

Abstract
The amyloid β-protein precursor (AβPP) is best recognized as the precursor to the Aβ peptide that accumulates in the brains of patients with Alzheimer's disease, but less is known about its physiological functions. Isoforms of AβPP that contain a Kunitz-type serine proteinase inhibitor (KPI) domain are expressed in brain and, outside the CNS, in circulating blood platelets. Recently, we showed that KPI-containing forms of AβPP regulates cerebral thrombosis in vivo (Xu et al., 2005, 2007). Amyloid precursor like protein-2 (APLP2), a closely related homolog to AβPP, also possesses a highly conserved KPI domain. Virtually nothing is known of its function. Here, we show that APLP2 also regulates cerebral thrombosis risk. Recombinant purified KPI domains of AβPP and APLP2 both inhibit the plasma clotting in vitro. In a carotid artery thrombosis model, both AβPP−/− and APLP2−/− mice exhibit similar significantly shorter times to vessel occlusion compared with wild-type mice indicating a prothrombotic phenotype. Similarly, in an experimental model of intracerebral hemorrhage, both AβPP−/− and APLP2−/− mice produce significantly smaller hematomas with reduced brain hemoglobin content compared with wild-type mice. Together, these results indicate that AβPP and APLP2 share overlapping anticoagulant functions with regard to regulating thrombosis after cerebral vascular injury.
Introduction
The amyloid β-protein precursor (AβPP), a type I transmembrane protein, is mostly recognized as the precursor to the amyloid β-peptide (Aβ) that accumulates in the brains of patients with Alzheimer's disease and related disorders (Hardy and Selkoe, 2002). Aβ is derived through sequential amyloidogenic proteolytic processing by β- and γ-secretase activities (Vassar et al., 1999; Wolfe et al., 1999). Alternatively, AβPP can undergo nonamyloidogenic proteolytic processing via a single cleavage through the Aβ domain by α-secretase resulting in release of the large extracellular domain of the protein into the external environment (Esch et al., 1990). AβPP can be derived from predominantly three alternatively spliced mRNAs of a common gene located on chromosome 21 giving rise to proteins of 695, 751, and 770 aa; the larger two isoforms contain an additional 56 aa domain that is structurally and functionally related to Kunitz-type serine proteinase inhibitors (KPI) (Ponte et al., 1988; Tanzi et al., 1988). Previously, we showed that secreted KPI domain-containing forms of AβPP are analogous to the cell-secreted proteinase inhibitor known as protease nexin-2 (PN2) (Van Nostrand et al., 1989).
Although much has been learned about the proteolytic processing of AβPP and generation of Aβ peptide, comparatively, little is known about its physiological functions. We and others have reported that both purified PN2/AβPP and its recombinantly expressed KPI domain are potent, tight-binding inhibitors of certain serine proteinases, most notably several prothrombotic enzymes including factor XIa, factor IXa, factor Xa, and tissue factor:factor VIIa complex (Smith et al., 1990; Van Nostrand et al., 1990; Schmaier et al., 1993; Mahdi et al., 1995). Recently, we showed that transgenic mice with specific and modest over-expression of PN2/AβPP either in platelets or in brain present with a significant antithrombotic phenotype suggesting that this protein has an important role in regulating cerebral thrombosis risk in vivo (Xu et al., 2005, 2007).
However, mice lacking AβPP exhibit significantly increased thrombosis in cerebral vascular injury models, although the impairment was not overly severe (Xu et al., 2005). However, this situation is confounded by the presence of the highly conserved amyloid precursor like protein 2 (APLP2) that also can contain a KPI domain that inhibits prothrombotic enzymes (Wasco et al., 1993; Slunt et al., 1994; Van Nostrand et al., 1994). Here, we show that the KPI domains of PN2/AβPP and APLP2 similarly inhibit the clotting of plasma in vitro. Accordingly, AβPP−/− mice and APLP2−/− mice exhibit a comparable prothrombotic phenotype using an in vivo model of carotid artery thrombosis. Moreover, AβPP−/− mice and APLP2−/− mice similarly produce significantly reduced pathology in experimental intracerebral hemorrhage. Together, these findings suggest that AβPP and APLP2 are a unique pair of proteolytic inhibitors that possess overlapping and shared activities in regulating thrombosis risk during cerebral vascular injury.
Materials and Methods
Recombinant expression and purification of the KPI domains of AβPP and APLP2.
Construction of the recombinant vectors, expression in Pichia pastoris, and purification of the KPI domains of AβPP and APLP2 was performed as described previously (Wagner et al., 1992; Van Nostrand et al., 1994).
Enzyme kinetics.
The ability of the KPI domains of AβPP or APLP2 to inhibit factor XIa or Xa (Hematological Technologies) hydrolysis of chromogenic substrate S2366 or S2222 (Diapharma), respectively, was determined as reported previously (Mahdi et al., 1995, Van Nostrand et al., 1990). Kinetic constants were calculated by nonlinear regression analysis to the integrated rate equation for slow-binding inhibitors (Huang et al., 1993).
In vitro plasma clotting assay.
Microtiter plate-activated partial thromboplastin time (APTT) assays were conducted by adding 30 μl of citrated pooled normal human plasma, 30 μl of APTT reagent, and 30 μl of Tris-buffered saline alone or in the presence of increasing concentrations of purified KPI–AβPP and/or KPI–APLP2 in triplicate microtiter plate wells followed by incubation for 10 min at 22°C (Pratt and Monroe, 1992). Clotting was initiated by adding 30 μl of 25 mm CaCl2, and the time to clot formation was monitored by absorbance change at 405 nm recorded every 5 s for 5 min in a Vmax kinetic microtiter plate reader (Molecular Devices).
AβPP and APLP2 gene knock-out mice.
All work with animals followed National Institutes of Health guidelines and was approved by the Stony Brook University Institutional Animal Care and Use Committee. Mice deficient for the AβPP gene (AβPP−/−) or the APLP2 gene (APLP2−/−) were obtained from The Jackson Laboratory. For genotyping purposes, the wild-type App allele was identified by PCR using sense primer, 5′-AGAGCACCGGGAGCAGAG-3′ and antisense primer, 5′-AGCAGGAGCAGTGCCAAG-3′, resulting in a 161 bp product. The homozygous App knock-out offspring were identified by PCR using sense primer, 5′-CTTGGGTGGAGAGGCTATTC-3′ and antisense primer, 5′-AGGTGAGATGACAGGAGATC-3′, resulting in a 280 bp product. Similarly, for genotyping purposes, the wild-type Aplp2 allele is identified by PCR using sense primer, 5′ GCCAAGCTTGAGTCGGTGTATCCGTGCT 3′ and the antisense primer, 5′ GCGACCGGAGGAGACGCAGATCGGGAGCTCGCC 3′, resulting in a 400 bp product. The homozygous Aplp2 knock-out offspring were identified by PCR using sense primer, 5′CCATTGCTCAGCGGTGCTG 3′, located within the 5′ promoter region of the pKGneo gene and antisense primer above, resulting in a 350 bp PCR product. All gene knock-out and wild-type mice were on pure C57BL/6 backgrounds and used at 3 months of age.
Carotid artery thrombosis.
This procedure was performed essentially as described by Eitzman et al. (2000) and induces a fibrin and platelet-rich clot. Briefly, mice (12 weeks of age) were prepared for surgery and anesthetized by intraperitoneal injection of sodium pentobarbital (70 mg/kg). A midline neck incision was made to expose the common carotid artery that was then cradled by a miniature flowmeter probe to record blood flow rate and ultimately determining presence of thrombosis. Then, 0.1 cc of Rose Bengal (4,5,6,7-tetrachloro-3′,6-dihydroxy-2,4,5,7-tetraiodospiro(isobenzo-furan-1(3H),9[9H]xanthan)-3-1-dipotassium salt) (50 mg/kg in 0.9% saline) was injected through the tail vein which is activated by laser light (540 nm) to generate a superoxide anion. The superoxide anion leads to endothelial cell damage with a transient thrombus and subsequent neointima formation. When the blood flow ceased for 20 min within the laser specific area, retrospectively, the time to occlusion was documented.
Experimental intracerebral hemorrhage.
This model was performed essentially as described by Clark et al. (1998). Briefly, mice (12 weeks of age) were prepared for surgery and anesthetized by intraperitoneal injection of sodium pentobarbital (70 mg/kg). A sagittal incision was made caudal to rostral allowing the scalp to be retracted and held in place with microclips. A small hole, 1.0 mm posterior and 3.0 mm lateral of bregma, was drilled to perforate the skull. A 1 μl Hamilton syringe was used to deliver 500 nl of bacterial collagenase/saline (150 U/ml) to the caudate/putamen at a depth of 4.0 mm unilaterally. After the injection of collagenase/saline (∼30 s), the needle remained in place for another 2 min to prevent reflux of fluid. The surgery was concluded with the closing of the scalp skin using 4-0 nylon sutures. Twenty-four hours after initiation of hemorrhage, the mice were perfused with PBS, the brains were harvested, 14 μm sections were prepared using a cryostat, and mounted on glass slides. Sections were stained with hematoxylin and an Olympus BX60 upright systems microscope with a digital camera was used to capture images. The hemorrhage volume was measured using the Stereologer software system. Alternatively, harvested perfused brains were divided midline sagittally, and the hemoglobin levels were determined in the hemorrhage and contralateral hemispheres using a spectrophotometric assay as a measure of the extent of hemorrhage in the lesioned hemispheres of the mice (Choudhri et al., 1997).
Statistical analysis.
The data were analyzed by one-way ANOVAs for each measure. Significant ANOVAs (p < 0.05) were followed by Fisher's post hoc tests, the results of which are reported in the corresponding figure legends.
Results
KPI domains of APLP2 and AβPP inhibit plasma clotting and factors XIa and Xa enzymatic activity in vitro
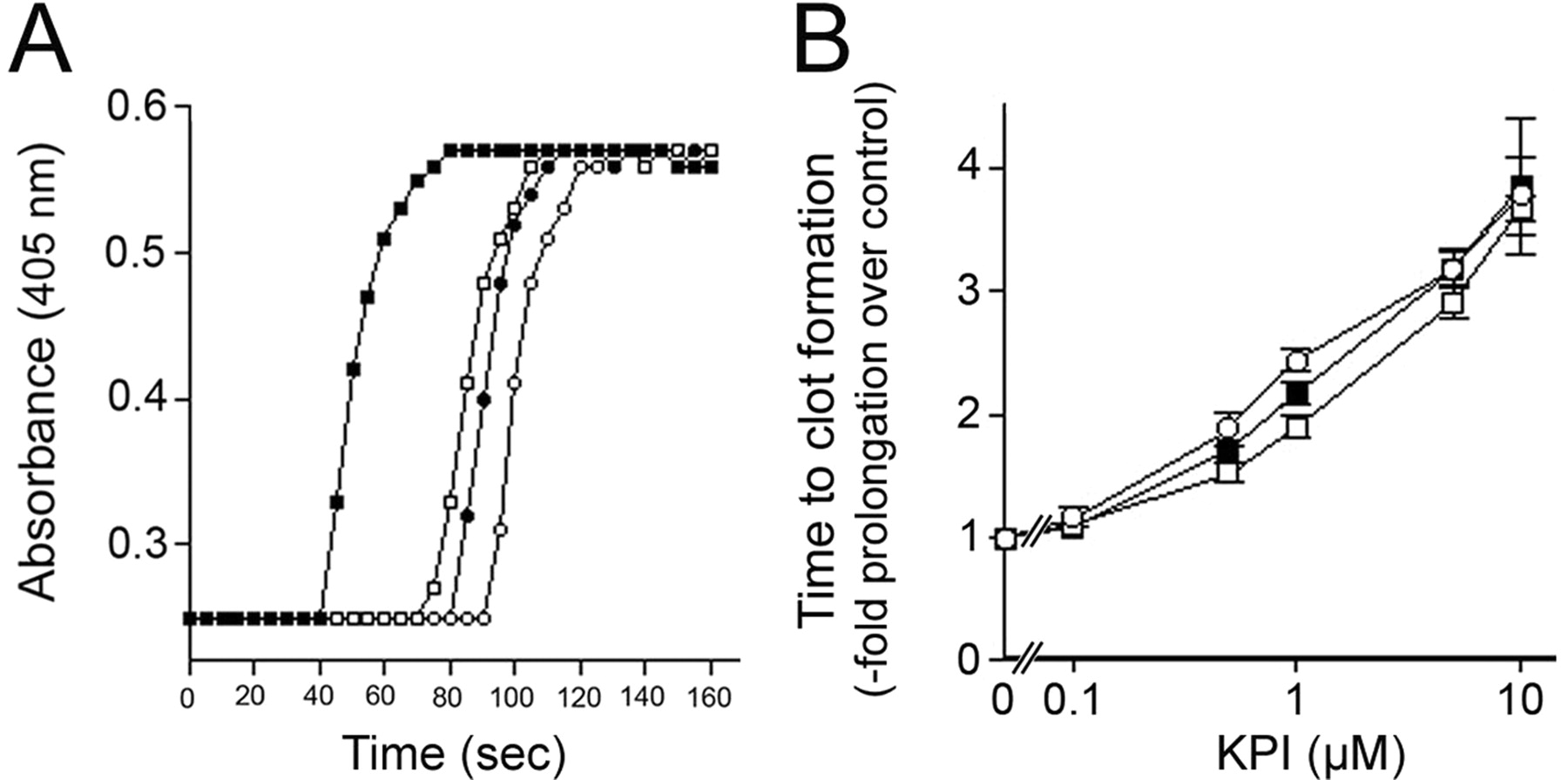
We first compared the activities of the purified KPI domains of APLP2 and AβPP in inhibiting specific clotting enzymes. The KPI domains of AβPP or APLP2 inhibited factor XIa with a Ki of 1.9 ± 0.7 or 46.6 ± 13.4 nm, respectively. KPI domain of AβPP (Ki = 14 ± 2 nm), but not that of APLP2, inhibited factor Xa. Neither KPI domain inhibited factor XIIa or thrombin. We next compared their abilities to inhibit coagulation in vitro in an APTT assay. Figure 1A shows that in the APTT assay at 1 μm, the KPI domains of APLP2 or AβPP delayed the time to change the absorbance. Both KPI domains showed a dose-dependent prolongation of the time for plasma to clot (Fig. 1B). A concentration of ≈0.6 μm AβPP KPI domain resulted in a doubling in the time for plasma to clot, whereas ≈1 μm of the APLP2 KPI domain was needed to obtain the same effect. An equimolar mixture of each KPI domain produced an intermediary effect with ≈0.8 μm needed to double the time for clot formation. These findings indicate that while both recombinant KPI domains could inhibit the clotting of plasma in vitro, the KPI domain of AβPP was somewhat better in this assay than the KPI domain of APLP2.

The KPI domains of APLP2 and AβPP similarly inhibit the clotting of plasma in vitro. A, Representative APTT assay measuring the time to plasma clot formation for normal human plasma in the absence (■) or presence of 1 μm KPI domain of APLP2 (□), 1 μm KPI domain of AβPP (○), or a combination of 0.5 μm of each KPI domain (●). B, Quantitation of the time to plasma clot formation in the presence of increasing concentrations of the KPI domain of APLP2 (□), KPI domain of AβPP (○), or an equimolar mixture of each KPI domain (●). Data shown are the mean ± SD of triplicate samples at each concentration of KPI domain and expressed as -fold prolongation to clot formation compared with plasma alone.
Loss of APLP2 or AβPP results in comparable enhancement of carotid artery thrombosis in mice
Since the above results suggest that increasing amounts of APLP2 or AβPP can similarly inhibit clotting in vitro, we next determined if the loss of either protein would exert a similar influence on cerebral thrombosis risk. A quantitative carotid artery thrombosis model was performed using APLP2−/− and AβPP−/− mice. Progression curves for the time of clot formation and cessation of carotid artery blood flow in wild-type mice, APLP2−/− mice, and AβPP−/− mice showing a prothrombotic phenotype in each of the gene knock-out mice compared with wild-type mice can be seen in Figure 2A. This assay performed in multiple mice of each genotype showed a highly significant 34 and 27% reduction in the time to cessation of blood flow in APLP2−/− mice and AβPP−/− mice, respectively, compared with wild-type mice (p < 0.001 and p < 0.0001, respectively) (Fig. 2B). These findings suggest that loss of either APLP2 or AβPP produces a similar prothrombotic phenotype in mice.

The lack of APLP2 or AβPP promotes cerebral thrombosis. A, Mice of different genotypes were injected with the photoactivated dye Rose Bengal at t = 0, and the carotid artery was exposed to a laser light. Blood flow through the carotid artery was monitored with a flow probe. Blood flow ceased because of thrombus formation in the vessel. Representative experiments showing wild-type mice (solid line), APLP2−/− mice (small dashes), and AβPP−/− (large dashes) mice. B, Quantitation of the time to cessation of carotid artery blood flow in mice of different genotypes. Data shown are the mean ± SD of n = 14 mice per each genotype. *p < 0.001; **p < 0.0001.
Loss of APLP2 or AβPP similarly reduces hemorrhagic pathology in mice
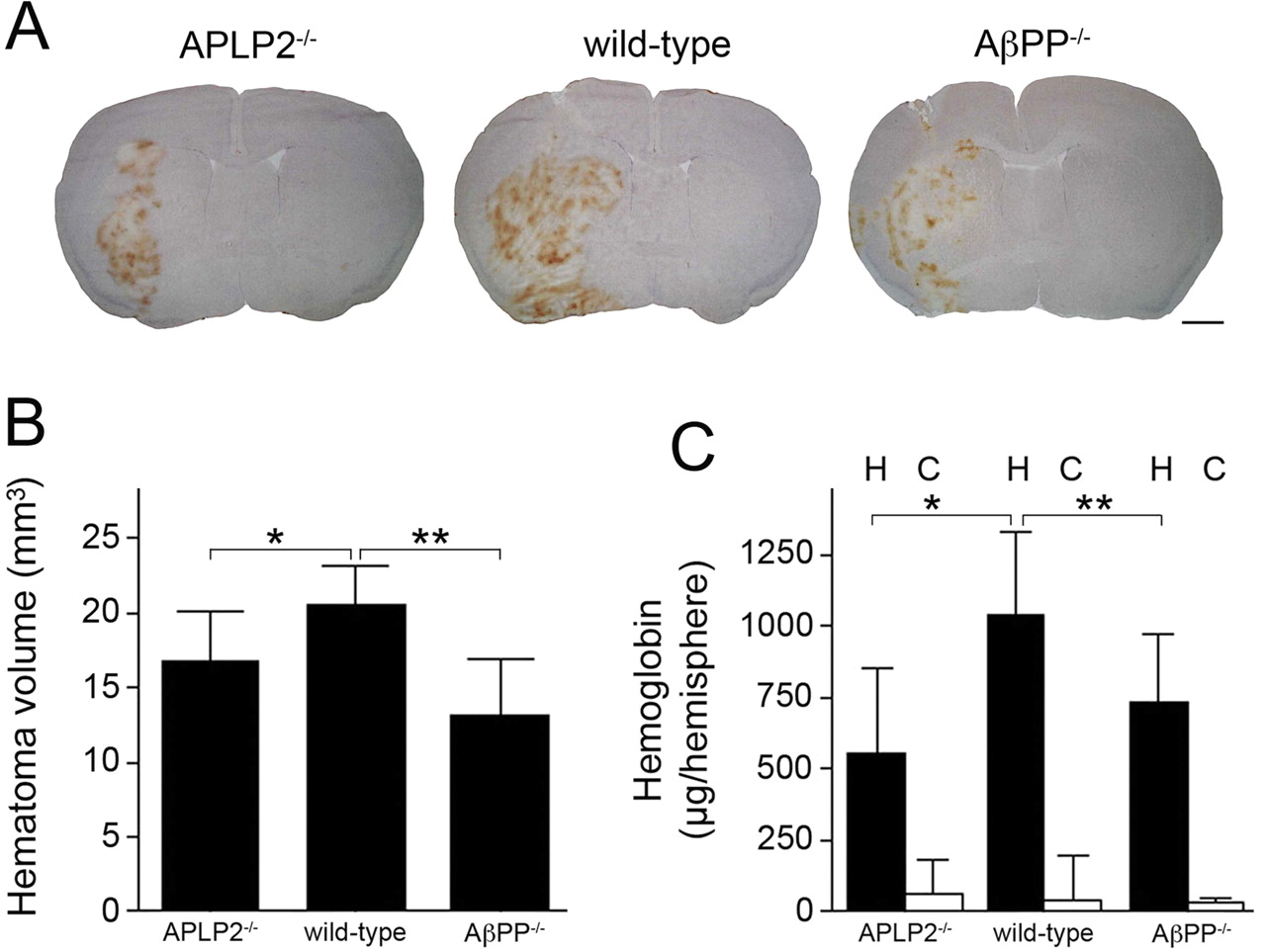
We next determined the consequences of the loss of APLP2 or AβPP in experimental intracerebral hemorrhage in mice, another model that evaluates cerebral thrombosis. Figure 3A shows representative hematomas in brain sections of APLP2−/− mice (left), wild-type mice (middle), and AβPP−/− mice (right). Figure 3B presents quantitation of the hematoma volumes from a number of mice of each genotype showing an ≈203% reduction in APLP2−/− mice and ≈35% reduction in AβPP−/− compared with wild-type mice (p < 0.005 and p < 0.001, respectively). As an independent measure of bleeding severity, we quantitated the hemoglobin content in the ipsilateral hemorrhagic and contralateral control hemispheres in mice of each genotype. As shown in Figure 3C, hemoglobin content was reduced by ≈40% in APLP2−/− mice and ≈30% in AβPP−/− mice compared with wild-type mice (p < 0.01 and p < 0.02, respectively). The hemoglobin content in the contralateral control hemispheres was similarly low in mice of each genotype. Together, these results indicate that loss of APLP2 or AβPP results in a similar prothrombotic phenotype leading to decreased severity in intracerebral hemorrhage.

The lack of APLP2 or AβPP similarly reduces the severity of intracerebral hemorrhage. A, Mice were stereotaxically injected with collagenase in the caudate/putamen. Twenty-four hours later, the brain was removed and analyzed for hematoma. Stained mouse brains sections from APLP2−/− (left), wild-type (middle), and AβPP−/− (right). B, Quantitation of hematoma volume from APLP2−/− mice, wild-type mice, and AβPP−/− mice at 24 h. The data presented are the mean ± SD of n = 13 mice for each genotype. *p < 0.005; **p < 0.001. C, Quantitation of hemoglobin levels in the hemorrhagic (H) and contralateral control (C) hemispheres from APLP2−/− mice, wild-type mice, and AβPP−/− mice at 24 h. The data presented are the mean ± SD of n = 8 mice for each genotype. *p < 0.01; **p < 0.02.
Discussion
AβPP is most commonly known as the precursor to the Aβ peptide; however, much less is known about its physiological functions and its potential participation in other pathological events. Although numerous and varied functional domains and activities have been identified for AβPP in vitro translation to observable effects in vivo has essentially been lacking. Previously, we provided in vivo experimental evidence that PN2/AβPP regulates cerebral thrombosis (Xu et al., 2005, 2007).
Physiological regulation of thrombosis is mainly accomplished by several key regulators including activated protein C, antithrombin III, and tissue factor pathway inhibitor (Griffin et al., 2002; Feistritzer and Wiedermann, 2007; Crawley and Lane, 2008). These anticoagulants are mainly effective in the nonneural, peripheral vascular beds of the body including heart, lung, liver, spleen, and gastrointestinal tract. Deletion of any of these particular genes in mice results in lethality attributable to unregulated thrombosis (Huang et al., 1997; Jalbert et al., 1998; Ishiguro et al., 2000). In the brain, there is a relative paucity of thrombomodulin, an essential cofactor for protein C activation (Maruyama et al., 1985). AβPP and, more recently, APLP2 have been proposed as cerebral anticoagulants. The severity of the prothrombotic risk of AβPP is not the same as protein C or antithrombin III. Although the deletion of the AβPP gene in mice produced a significant prothrombotic phenotype, it was neither lethal nor overly severe. The function of AβPP is complicated by the presence of the highly homologous APLP2. The APLP2 protein also possesses a KPI domain that is structurally and functionally very similar to the KPI domain contained in PN-2/AβPP (Wasco et al., 1993; Slunt et al., 1994). In the present study, we show that both purified recombinant KPI domains inhibit similar coagulation enzymes and exhibit dose-dependent inhibition of clotting in vitro. Earlier work showed that the proteinase inhibitory properties of the purified recombinant KPI domain of AβPP were comparable with that of purified secreted PN2/AβPP protein, indicating that this activity of AβPP, and likely APLP2, resides solely within their respective KPI domains (Wagner et al., 1992; Schmaier et al., 1993). Moreover, APLP2, like PN2/AβPP, is found in many tissues and is abundant in brain and in platelets (Webster et al., 1995; Sisodia et al., 1996). This suggests that each of these proteins may have shared and overlapping activities in regulating thrombosis during cerebral vascular injury. Here, we show that using a model for carotid artery thrombosis, which is a measure of intraluminal thrombosis, that deletion of either APLP2 or AβPP in mice results in a similar increase in prothrombotic risk in vivo. Previously, we showed that platelet-poor plasma prepared from AβPP−/− mice exhibited the same clotting times as platelet-poor plasma prepared from wild-type mice indicating that the plasma itself from the AβPP−/− animals was not functionally different (Xu et al., 2005). AβPP and APLP2 are not plasma proteins. However, platelet-rich plasma prepared from AβPP−/− mice exhibited significantly shorter times for clotting compared with platelet-rich plasma prepared from wild-type animals. These data indicate that the presence of AβPP in the platelets influences the clotting of platelet-rich plasma, suggesting that in the absence of AβPP or APLP2, there is shortened arterial thrombosis times attributable to the loss of these proteins from platelets and other cells.
It was previously reported that mice that are deficient for both APLP2 and AβPP exhibit an early postnatal lethal phenotype (von Koch et al., 1997). Although the precise basis for the lethality in the APLP2/AβPP double gene knock-out mice is presently unknown, further study is needed to determine if it involves the overlapping prothrombotic proteinase inhibitory roles or some other redundant function between the two proteins. Although a defined role for either APLP2 or AβPP in cerebral thrombotic disorders is not currently known, several lines of evidence suggest that these two inhibitory proteins may participate in cerebral vascular homeostasis and thrombosis at some level. For example, adults with Down's syndrome, a disease caused by an extra copy of chromosome 21 that harbors the AβPP gene, have been reported to express higher levels of AβPP (Cheon et al., 2008). There are reports of middle cerebral artery stenosis with subarachnoid hemorrhage in Down's syndrome (Donahue et al., 1998). Furthermore, Moyamoya disease, a rare disorder characterized by progressive intracranial vascular stenosis with successive ischemic and hemorrhagic events, is more frequently observed in Down's syndrome (Jea et al., 2005; Rafay et al., 2006; Chaanine et al., 2008). However, the defect in Moyamoya disease appears to be structural, and the hemorrhage associated with it may be anatomically based, rather than that seen in Down's syndrome, which may arise because of excessive local anticoagulation attributable to increased local expression of AβPP. Last, a familial form of intracranial aneurysm and hemorrhage has been linked to chromosome 11q24–25 (Ozturk et al., 2006), the same region where the APLP2 gene resides (Leach et al., 1999). However, a possible genetic link between AβPP and APLP2 with intracranial stenosis, aneurysm, and hemorrhage needs to be further explored.
Previously, it was shown that fibrillogenic forms of Aβ can stimulate the expression and cell surface accumulation of AβPP and APLP2 in cultured human cerebrovascular smooth muscle cells (Davis-Salinas et al., 1995; Saporito-Irwin et al., 1997; Van Nostrand et al., 1998). Aβ fibrils bind AβPP through a specific N-terminal domain and can stimulate its activity toward inhibiting certain prothrombotic enzymes (Wagner et al., 2000; Van Nostrand et al., 2002). In this regard, with cases of cerebral amyloid angiopathy, it was shown that there is pathologic accumulation of AβPP and APLP2 in cerebral blood vessel walls (Rozemuller et al., 1993; McNamara et al., 1998). This event could result in an increase in the antithrombotic potential of affected vessels and could contribute to the hemorrhagic condition that is characteristic in severe cases of cerebral amyloid angiopathy. In any case, the present findings provide significant in vivo evidence that AβPP and APLP2 comprise a highly homologous pair of proteinase inhibitors that share redundant and overlapping activities with respect to regulating cerebral thrombosis.
Footnotes
-
This work was supported by National Institutes of Health Grants RO1-NS052533 and RO1-HL052779.
- Correspondence should be addressed to Dr. William E. Van Nostrand, Department of Medicine, HSC T-15/083, Stony Brook University, Stony Brook, NY 11794-8153. William.VanNostrand{at}stonybrook.edu





{kind=link}
{kind=link}
{kind=link}