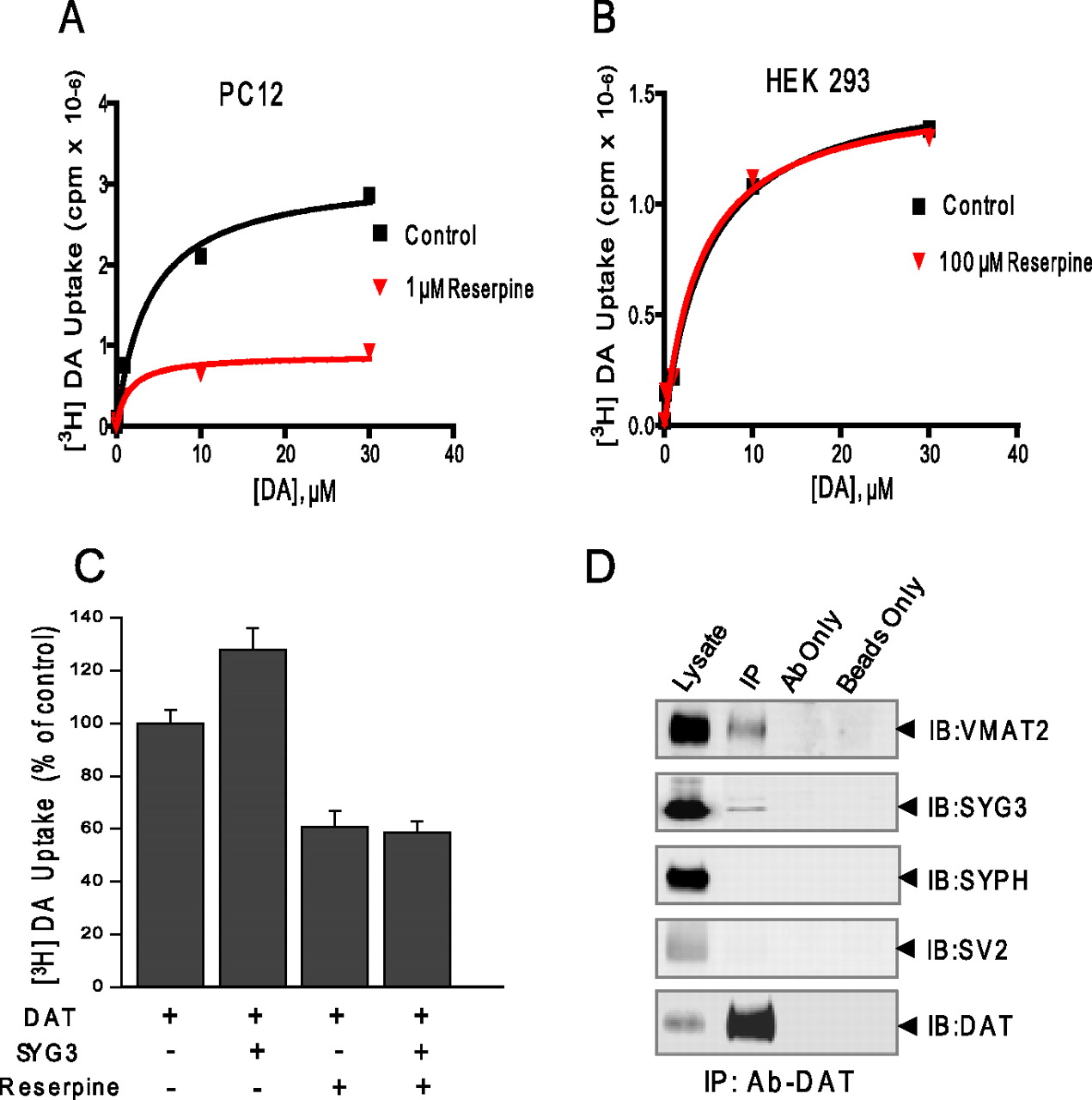
The vesicular DA storage system has a role in the effect of synaptogyrin-3 on DAT activity. A, The VMAT2 inhibitor reserpine (1 μm) reduces DAT activity and prevents the effect of synaptogyrin-3 on DAT activity in PC12 cells transiently cotransfected with DAT and synaptogyrin-3 cDNAs. Uptake assays were performed in the presence (red) or absence (black) of 1 μm reserpine. The data are representative of three independent experiments. B, Reserpine (100 μm) (red) fails to reduce DAT activity in HEK-293 cells transiently cotransfected with DAT and synaptogyrin-3 cDNAs compared with uptake assays performed in the absence of reserpine (black). The data are representative of three independent experiments. C, Synaptogryin-3 fails to increase DAT activity in the presence of 1 μm reserpine in PC12 cells. [3H]DA uptake was performed 96 h after transfection in the presence of 10 μm unlabeled DA. The data represent the means ± SE of three independent experiments. D, Immunoprecipitation of DAT results in the coprecipitation of synaptogyrin-3 and VMAT2 from mouse striatum. The cross linking agent, DSP, was used. Synaptophysin (SYPH) and SV2 did not coprecipitate with DAT, showing that all synaptic vesicle proteins were not cross-linked with DAT. Control experiments show that beads only or DAT antibody alone does not precipitate any of the tested antibodies.
A biochemical complex involving DAT, synaptogyrin-3, and VMAT2
Since our data lead us to speculate that synaptogyrin is capable of facilitating a physical and functional interaction between DAT and synaptic vesicles, we examined the possibility that the plasma membrane transporter and the vesicular monoamine transporter were actually interacting. Using striata lysate generated in the presence of DSP, coimmunoprecipitation experiments were performed using the anti-DAT antibody in the presence of detergent. Consistent with our previous findings, synaptogyrin-3 coprecipitated with DAT (Fig. 11D). More importantly, VMAT2 also successfully coprecipitated with DAT. To confirm that this interaction between VMAT2 and DAT was not a result of cross-linking all vesicular proteins with DAT, we also show that the synaptic vesicle proteins SV2 and synaptophysin fail to coprecipitate with DAT. As additional specificity controls, immunoprecipitations were also performed in the same lysate using only anti-DAT antibody or beads alone, and DAT, VMAT2, SV2, or synaptophysin was not detected in either of these conditions (Fig. 11D). This result indicates that a biochemical complex between DAT, synaptogyrin-3, and VMAT2 may exist and together with our functional data suggests a link between plasma membrane transporter and the vesicular storage system for DA.
Discussion
Synaptogyrin-3 belongs to a multigene family of integral synaptic vesicle proteins including at least two other isoforms (i.e., synaptogyrin-1 and cellugyrin). All members of this family share the same topology, four membrane-spanning domains and both the N and C termini facing the cytoplasmic side of the synaptic membrane. Synaptogyrin-1 and -3 are present in membranes of synaptic vesicles as demonstrated by subcellular fractionation studies, immunoprecipitation of synaptic vesicles from mouse brain, and immunofluorescent staining of brain tissue and neurons in culture (Takamori et al., 2006; this study). Synaptogyrin-3 is expressed in several brain areas including hippocampus, cerebellum, and midbrain DA neurons (Belizaire et al., 2004). It has been speculated that synaptogyrin-3 might have a role in exocytosis since overexpression of this protein in PC-12 cells inhibits the release of cotransfected human growth hormone (Sugita et al., 1999). However, to our knowledge, there are no additional studies examining the function of synaptogyrin-3. Here, we identified synaptogyrin-3 as a DAT interacting protein using the mating-based split ubiquitin yeast two-hybrid system. We demonstrate that DAT can specifically interact with synaptogyrin-3 by coimmunoprecipitation experiments from mouse brain tissue and heterologous cells, FRET assays in live rat hippocampal neurons, and GST coprecipitation experiments. The intracellular N terminus of DAT and the cytoplasmic N-terminal domain of synaptogyrin-3 are sufficient for this interaction. To our knowledge, this is the first study reporting an interaction between a synaptic vesicle protein and a plasma membrane neurotransmitter transporter.
Furthermore, our data show that the N terminus of DAT is capable of interacting with synaptic vesicles (Fig. 5). This physical link between synaptic vesicles and DAT, which might be facilitated by synaptogryin-3, implies that the DAT/synaptogyrin-3 interaction likely occurs in a trans orientation (i.e., the two proteins exist in two separate membrane compartments). Alternatively, we cannot rule out the possibility that DAT and synaptogryin-3 interact in a cis orientation (i.e., the two proteins interact on the same plane of the membrane). This could occur if the membrane of the synaptic vesicle fused with the membrane of the plasma membrane. Indeed, biotinylation experiments paired with coimmunoprecipitation may have provided interesting information regarding these possibilities; however, our efforts were inconclusive because DAT and synaptogyrin-3 did not coimmunoprecipitate in either the membrane fraction or the intracellular fraction. This result may be attributable to two causes. First, the biotinylation process may leave insufficient levels of DAT to be subsequently immunoprecipitated, or alternatively, the conditions used for biotinylation may not be compatible with those for immunoprecipitation. Indeed, we are not aware of any studies in which both these techniques have been used jointly. Additional studies will be needed to conclude unequivocally whether the DAT/synaptogyrin-3 interaction occurs in one or both of these manners.
Our results also show that synaptogyrin-3 expression correlates with DAT activity in MN9D and PC12 cells. Three potential mechanisms were examined to dissect how synaptogyrin-3 overexpression increases DAT activity: (1) increased DAT levels at the plasma membrane, (2) a direct effect of synaptogyrin-3 on transporter function through protein–protein interactions, or (3) a contribution of the vesicular DA system. Indeed, when synaptogyrin-3 was overexpressed, DAT uptake was increased in PC12 and MN9D-DAT cells, but not in HEK-293 cells. Since synaptogyrin-3 and DAT coimmmunoprecipitate in all three cell lines, including HEK-293 cells, the interaction alone is not sufficient to change transporter activity. In agreement with this idea, DAT uptake was decreased when synaptogyrin-3 levels were downregulated by siRNA in PC12 cells. Second, the observed changes in DAT activity by synaptogyrin-3 overexpression or knockdown were not attributable to changes in cell surface levels of DAT, thus ruling out trafficking mechanisms. Instead, the effect of synaptogyrin-3 on DAT activity was abolished in the presence of the VMAT2 inhibitor reserpine, suggesting that a functional vesicular pool was required for the effect of synaptogyrin-3. Finally, synaptogyrin-3 failed to physically or functionally interact with NET in PC12 cells, supporting the specificity its effect on DAT function. Our interpretation of these findings is that a physical link of DAT with synaptic vesicles through synaptogyrin-3 might accelerate uptake by rapidly dissipating the DA concentration gradient. Thus, the transporter would function more efficiently when DA is removed from the cytosolic microenvironment to nearby storage vesicles. Although we are cautious about this interpretation, it is consistent with a recent study demonstrating that pharmacological or genetic inhibition of VMAT2 results in decreased DAT activity (Yamamoto et al., 2007). This decrease in DAT activity was reported to be independent of trafficking mechanisms and substrate inhibition. Interestingly, these authors also found that, in DA neurons from VMAT2 knock-out mice, synaptogyrin-3 expression was also dramatically downregulated (Yamamoto et al., 2007).
Based on our data, it is tempting to speculate that synaptogyrin-3 might facilitate a physical and functional link between the DAT and the DA storage system. Thus, the physical interaction between DAT and synaptogyrin-3 might play a role in docking synaptic vesicles at the plasma membrane near DAT to facilitate a more efficient loading of the vesicles with extracellular DA after release. Indeed, our studies also show an interaction between DAT and VMAT2, the vesicular transporter primarily responsible for loading synaptic vesicles with DA. Molecular interactions that link DAT to proteins of the synaptic vesicle machinery could contribute to a rapid and efficient coupling between plasma membrane DA uptake and vesicular refilling by optimally locating vesicles near the plasma membrane transporter.
Electron microscopy studies have shown that DAT is located at perisynaptic areas away from sites for neurotransmitter release (Hersch et al., 1997; Nirenberg et al., 1997). Thus, our findings raise questions as to whether it is plausible that synaptic vesicles would interact with a transporter located at perisynaptic sites. Release of neurotransmitters at the active zone is followed by retrieval of the synaptic vesicles to refill them for subsequent use. Indeed, ample evidence indicates that synaptic vesicles are organized into at least two distinct functional groups: a readily releasable pool and a reserve pool (for review, see Fon and Edwards, 2001). The readily releasable pool is located at the active zone near the plasma membrane, whereas the reserve pool is distributed throughout the terminal. The latter is clearly evident in experiments using electron microscopy of nerve terminals (Schikorski and Stevens, 2001). Even in DA nerve terminals, it is clear that synaptic vesicles are distributed uniformly throughout the terminal (Hersch et al., 1997; Nirenberg et al., 1997; Melchitzky et al., 2006). In addition, there is evidence for at least three distinct retrieval mechanisms: kiss and run, kiss and stay, and clathrin-mediated endocytosis. Additional evidence indicates that the machinery involved in synaptic vesicle recycling for clathrin-mediated endocytosis is away from the active site (Yao et al., 2005, and references therein). Thus, it is plausible that synaptic vesicles come in contact with sites near DAT at the perisynaptic areas when recycling occurs or that a pool of vesicles resides near DAT ready for refilling after DA is released. Additional studies are needed to distinguish between these two possibilities.
Indeed, protein–protein interactions between plasma membrane signaling proteins, including ion channels and receptors, with synaptic vesicle proteins have been described previously (Jarvis and Zamponi, 2001). The most studied and well understood interaction is that of N-type and P/Q-type voltage-gated calcium channels with key proteins of the vesicle release machinery including synaptotagmin I (Catterall, 1999; Atlas et al., 2001). These interactions play an important role in docking synaptic vesicles in close proximity to channels to ensure a local and temporal coupling between calcium transients and neurotransmitter release. Voltage-sensitive sodium channels have also been shown to be associated with synaptotagmin I, although the physiological significance of this interaction has not been elucidated (Sampo et al., 2000). Recently, the synaptic vesicle protein CAPS1 was identified as a D2 DA receptor interacting protein and a functional interaction between DA receptors with components of the exocytotic machinery was suggested (Binda et al., 2005).
Thus, our study opens a new series of questions regarding the role of DAT in the regulation of DA homeostasis. Although our data show that the N termini of both DAT and synaptogyrin-3 are sufficient for the DAT/synaptogyrin-3 interaction, additional studies will be necessary to consider whether additional domains of either protein are involved in this interaction and whether other protein–protein interactions allow the plasma membrane transporter to interact with the vesicular DA storage system. Likewise, it will be important to consider the contribution of the DAT/synaptogyrin-3 interaction in the effect of psychostimulants and vice versa whether psychostimulants affect this interaction. Indeed, additional identification of synaptic vesicle proteins as DAT interactors along with the molecular bases and physiological significance of such interactions will result in a better understanding of the role DAT plays in regulating DA homeostasis in the brain.
Footnotes
-
This work was supported by the National Institute on Drug Abuse–National Institutes of Health Grant DA 016710-02 (G.E.T.), the National Alliance for Schizophrenia and Depression (G.E.T.), the National Science Foundation (G.E.T.), and the Austrian Science Fund/Fonds zur Förderung der Wissenschaftlichen Forschung Grants P-17076 and SFB35 (H.H.S.). We thank members of the Amara and Torres Laboratory for helpful discussions. We are also grateful to Drs. Alfred Heller and Lisa Won for providing the MN9D cells and Dr. Carl Lagenaur for help with the velocity gradient experiments. G.E.T. dedicates this work to Dr. Marc G. Caron, Duke University.
- Correspondence should be addressed to Dr. Gonzalo E. Torres, Department of Neurobiology, University of Pittsburgh School of Medicine, Room 6061, BST3, 3501 Fifth Avenue, Pittsburgh, PA 15261. gtorres{at}pitt.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}