

Abstract
The CNS-restricted versican splice-variant V2 is a large chondroitin sulfate proteoglycan incorporated in the extracellular matrix surrounding myelinated fibers and particularly accumulating at nodes of Ranvier. In vitro, it is a potent inhibitor of axonal growth and therefore considered to participate in the reduction of structural plasticity connected to myelination. To study the role of versican V2 during postnatal development, we designed a novel isoform-specific gene inactivation approach circumventing early embryonic lethality of the complete knock-out and preventing compensation by the remaining versican splice variants. These mice are viable and fertile; however, they display major molecular alterations at the nodes of Ranvier. While the clustering of nodal sodium channels and paranodal structures appear in versican V2-deficient mice unaffected, the formation of the extracellular matrix surrounding the nodes is largely impaired. The conjoint loss of tenascin-R and phosphacan from the perinodal matrix provide strong evidence that versican V2, possibly controlled by a nodal receptor, organizes the extracellular matrix assembly in vivo.
Introduction
Neuronal and glial cells of the adult CNS are surrounded by specialized extracellular matrices (ECMs) that assemble during maturation and replace the loose meshwork of the late embryonic and early postnatal phase [for review, see Bandtlow and Zimmermann (2000), Yamaguchi (2000), Rauch (2004), and Zimmermann and Dours-Zimmermann (2008)]. Both juvenile and mature forms of the CNS matrix are composed of chondroitin sulfate proteoglycans (CSPGs), tenascins, link proteins, and hyaluronan. The majority of the CSPGs belongs to the lectican family that includes brevican, neurocan, aggrecan, and versican. Lecticans share N- and C-terminal globular structures (G1 and G3, respectively) separated by unique glycosaminoglycan (GAG) carrying middle portions of variable size. Moreover, alternative incorporation of the two glycosaminoglycan attachment regions, GAG-α and GAG-β, results in four versican splice variants (V0 contains the GAG-α and GAG-β domain, V1 only GAG-β, V2 only GAG-α, and V3 neither of these domains) (Dours-Zimmermann and Zimmermann, 1994; Zako et al., 1995).
All lecticans bind via their G1 domain to hyaluronan. This interaction is stabilized by one of four link proteins (HAPLN1–4) (Spicer et al., 2003). In addition, the lectin-like element in the C-terminal G3 domain displays in vitro moderate-to-high binding affinities toward tenascin-R (TnR) (Aspberg et al., 1997), tenascin-C (TnC) (Day et al., 2004), and sulfated glycolipids (Miura et al., 1999). The trimeric TnR and the hexameric TnC glycoproteins may cross-link the G3 domains of lecticans and, thus, tie up the extracellular network (Lundell et al., 2004). Finally, phosphacan, a secreted CSPG form of the receptor-like protein tyrosine phosphatase β (RPTPβ) (Maurel et al., 1994), joins the complex.
In the adult CNS, versican V2 and HAPLN2/Bral1 are the prominent components of the white matter ECM and particularly accumulate at the nodes of Ranvier (Schmalfeldt et al., 1998, 2000; Oohashi et al., 2002; Melendez-Vasquez et al., 2005). Conversely, aggrecan, HAPLN4/Bral2, and neurocan associate with perineuronal nets (Brückner et al., 2000; Bekku et al., 2003). Brevican, hyaluronan, TnR, and phosphacan are observed in both of these meshworks.
Since lecticans strongly inhibit axonal growth in vitro, these specialized ECMs might participate in consolidating myelinated fiber tracts, limiting structural plasticity, and restricting regeneration in the maturating CNS (Yamaguchi, 2000; Zurn and Bandtlow, 2006; Galtrey and Fawcett, 2007; Zimmermann and Dours-Zimmermann, 2008). Furthermore, they may regulate the assembly of axoglial complexes and facilitate the induction and propagation of action potentials at the axon initial segments (AISs) and the nodes of Ranvier (Brückner et al., 1993, 2006; Poliak and Peles, 2003; Salzer, 2003; Sherman and Brophy, 2005; Hedstrom and Rasband, 2006), or they could fulfill neuroprotective functions (Morawski et al., 2004).
To further explore these potential roles in vivo, we have now suppressed the expression of versican V2, one of the main constituents of the mature neural ECM. We avoided the early embryonic lethality of the complete gene knock-out (Mjaatvedt et al., 1998) by using an unconventional isoform-specific gene targeting strategy. The newly generated mouse strain is viable and fertile, but displays major aberrations in the matrix assembly.
Materials and Methods
Generation of versican V0/V2 KO mice.
Using a targeting vector containing a floxed neomycin-thymidine kinase (neo-tk) selection cassette under the control of the HSV-tk promoter, we introduced an ER-retention signal followed by an early translational stop codon into the GAG-α encoding exon VII of the mouse versican gene (codon-insertion KDEL-stop after E749, Swiss-Prot Q62059) (Fig. 1). The two 3.9-kb-long genomic arms of the construct included corresponding parts of exon VII plus adjacent intron sequences, previously cloned from syngeneic DNA by PCR (primer sequences and PCR conditions available on request). Electroporation of the linearized construct into R1 129Sv embryonic stem (ES) cells (Nagy et al., 1993), selection, and screening of ES cell clones were done as previously described (Talts et al., 1999). Homologous recombinants were identified by Southern blot analysis using a digoxigenin (DIG)-labeled probe (Roche Applied Science) downstream of the target site. The ES cells were injected into C57BL/6J blastocysts and transferred into a foster mother. The chimeric offspring were cross-bred with C57BL/6J WT mice. Germline transmission of the mutated versican allele was verified by Southern blotting and long-distance PCR.
The floxed neo-tk selection cassette was removed in vivo by cross-breeding heterozygous V0/V2 neo-tk animals with a mouse strain expressing the CRE transgene under the control of the cytomegalovirus promoter (Schwenk et al., 1995). Correct target integration and recombination was verified by PCR and sequencing of the modified allele. The mutant mice, named VCAN(tm1Zim), were backcrossed to establish 129Sv inbred and C57BL/6J outbred strains.
Northern blotting and quantitative RT-PCR.
Primary fibroblasts were isolated from E14.5 embryos and maintained in culture (Talts et al., 1999). Total RNA extraction, Northern blotting, and DIG labeling of a riboprobe hybridizing with the hyaluronan-binding region of the mouse versican mRNA (HABR, positions 458–1203, GenBank D28599) were done as described previously (Zimmermann et al., 1994).
To determine relative amounts of versican mRNA expression in the mutant versus WT mouse brains, quantitative RT-PCR was performed with an ABI PRISM 7700 Sequence Detection System using the QuantiTect SYBR Green RT-PCR Kit (Qiagen). Total RNAs from brains of littermates were extracted with RNeasy Protect kit (Qiagen). Primers detecting the different splice variants of versican were designed to cover the isoform-specific exon–exon boundaries (supplemental Table 1, available at www.jneurosci.org as supplemental material). Threshold cycle (CT) values from at least three animals per genotype and per developmental time point were normalized against glyceraldehyde-3-phosphate dehydrogenase mRNA (mGAPDH, GenBank M32599) as endogenous control. Relative mRNA amounts (2−ΔΔCT) were calculated on the basis of the comparative CT method, applying the formula ΔΔCT = (ΔCT KO − ΔCT WT). The ΔCT of each measurement was determined by subtracting the GAPDH CT from the versican CT value (ΔCT = VC CT − GAPDH CT).
Antibodies.
All polyclonal antibodies have been raised against recombinant core protein fragments as described previously (Zimmermann et al., 1994). The GAG-α-specific antibodies (Schmalfeldt et al., 2000) recognize the N-terminal portion of this domain of mouse versican (amino acids 362–585, Swiss-Prot Q62059), also present in the potentially translated truncated V0/V2 polypeptide of the mutant mice. The GAG-β antigen comprised amino acids 2750–3040. Both versican fragments were used to generate rabbit and guinea pig antisera in parallel. Recombinant portions of aggrecan (mAC-IGD: residues 368–481 and mAC-GAG: 1678–1896; Swiss-Prot Q61282) and neurocan (mNC-C: residues 645–944; Swiss-Prot P55066) served as antigens for the immunization of guinea pigs. Rabbit antisera against rat brevican (Thon et al., 2000) and TnR (Day et al., 2004) were kind gifts of Takako Sasaki (University of Erlangen, Erlangen, Germany) and Anders Aspberg (University of Lund, Lund, Sweden), respectively.
The anti-phosphacan monoclonal antibody 3F8 (Rauch et al., 1991) was purchased from the Developmental Studies Hybridoma Bank (University of Iowa). Other commercial antibodies used were as follows: goat anti-human contactin-1 polyclonal antibodies (R&D Systems), mouse monoclonal antibody K14/16 against the Kv1.2 α-subunit (Millipore), rabbit anti-rat-voltage gated sodium channel 1.6 (Nav1.6) polyclonal antibodies (Alomone Labs), mouse monoclonal antibody anti-Caspr clone K65/35 (obtained from the UC Davis/NIH NeuroMab Facility, University of California), rabbit anti-human myelin basic protein (MBP) polyclonal antibodies (Dako), mouse anti-human MBP monoclonal antibody 67–74 (Millipore Bioscience Research Reagents), rabbit polyclonal antibodies against bovine glial fibrillary acidic protein (GFAP) (Dako), mouse (pan-)sodium channel monoclonal antibody K58/35 (Sigma), and mouse anti-neurofilament-200 monoclonal antibody NE14 (Sigma).
Protein extraction and immunoblots.
Brain extracts were prepared as described previously (Schmalfeldt et al., 1998); each step of sample preparations included Pefabloc and “complete protease inhibitors” (Roche Applied Science). In brief, tissues were disrupted in a Polytron homogenizer with 4 volumes of extraction buffer (0.5 m NaCl, 50 mm Tris, pH 7.5, 25 mm EDTA, 0.5% Nonidet P-40) and stirred overnight at 4°C. The homogenates were centrifuged at 100,000 × g for 1 h and the supernatants dialyzed against chondroitinase buffer (40 mm Tris, 40 mm sodium acetate, 10 mm EDTA, pH 8). The protein concentration was determined with the BCA System (Pierce).
Samples were run on 4–15% PHAST polyacrylamide gels (GE Healthcare) under reducing conditions and blotted onto Immobilon-P membranes (Millipore) by diffusion transfer at 70°C for 20 min. Alkaline phosphatase-conjugated secondary antibodies (Biosource or Jackson ImmunoResearch Laboratories) and color reaction with Western Blue substrate (Promega) were used. To detect lecticans, samples were digested with chondroitinase ABC (Seikagaku) (1–2 mU/μg protein) at 37°C overnight before electrophoresis. Spent medium from primary embryonic fibroblast culture was extensively dialyzed against chondroitinase buffer before chondroitinase ABC digestion.
Immunohistochemistry and immunofluorescence.
All animal studies were approved by our institutional review boards. Animals were killed by CO2 inhalation and decapitated. The tissues were dissected, immediately fixed in 4% paraformaldehyde (PFA) in PBS, paraffin embedded, and sectioned according to routine protocols. If needed, unmasking of the antigen before staining was done in a controlled antigen-retrieval device (FSG 120-T/T from Milestone), filled with appropriate buffer (10 mm trisodium citrate, pH 6.0, or 2 mm Tris, 1.7 mm EDTA, 1 mm trisodium citrate, pH 7.8).
In some cases, mice were anesthetized with a lethal dose of pentobarbital and transcardially perfused with PBS, followed by perfusion–fixation with 4% PFA in PBS as described previously (Beggah et al., 2005). Brains and spinal cords were removed, postfixed overnight at 4°C, and either paraffin embedded or cryoprotected and rapidly frozen in O.C.T. compound (Tissue-Tek).
Immunohistochemistry followed the avidin-biotinylated peroxidase method (VECTASTAIN Elite ABC Reagent, Vector Laboratories) using biotinylated secondary antibodies from Jackson ImmunoResearch Laboratories [biotin-SP-conjugated donkey anti-goat, guinea pig, or rabbit IgG (H+L)]. Counterstaining was done with Mayer's hematoxylin. With mouse monoclonal primary antibodies, the M.O.M. kit (Vector Laboratories) was used to avoid nonspecific reactions.
To stain hyaluronan in tissue sections, biotinylated hyaluronic acid-binding protein (B-HABP) (Seikagaku) was applied as 0.5 μg/ml solution in PBS overnight at 4°C, then detected with the ABC method.
Hyaluronan was eliminated from the tissue sections by pretreatment with hyaluronate lyase from Streptomyces hyalurolyticus (Sigma) in 30 mm sodium acetate, 125 mm NaCl, pH 5.2, containing “complete protease inhibitors.” Digestions went on for 2 h at 37°C in a humid chamber. Control sections were processed in parallel with buffer alone. Sections were washed with PBS before blocking and immunostaining.
Immunofluorescence staining by standard procedures used Alexa secondary antibodies: Fluor-488 or Fluor-594 highly cross-adsorbed goat anti-rabbit, -guinea pig, or -mouse IgG (H+L), or Fluor-594 donkey anti-goat. For triple immunofluorescence, Alexa Fluor-488 donkey anti-rabbit, Alexa Fluor-555 donkey anti-goat, and Alexa Fluor-647 donkey anti-mouse IgGs were used. Counterstaining was with Hoechst H33258 bis-benzimide (all from Invitrogen). Blocking reagent of the M.O.M. kit was used for mouse primary antibodies.
Photomicrographs were taken with an Olympus BX61 microscope equipped with an F-view camera. The image capture was controlled with the AnalySISPro software (Soft Imaging System). Confocal laser scanning microscopy was done with a Leica TCS4D, and 3D image processing using IMARIS software (Bitplane).
Electrophysiology.
Analysis of compound action potentials (CAPs) of mouse optic nerves were essentially done as described previously (Allen et al., 2006). Measurements were performed on optic nerves from seven wild-type and four V0/V2-knock-out mice. All animals were adults and deduced from the 10th generation of the same C57BL/6J backcrossing. Optic nerves were carefully dissected, cut behind the orbit and in close vicinity to the optic chiasma. They were gently freed from surrounding connective tissue, placed in a circulation system, and superfused with artificial CSF (aCSF) containing 126 mm NaCl, 3 mm KCl, 2.5 mm CaCl2, 4 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm glucose (Stys et al., 1991). The aCSF was continually oxygenated with a mix of 95% O2 and 5% CO2. The nerves were allowed to equilibrate for ∼1 h before starting the experiment. Suction electrodes (A-M Systems) back-filled with aCSF were used for stimulation and recording. One electrode was attached to the rostral end of the nerve for stimulation and the second suction electrode attached to the proximal end served to record the CAP. All recordings were orthodromic. A square stimulation pulse (30 μs duration) was generated by the software QTRAC (Institute of Neurology, University College London, London, UK) and converted to current using an isolated linear bipolar constant current stimulator (Digitimer DS-5 Isolated Bipolar Constant Current Stimulator). The current was stepwise adjusted to determine maximum CAP signal and then further increased by 25% (supramaximal stimulation). During an experiment, the supramaximal CAP was elicited every 10 s. Generated action potentials were recorded with an isolated bio-amplifier (ISO, World Precision Instruments), amplified 100× (filter settings: high, 1 kHz; low, 300 Hz). Data were digitized by a data acquisition unit (CED micro1401 MK II, Cambridge Electronic Design) using a sampling rate of 100 kHz. Data were acquired and analyzed with the QTRAC software (Institute of Neurology, University College London). The length of the conduction distance between the two glass electrodes was measured with a digital camera and an internal size standard.
Results
Generation of versican V0- and V2-specific knock-out mice
The constitutive knock-out of all versican isoforms is lethal at an early embryonic stage (Mjaatvedt et al., 1998). We therefore chose a splice-variant-specific gene inactivation approach to explore the function of versican V2 in the adult mouse CNS in vivo. For this purpose, we introduced a translational stop codon into exon VII of the versican gene (VCAN) preceded by an ER-retention signal (Fig. 1). This minimal modification, supplemented only by a loxP site, was sought to circumvent potential splicing errors or compensatory expression of other isoforms and to suppress the secretion of a truncated protein in the recombinant mice. Due to the splicing modality of the versican gene, the elimination of the expression of the largest V0 isoform conjointly with versican V2 was unavoidable, however.

Generation of versican V0/V2 KO mice. A, Intron/exon organization of the mouse versican gene giving rise to four different versican isoforms (V0–V3) due to alternative splicing of exons VII and VIII encoding the GAG-attachment domains (GAG-α and GAG-β). Gray box shows region of interest displayed in B. B, Locus diagram and targeting strategy: introduction of an ER retention signal, a translational stop codon, and a floxed neo-tk cassette into exon VII of the versican gene (V0/V2 neo-tk). Removal of selection cassette by cross-breeding with a CRE-deleter strain leaves only the KDEL coding sequence and a stop codon plus one loxP site in exon VII of the mutated versican allele (V0/V2−). Corresponding restriction fragments detected in Southern blot are depicted below. C, Southern blot analysis of genomic DNA from mice and ES cells: the DIG probe downstream of the targeting locus hybridizes to the 17 kb EcoRI fragment of the WT allele, which converts to a 6.6 kb fragment in versican V0/V2-null mutants. BamHI restriction allows a distinction between WT, V0/V2 neo-tk−, and V0/V2− alleles, confirming the successful targeting.
Heterozygous and homozygous mice of the newly generated knock-out mouse strain VCAN(tm1Zim) are viable and fertile, have a normal life span, exhibit in comparison to their wild-type littermates no immediately obvious phenotype in the nervous system and display within the cage environment an inconspicuous behavior. Litters of heterozygous breeding pairs follow a normal Mendelian inheritance indicating no increased lethality in association with the versican mutant genotype.
Expression of the versican transcripts in V0/V2 knock-out mice
In line with our targeting strategy to abrogate the versican V0 and V2 production only on the protein level and to preserve correct splicing and normal expression of versican transcripts, we could find mRNAs of all splice variants in knock-out animals. Nevertheless, the ratios of the different versican transcripts were to some extend affected in the mutant mice. For instance, Northern blot analysis of RNA isolated from E14.5 embryonic fibroblasts showed that versican V0 transcripts were reduced in cells from heterozygous mice or even evaded detection when they were isolated from homozygous animals (Fig. 2A). In both cases the level of the V1 message remained unchanged in embryonic fibroblasts. A more sensitive approach using RT-PCR with isoform-specific primers confirmed however, the presence of versican V0 transcripts in all mutant fibroblast cultures (data not shown). Relative quantification of versican transcripts in brains of homozygous versus wild-type littermates by real-time RT-PCR finally demonstrated that the total amount of versican mRNA was diminished in knock-out animals of all ages examined (P0, P30, and P100). In homozygous mice this change in expression resulted from a 90% reduction of versican V2 mRNA (all ages tested) and a decline of V0 mRNA to ∼20% (P0) or 50% (P30, P100) of the level measured in the wild-type animals (Fig. 2B). In contrast, the relative expression level of versican V1 mRNA in brain was not influenced by the mutation, and also the V3-transcript stayed rather stably low.

RNA expression analysis. A, Northern blot of total RNA from E14.5 embryonic fibroblasts hybridized with a riboprobe detecting all versican splice variants. The ∼10 kb and 13 kb bands correspond to mRNAs of versican V1 and V0, respectively. Minor fragments are products of alternative polyadenylation sites. Loading control: 28S ribosomal RNA. B, Relative expression of alternative versican transcripts in brains of KO and WT mice at postnatal stages P0, P30, and P100 using quantitative RT-PCR. mRNA ratios of mutants versus WT (2−ΔΔCt) were calculated as described in Materials and Methods. C, Immunoblot of brain extracts using GAG-α domain-specific antibodies demonstrates the reduction and the complete loss of versican V2 in heterozygous V0/V2+/− and homozygous V0/V2−/− animals, respectively. Note that the main portion of mouse versican V2 from WT mouse brains seems to be devoid of chondroitin sulfate (band also visible before chondroitinase ABC digestion). The lower V2 core protein band may represent intracellular precursor protein before full O-glycosylation. D, Immunoblot with antibodies against the GAG-β domain. Supernatants from embryonic fibroblast cultures lack the V0 isoform in cells from homozygous mice. Samples in C and D were loaded with (+) or without (−) preceding treatment with chondroitinase ABC (Ch'ase).
Proof of absence of versican V0 and V2 core proteins in homozygous mutants
The efficient elimination of the V0 and V2 core protein expression in homozygous animals was demonstrated in comparisons with adult wild-type and heterozygous littermates. Immunoblots of brain extracts with GAG-α domain-specific antibodies showed that versican V2 protein was significantly reduced in heterozygous and abolished in homozygous mice (Fig. 2C). Moreover, the absence of the truncated form of V0 and V2 provided indirect evidence for a successful intracellular degradation in neural cells. Similarly, immunoblot of spent medium from primary cultures of E14.5 mutant embryonic fibroblasts with anti-GAG-β domain antibodies revealed that homozygous fibroblasts secreted normal levels of the V1 isoform, but completely lacked the versican V0 core protein (Fig. 2D). The genetic manipulation neither impaired viability nor altered the cellular growth rate (data not shown), confirming the effectiveness of our targeting approach also in mesenchymal cells.
The complete absence of the V0 and V2 core proteins in the homozygous mice could ultimately be proven by comparative immunohistological analyses of wild-type animals and mutants using our GAG domain-specific antibodies. In wild-type mice, versican V0 is normally present during early CNS development, mostly disappearing after birth (data not shown). In contrast, versican V2 expression initiates postnatally, increasing continuously until it reaches a plateau (mRNA level around P30/protein level around P90). At this time point, versican V2 has become a major component of the brain ECM, being associated with the white matter of the CNS (Figs. 3, 4). In the knock-out strain, neither the core proteins of versican V0 (includes GAG-α and GAG-β) nor those of versican V2 (contains only GAG-α) could be detected by immunohistochemical stainings of embryonic (data not shown) or postnatal tissues, confirming their effective abrogation (Figs. 3A, 4, 6, and 7 display examples of cerebellum, cerebrum, optic nerve, and spinal cord). Intracellular accumulations of truncated products, which would still be recognized by our antibodies, were not observed, while apoptotic cells stained by TUNEL technique were at P20 (after the onset of normal versican V2 expression) similarly scarce in brain sections of mutant and wild-type mice (data not shown).
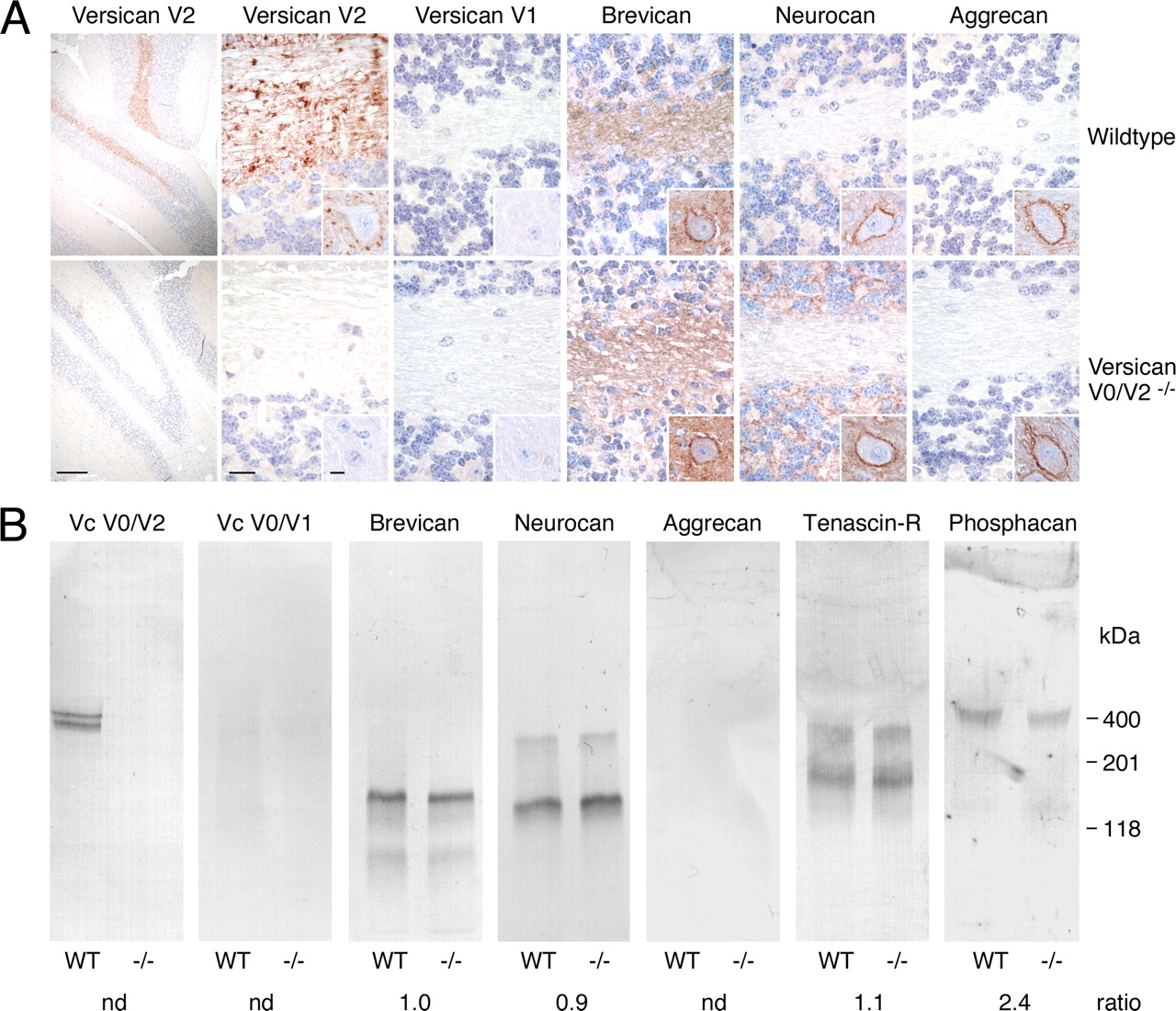
Absence of compensatory expression of other ECM components in versican V0/V2-deficient mice. A, Immunohistochemistry of cerebellar sections of 2-month-old littermates with antibodies against versican GAG-α (versican V2) and GAG-β domains (versican V1), brevican, neurocan-C, and aggrecan reveal no compensatory overexpression of versican V1 and other lecticans in V0/V2-deficient mice. In WT mice, versican V2 and brevican are both present along myelinated fibers, but only versican V2 accumulates at nodes of Ranvier (punctate staining). Brevican and aggrecan, but not versican V2, form part of perineuronal nets in the deep cerebellar nuclei, while neurocan-C is associated with perineuronal nets of brainstem nuclei (small insets). Scale bars: overview on the left, 500 μm; insets, 10 μm; remaining images, 20 μm. B, Immunoblots corroborate the basically unaltered content of lecticans and the slight or moderate reductions of TnR and phosphacan in brain extracts of P30 WT and versican V0/V2-deficient mice. Aggrecan and versican V1 evade detection due to their limited expression in mature brain tissues. The values below the blots represent mean ratios from densitometric measurements of immunoblots of brain extracts from three wild-type versus three V0/V2-null mice. nd, Not done.

Comparative analysis of myelinated fiber tracts in WT and versican V0/V2−/− mutants. Coronal brain sections of postnatal stages P10, P20, and P90 stained with antibodies against MBP display no morphological aberrations despite the complete absence of versican V2 in the KO mice. Scale bar, 1 mm.
Effects of the versican V0/V2 knock-out on the expression of other lecticans
In accordance with our strategy to minimize the impact on the transcriptional level and to evade potential feedback mechanisms, the lack of versican V2 in the adult CNS of homozygous mutants was not compensated by increased expression and deposition of the remaining V1 and V3 splice variants (Figs. 2B, 3). In fact, immunostaining with GAG-β-specific antibodies revealed that versican V1 stayed in wild-type and knock-out littermates confined to a few tissues, such as the choroid plexus, meninges, and blood vessels (data not shown). Also other lecticans retained their normal protein levels and distribution in the absence of versican V2. This is exemplified by immunohistochemical stainings of cerebellar white matter and granular cell layer and by immunoblotting of whole brain extracts using antibodies against brevican, neurocan, and aggrecan (Fig. 3A,B, respectively). Similarly, brain ECM structures typically devoid of versican V2 (e.g., the specialized perineuronal nets in the deep cerebellar nuclei and/or the brainstem) displayed in the knock-out mice their characteristic staining patterns for aggrecan, brevican, and neurocan (Fig. 3, insets). Finally, no compensatory upregulations, but rather slight or moderate reductions of the other brain ECM molecules TnR and its binding partner phosphacan, respectively, could be observed in the initial immunoblotting experiments (Fig. 3B).
Histological and molecular analysis of myelinated fiber tracts
Despite the complete absence of versican V2 and the lack of a compensatory expression of a related lectican, postnatal brain development seemed to be mostly unaffected in knock-out mice (Fig. 4). Hematoxylin-eosin stainings revealed no significant differences in comparisons of brain sections from wild-type and mutant littermates aged P10 or older (data not shown). Also the formation of the major fiber tracts and myelination appeared normal, since no alterations could be observed by light microscopical examination of Nissl-stained sections or after immunohistochemistry with antibodies against neurofilament-200 (data not shown) and MBP (Fig. 4). Likewise, the distribution of GFAP-positive astrocytes was comparable in wild-type and in knock-out mice (data not shown).
Although the CNS-specific versican V2 is in wild-type mice present all along the myelinated fibers, it is preferentially deposited at the nodes of Ranvier. Therefore, we focused next onto the nodal region. For this purpose, we prepared double- and triple-immunofluorescence stainings of cerebellum, spinal cord, and optic nerve sections using antibodies against nodal, paranodal, and juxtaparanodal markers. Our confocal images displayed in the CNS of wild-type mice a versican V2- and TnR-enriched extracellular matrix that engulfs the nodes between the compact MBP-positive myelin sheaths (Fig. 5). Surprisingly, the absence of versican from this structure had in the mouse mutants no effect on the distribution of MBP (data not shown), the juxtaparanodal voltage-gated potassium channel 1.2 (Kv1.2), or the paranodal Caspr and contactin (Figs. 6, 7). Also the compact clustering of the voltage-gated sodium channel 1.6 (Nav1.6) embedded in the axolemma at the node of Ranvier appeared unaltered in versican V0/V2-null mice. A limited electron microscopical analysis of the nodes in the optic nerve revealed a normal organization of the paranodal loops and regular septate-like junctions at their axoglial interface (data not shown).

Visualization of the ECM around the CNS nodes of Ranvier. Double immunofluorescence of cerebellar sections reveals in wild-type mice the nodal accumulation of versican V2 (VcV2) in relation to the myelin component MBP (A, VcV2, green; MBP, red; sagittal view) and demonstrates its colocalization with TnR (B–D, VcV2, green; TnR, red; transverse view) intermingling with versican in the perinodal ECM (E, 3D reconstruction based on confocal image stack). Scale bars: A, B (for B–E), 1 μm.

Effect of the versican V2 deficiency on the distribution of nodal, paranodal, and juxtaparanodal components. In triple/double immunofluorescence of optic nerve tissue from wild-type (A, C, E) and versican V2-deficient mice (B, D, F), no difference in the distribution of paranodal contactin (Cntn: red in A–D) and Caspr (red in E and F) or juxtaparanodal potassium channels Kv1.2 (turquoise in A–D) can be observed (VcV2: green in A and B; Hoechst nuclear staining: blue in A–F). Also the clustering of the sodium channel Nav1.6 (green in C–F) is not impaired. The localization of key components of the CNS nodes of Ranvier is depicted in the sketch (G) modified from Rasband and Trimmer (2001). Scale bars: 10 μm; insets, 2.5 μm.

Simultaneous loss of TnR and phosphacan from nodes of Ranvier in versican V2 mutants. Immunohistochemical and immunofluorescence analysis of cerebellum and spinal cord white matter, respectively, with antibodies against versican V2 (GAG-α domain), TnR, and phosphacan demonstrates their colocalization in the perinodal ECM in wild-type mice. In versican V2-deficient mice, TnR and phosphacan are both absent from the CNS nodes of Ranvier, while keeping their normal distribution in perineuronal nets of deep cerebellar nuclei. Paranodal contactin (red) and juxtaparanodal potassium channel Kv1.2 (turquoise) were immunostained in spinal cord for node localization. Scale bars: cerebellum, 50 μm; spinal cord, 5 μm.
Disturbed assembly of the extracellular matrix around the node of Ranvier
In contrast to the normal localization of these membranous components, the molecular organization of extracellular matrix surrounding the myelin-free portion of the CNS nodes is in the V0/V2 knock-out animals severely affected. This rather compact ECM forms a 1- to 2-μm-wide ring with an outer diameter of 3–4 μm (Figs. 5⇑–7). It includes, apart from the tightly intermingled versican V2 and TnR, also phosphacan (the secreted proteoglycan isoform of RPTPβ) and hyaluronan. Intriguingly, this specialized meshwork structure is practically absent in our mutant mice, as the selective ablation of the V2 isoform is accompanied by the complete loss of tenascin-R and phosphacan, suggesting a key regulatory function of versican in the assembly of the perinodal extracellular matrix. Nevertheless, TnR and phosphacan maintain their normal distribution in locations where versican V2 is usually not present. This includes the perineuronal nets of the deep cerebellar nuclei (TnR and phosphacan) (Fig. 7) as well as the molecular layer of the cerebellum (phosphacan/RPTPβ, data not shown). This specific loss of tenascin-R and phosphacan from the nodal ECM of versican V0/V2 knock-out mice could also be corroborated in other CNS tissues, such as spinal cord (Fig. 7) and optic nerve (data not shown).
To confirm that versican V2 recruits tenascin-R to the nodal matrix and not vice versa, we made a comparative analysis of cerebellar sections from a TnR knock-out strain (Rauch et al., 2005) with tissue sections from our versican V0/V2 mutant. These immunohistological experiments indeed demonstrated that versican V2 incorporated normally into the extracellular matrix at the nodes of Ranvier, even in the absence of tenascin-R, while TnR implicitly required versican V2 for its deposition at this location (Fig. 8). Interestingly, phosphacan was missing from the nodal matrix of both knock-out strains indicating that its loss in the CNS of versican V0/V2 knock-out mice is caused indirectly by the absence of TnR (data not shown).

Perinodal localization of versican V2 is not affected by absence of TnR. Immunohistochemistry of cerebellar sections demonstrates that TnR is selectively lost from the nodes of Ranvier in versican V2-deficient animals, while versican V2 maintains its normal distribution in TnR−/− mice. Scale bar, 10 μm.
Previous in vitro experiments have demonstrated that the C-type lectin domain within the C-terminal G3 region of versican mediates the binding to tenascin-R (Aspberg et al., 1997), whereas the N-terminal G1 domain interacts with hyaluronan (LeBaron et al., 1992). Because versican V2 is recruited to the nodal matrix independently of tenascin-R, we subsequently tested whether hyaluronan, its second ligand at the node of Ranvier, could be responsible for anchoring the large molecular complex of versican V2, TnR, and phosphacan. We therefore removed hyaluronan from wild-type cerebellar sections by cleavage with the highly specific Streptomyces hyalurolyticus hyaluronate lyase. Despite the effective elimination of hyaluronan, the distribution of versican V2, tenascin-R, and phosphacan remained unchanged (Fig. 9). The versican V2 immunostaining was also not affected in hyaluronan-depleted cerebellum sections of tenascin-R-null mutants, demonstrating that neither ligand is absolutely necessary for its anchorage in the ECM (data not shown).

Hyaluronan is not responsible for anchoring the perinodal matrix. Complete removal of hyaluronan by treating cerebellar sections with Streptomyces hyalurolyticus hyaluronate lyase (HA-lyase) has no effect on the distribution of versican V2 and TnR in WT mice. Sections were stained with corresponding antibodies to reveal the localization of the ECM proteins and with biotinylated hyaluronic acid-binding protein (B-HABP) to visualize hyaluronan. Scale bar, 20 μm.
Cell surface sulfatides are other neural components interacting in vitro with the C-terminal globular domain of lecticans, including versican (Miura et al., 1999). We therefore also analyzed the distribution of the V2 isoform in the myelinated fiber tracts of brains from mice deficient in UDP-galactose ceramide galactosyltransferase (CGT) (Bosio et al., 1996). CGT is a key enzyme in the biosynthesis of galactocerebroside (GalC) and its sulfated derivative, sulfatide, both major lipid constituents of myelin. Although CGT-knock-out mice completely lack axoglial junctions in the CNS paranodes and die shortly after onset of myelination, they displayed ∼19 d after birth no abnormal distribution of versican V2 and tenascin-R in cerebrum and cerebellum (Fig. 10). Even additional enzymatic removal of hyaluronan from the brain sections of CGT-null mice did not result in the release of versican V2 from the newly formed nodes of Ranvier. These experiments indicate that the cell surface sulfatide lipids are, unlike versican V2, not directly involved in the assembly of the nodal extracellular matrix.

The perinodal accumulation of versican V2 does not depend on sulfatide-mediated cell surface binding. Immunohistochemical analysis of cerebellar sections reveals the normal deposition of versican V2 and TnR around the newly formed nodes of Ranvier in 19-d-old (CGT−/−) mice despite their deficiency in the biosynthesis of sulfatide, a glycolipid ligand of versican in vitro. Scale bar, 20 μm.
Conduction velocity is not affected in absence of versican V2
To test whether the absence of versican V2 from the perinodal matrix in the CNS would affect the conductivity of the myelinated fiber tracts in our knock-out strain, we determined the electrophysiological characteristics of CAPs at 26°C and 37°C (Fig. 11). We chose these two temperatures because the optic nerves of C57BL/6J background mice are very short and the discrimination of different populations of nerve fibers by their conduction velocity becomes clearer at lower than physiological temperatures. The net conduction distances were in our set-up finally 3.75 ± 0.14 mm for wild-type and 3.59 ± 0.18 mm for V0/V2 knock-out mice (mean ± SEM; n = 13 and n = 6, respectively). For both mutant and wild-type strains, all recorded CAPs showed a three-peak shape, which is characteristic for rodent optic nerves (Fig. 11A). With increasing stimulus intensities, the CAP profile changed. The peaks and the nadir of the CAP successively shifted to slightly lower latencies. Additionally, the stimulus artifact became more pronounced, affecting in a few recordings of very short nerves the shape of peak 1 and rarely peak 2. In these cases, the corresponding latencies were excluded from the calculations. In complete agreement with a previous report (Allen et al., 2006), the peak with the shortest latency appeared first, followed by peaks 2 and 3 with increasing stimulation strength. Compared with wild-type control, the current needed to elicit a maximal CAP (wild type: 6.37 ± 0.47 mA; V0/V2−/−: 5.96 ± 0.72 mA, mean ± SEM) was not different in V0/V2−/− animals.

Electrophysiological characteristics of CAPs of optic nerves at 26°C and 37°C. A, Four representative CAPs from wild-type and V0/V2−/− mice are displayed. The 0.03 ms square-wave stimulus was applied at time point 0 on the time axis. Four sweeps per CAP show the response to four increasing stimulus strengths. The lowest sweep is the response to a stimulus of 10% of the current needed to elicit the maximal CAP. The last sweep shows the maximal CAP. The stimulus artifacts appear as very narrow spikes at ∼0 ms. With increasing stimulus current, peaks with different latencies develop, forming the typical three-peak shape of a rodent optic nerve CAP (calibration, 1 mV). No apparent difference between wild-type and the V0/V2−/− mice was observed. B, Conduction velocities of the three peaks and the nadir were determined by dividing the conduction distance of the recorded nerve by the latency, measured from the stimulus onset to the peak. At both temperatures, the conduction velocities for peaks and the nadir were virtually identical for wild-type and V0/V2−/− mice (mean ± SEM).
We subsequently determined conduction velocities of the three peaks and the nadir by dividing the conduction distance of the recorded nerve by the latency measured from the stimulus onset to the peak. In both groups, the conduction of all peaks slowed equally down, when the temperature was reduced from 37°C to 26°C; wild type: 34.7% slowing, V0/V2−/−: 33.8% slowing (Fig. 11B). The variability of the conduction velocity between the different optic nerves was very small for peaks 2 and 3. It was slightly higher in regard to peak 1, which is most probably due to an increased influence of the stimulus artifact. Finally, the mean conduction velocities of the three peaks were highly similar for wild-type and mutant animals independent of the temperature, at which the CAPs were measured.
Discussion
Our previous studies, first identifying the versican isoform V2 as major constituent of adult brain extracellular matrices (Dours-Zimmermann and Zimmermann, 1994; Schmalfeldt et al., 1998), then revealing its characteristic expression pattern in myelinated fiber tracts of the CNS and subsequently demonstrating its potent inhibitory properties on axonal growth (Schmalfeldt et al., 2000) in vitro, prompted us to generate a knock-out mouse model to further explore the function of this CNS-specific lectican in vivo. Due to the early intrauterine lethality caused by the constitutive elimination of all versican splice variants in hdf mice (Mjaatvedt et al., 1998), we had to choose between (1) a conditional knock-out approach, potentially facing difficulties with efficiency, timing, and tissue specificity of the versican gene ablation, or (2) an isoform-selective knock-out, which would only affect the alternatively spliced exon VII, but would also abolish the expression of the more widely distributed V0 variant along with the CNS-restricted versican V2. Since versican V0 appears to be always coexpressed with versican V1 (Dours-Zimmermann and Zimmermann, 1994; Zimmermann et al., 1994; Landolt et al., 1995; Dutt et al., 2006), we considered the collateral elimination of versican V0 the minor problem, and finally opted for the latter approach. For this purpose, we designed a novel strategy that ultimately left the expression of versican V1 and V3 intact, effectively prevented compensation by other versican splice variants and successfully circumvented the secretion of an artificially truncated core protein that potentially could cause a dominant side effect. Although we originally attempted to maintain normal mRNA levels of all versican splice variants in the genetically manipulated VCAN(tm1Zim) strain and to disrupt V0 and V2 production and secretion only via a premature stop codon and an ER-retention signal, instead, the complete annihilation of versican V0 and V2 in the mutants has most probably to be attributed to combined action of a nonsense-mediated decay mechanism reducing the proportion of V0 and V2 transcripts (for review, see Kuzmiak and Maquat, 2006) and efficient intracellular clearance of the few truncated core proteins translated from the mutated mRNAs.
The normal reproduction behavior, litter size, and Mendelian distribution of wild-type, heterozygous, and homozygous progeny indeed confirmed our prediction that the early embryonic lethality of hdf mice is mostly caused by the absence of versican V1 and consequently could be overcome by keeping the expression of this isoform unchanged in our mutants. Similar to the single gene inactivation of other prominent myelin-derived inhibitors of axonal growth like Nogo, MAG (myelin-associated glycoprotein), and oligodendrocyte myelin glycoprotein (OMgp) (Zurn and Bandtlow, 2006), the abolition of the CNS-restricted versican V2 alone had no apparent effect on the formation of fiber tracts. Versican V2 deficiency neither led to excessive axonal sprouting nor caused myelination defects in the developing CNS. Albeit very difficult to discern from immunofluorescence experiments, increased collateral axon branching at the nodes of Ranvier or enlargement of the nodal region was not evident. Since the proper formation and stabilization of fiber tracts and synaptic contacts in the CNS certainly involves inhibitory activities of many cell surface and ECM molecules, functional redundancy may explain why dramatic histological changes are only rarely observed in single mutants. These include also the brevican and neurocan knock-out strains, which both displayed no alterations upon histological examination (Zhou et al., 2001; Brakebusch et al., 2002). While neurocan forms mostly part of the embryonic and perinatal CNS matrix, brevican expression initiates postnatally and overlaps to a certain extent with versican V2 in the spaces between myelinated fibers. It is therefore conceivable that a lack of versican V2 would at least partly be compensated by brevican. Hence, eliminating both of these “late” lecticans in double knock-out mutants may be required to clarify their functional involvement in fiber tract stabilization.
Redundancy may also obscure the postulated function of versican V2 in forming a perinodal ion pool for rapid generation and propagation of action potentials, as CAP measurements of optic nerves revealed no abnormalities in our single mutants. This is somewhat surprising, as a reduced conduction velocity has been reported for mice lacking TnR (Weber et al., 1999), which is also absent from the nodal matrix in our knock-out strain. The discrepancy may be connected to the fact that the strain background in those comparative experiments was not uniform and that the resolution of nowadays CAP recordings permits an individual analysis of the three timely spaced signals also in mice.
Brevican, aggrecan, and neurocan concentrate along with TnR in perineuronal nets engulfing perikaryon, proximal dendrites, and particularly AISs of large CNS neurons. Conversely, versican V2, is predominantly deposited in an analogous ECM structure at the nodes of Ranvier. Despite this intriguing accumulation occurring already during the establishment of the nodes (Oohashi et al., 2002), versican V2 may not be directly involved in controlling the initial steps of their formation. Unaffected Nav channel clustering at the CNS nodes and accurate localization of paranodal and juxtaparanodal components in our mutants argue against a role of versican V2 as functional analog of gliomedin, the glial organizer of the nodal structure in the peripheral nervous system (Eshed et al., 2005). Furthermore, the concomitant absence of node-associated TnR and phosphacan and the lack of compensatory expression of aggrecan, neurocan, or other versican isoforms in our mice make it unlikely that any of these ECM proteins are related to the oligodendrocyte-derived soluble factors reported to induce node-like Nav clustering in vitro (Kaplan et al., 1997). Nevertheless, we cannot entirely exclude that protein complexes of the paranodal junctions or brevican, which may also accumulate at a few large diameter nodes (Bekku et al., 2009), could substitute such a hypothetical clustering activity of versican V2 in the CNS.
Instead, the data from our versican V0/V2 knock-out strain clearly point toward a key role of versican V2 in the assembly of the extracellular structure surrounding and possibly stabilizing the CNS nodes. The conjoint loss of versican V2, tenascin-R, and phosphacan from the nodal matrix in our mutants now confirms in vivo macromolecular interactions formerly postulated on the basis of in vitro findings. This includes interactions between versican V2 and tenascin-R (Aspberg et al., 1997) and between tenascin-R and phosphacan (Xiao et al., 1997; Milev et al., 1998). Since phosphacan is also absent from the nodes of Ranvier in TnR-null mice (Weber et al., 1999), we conclude that versican V2 first recruits tenascin-R, which in turn integrates phosphacan into the matrix. Additional components joining the complex are hyaluronan and its binding partner Bral1, which shortly follows versican V2 expression during postnatal development (Oohashi et al., 2002). Bral1 possibly stabilizes the interaction between hyaluronan and the N-terminal G1 domain of versican, whereas TnR may cross-link the versican G3 domain at the other end of the core protein. Hyaluronan certainly forms the backbone for versican aggregates. Yet, hyaluronan is unlikely to increase the versican deposition around the nodes because of its uniform distribution along the myelinated fiber tracts. Moreover, our hyaluronan-degradation experiments demonstrate that the binding of versican V2 is not crucially dependent on interactions with hyaluronan and TnR alone or in combination. Considering all these observations, we propose a model, in which the accumulation of versican V2 around the CNS nodes of Ranvier forms the extracellular nucleation point for the subsequent assembly of the perinodal matrix.
Accordingly, the deposition of the versican-enriched matrix at the nodes is likely to be initiated by a firm interaction of versican V2 with the nodal axolemma or, less probable, with the paranodal structure, while previously reported binding activities of its partner TnR and/or phosphacan to paranodal contactin (Brümmendorf et al., 1993; Peles et al., 1995) and neurofascin-155 (NF-155) (Volkmer et al., 1998) or to the nodal sodium channels (Srinivasan et al., 1998; Ratcliffe et al., 2000) may play either no or only subordinate roles in matrix anchoring. The prominent paranodal components contactin, NF-155, Caspr, and sulfatide [interacts with versican in vitro (Miura et al., 1999)] seem also not necessary for the selective deposition and binding of versican V2 at the nodes. In CGT-null mice, they are all virtually absent from the severely disturbed terminal glial loops of the paranodes (Poliak et al., 2001; Marcus et al., 2002). Nevertheless, versican V2 and TnR accumulate normally in CGT mutants, as demonstrated by our immunohistochemical analyses. Hence, cell surface proteins selectively localized within the nodal axolemma are more promising candidates for the versican-dependent anchoring of the perinodal extracellular matrix. In the adult CNS, potential membrane receptors of versican are voltage-gated sodium channels (mainly Nav 1.6) and neurofascin-186 (NF-186). Hedstrom et al. (2007) recently showed that a knockdown of NF-186 by RNA interference abolished the recruitment of the close versican-relative brevican to the perineuronal net covering the AIS of several subsets of CNS neurons. Since NF-186 is also crucially involved in the nodal assembly (Zonta et al., 2008), it seems conceivable that it similarly accumulates and immobilizes the ECM at the CNS nodes of Ranvier via a versican V2-mediated interaction. Yet, direct binding of NF-186 to brevican and/or versican V2 has still to be demonstrated.
Footnotes
-
This work was financed in part by grants from the Swiss National Science Foundation and the Velux Foundation to D.R.Z. We thank Anders Aspberg and Takako Sasaki for the generous gift of antibodies, Marie-Therese Abdou, Belinda Senn, and Karin Hansen for preparing tissue sections, Mathias Hoechli and Nicole Schaeren-Wiemers for technical advice, and Holger Moch for support.
- Correspondence should be addressed to Dieter R. Zimmermann, Institute of Surgical Pathology, University Hospital Zurich, Schmelzbergstrasse 12, CH-8091 Zürich, Switzerland. dieterzi{at}pathol.uzh.ch
This article is freely available online through the J Neurosci Open Choice option.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}