

Abstract
Survival and integration of new neurons in the hippocampal circuit are rate-limiting steps in adult hippocampal neurogenesis. Neuronal network activity is a major regulator of these processes, yet little is known about the respective downstream signaling pathways. Here, we investigate the role of cAMP response element-binding protein (CREB) signaling in adult hippocampal neurogenesis. CREB is activated in new granule neurons during a distinct developmental period. Loss of CREB function in a cell-autonomous manner impairs dendritic development, decreases the expression of the neurogenic transcription factor NeuroD and of the neuronal microtubule-associated protein, doublecortin (DCX), and compromises the survival of newborn neurons. In addition, GABA-mediated excitation regulates CREB activation at early developmental stages. Importantly, developmental defects after loss of GABA-mediated excitation can be compensated by enhanced CREB signaling. These results indicate that CREB signaling is a central pathway in adult hippocampal neurogenesis, regulating the development and survival of new hippocampal neurons downstream of GABA-mediated excitation.
Introduction
A remarkable form of structural plasticity in the adult brain is the generation of new functional neurons from adult neural stem and progenitor cells. There is accumulating evidence that neurogenesis in the adult hippocampus contributes to learning and memory, but its precise physiological role remains elusive (Drapeau and Nora Abrous, 2008; Zhao et al., 2008). Moreover, increasing, albeit conflicting, evidence supports the notion that decreased adult hippocampal neurogenesis might be an etiological factor for depression (Thomas and Peterson, 2008) and that adult hippocampal neurogenesis might be an essential target for the behavior effects of distinct antidepressants (Santarelli et al., 2003; Warner-Schmidt and Duman, 2007; Sahay and Hen, 2008).
Survival and integration of newborn neurons are the main rate-limiting steps to the level of hippocampal neurogenesis (Kempermann et al., 2006). A large fraction of newborn cells in the adult dentate gyrus dies within 1–2 weeks after their birth (Dayer et al., 2003; Kempermann et al., 2003). The rate of survival can be strongly modulated by complex behavioral stimuli, such as exposure to an enriched environment (van Praag et al., 1999; Tashiro et al., 2007) and hippocampus-dependent learning paradigms (Gould et al., 1999; Dupret et al., 2007; Kee et al., 2007; Aimone et al., 2009), all of which are considered to augment hippocampal network activity (van Praag et al., 2000). Neurotransmitter-mediated receptor activation appears to provide a mechanism through which hippocampal network activity could regulate neurogenesis to recruit the precise number of new neurons for activity-dependent modification of the hippocampal circuitry (Tashiro et al., 2006b; Ge et al., 2007; Aimone et al., 2009). Indeed, recent studies have revealed that GABA-mediated excitatory input (S. Ge et al., 2006) and NMDA-receptor-mediated input (Tashiro et al., 2006b) are required for dendrite outgrowth and survival of newborn neurons in the adult dentate gyrus, respectively. However, the intracellular signaling pathways downstream of these signals are largely unknown.
The transcription factor cAMP response element-binding protein (CREB) is a rapidly responding intracellular effector of neurotransmitter signaling and growth factor signaling in mature neurons (Lonze and Ginty, 2002). There is evidence that CREB signaling is regulated in an activity-dependent manner during adult subventricular zone/olfactory bulb neurogenesis, where it may participate in the regulation of neuronal maturation (Giachino et al., 2005). Previous studies have provided conflicting results regarding the role of CREB signaling in adult hippocampal neurogenesis. The expression of a dominant negative CREB protein in mature granule neurons or throughout the dentate gyrus decreases the proliferation of hippocampal progenitor cells (Nakagawa et al., 2002a) and the morphological maturation of neurons (Fujioka et al., 2004), respectively. Conversely, rolipram, an inhibitor of phosphodiesterase IV, and the antidepressant fluoxetine, a 5-hydroxytryptamine reuptake inhibitor, which both stimulate CREB signaling, promote precursor proliferation, survival, and dendrite outgrowth of newborn neurons in the adult hippocampus (Nakagawa et al., 2002b; Fujioka et al., 2004). These findings are in stark contrast to a recent study that demonstrated that CREB αΔ mice, which are deficient in CREB expression, have increased levels of hippocampal neurogenesis (Gur et al., 2007). Thus, the precise role of CREB signaling in adult neurogenesis remains unknown.
In an effort to specifically characterize the cell-autonomous function of CREB signaling in the adult hippocampal neurogenic lineage, we used an in vivo retroviral strategy to manipulate CREB signaling exclusively in newborn cells of the adult dentate gyrus (Tashiro et al., 2006a). Moreover, we applied a retrovirus-mediated gain- and loss-of-function strategy to connect neurotransmitter-induced signaling to the CREB-signaling pathway in the regulation of survival and morphological development of new neurons in adult hippocampal neurogenesis.
Materials and Methods
Retroviral vectors and virus preparation.
The retroviral vectors CAG-GFP (cytomegalovirus immediate early enhancer-chicken β-actin hybrid-green fluorescent protein) (Zhao et al., 2006), CAG-RFP (red fluorescent protein) (Tashiro et al., 2006b), CAG-IRES (internal ribosomal entry site)-GFP (Jessberger et al., 2008), and short hairpin (sh) RNA-Na+-K+-Cl− cotransporter (NKCC1)-1 (shNKCC1) and shRNA-DSRED (Discosoma sp. red fluorescent protein) (shCTR) (S. Ge et al., 2006) were previously described. CAG-TagRFP, CAG-GFP-IRES-CRE, and CAG-IRES-DSRED were generated from the original CAG-GFP vector (Zhao et al., 2006) by replacing the GFP coding sequence with the respective expression cassettes. For CREB gain- and loss-of-function experiments, cDNA encoding for CREB-Y134F (Du et al., 2000), and acidic CREB (ACREB) (Ahn et al., 1998) were cloned into the CAG-IRES-DSRED and CAG-IRES-GFP vectors, respectively. Functionality of the vectors with regard to stimulation and inhibition of the CREB-signaling pathway was confirmed via dual luciferase assays in 293T cells, which were transfected with a CREB-luciferase reporter construct (EVX4-Luciferase) and an internal control (hELF1-Renilla-luciferase) and were subsequently treated with forskolin (Fsk; 10 μm) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In addition, immunohistochemical analysis revealed that ACREB-transduced cells in the adult subgranular zone (SGZ) were devoid of nuclear pCREB, indicating that ACREB-encoding retroviruses efficiently inhibited CREB signaling (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Retroviruses were generated using an all-transient transfection approach. 293T cells were transfected with a mixture containing three separate plasmids, including capsid (CMV-VsVg), viral proteins (CMV-gag/pol), and retroviral plasmid (CAG-vectors, or short hairpin vectors) using Lipofectamine 2000 (Invitrogen). Virus-containing supernatant was harvested twice, 48 and 96 h after transfection, and concentrated by two rounds of ultracentrifugation (Tashiro et al., 2006a). Viral titers ranged between 0.5 and 5 × 107 colony-forming units (cfu) ml−1.
Animals and stereotactic injections.
All experiments were performed in accordance with the European Communities Council Directive (86/609/EEC). Stereotactic injections of retroviruses into the brain of adult C57BL/6 mice were approved by the Government of Upper Bavaria. Stereotactic injections of retroviruses into the brain of adult NR1fl/fl mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Salk Institute for Biological Studies.
For all experiments 8- to 11-week-old female C57BL/6 mice were used. Animals were group housed and were kept under a 12 h light/dark cycle. Mice were anesthetized with a mixture of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight). Titers of injected viruses were adjusted for each experiment to the retroviral preparation with the lower titer. Mice were stereotactically injected with 1 μl of the CAG-GFP, CAG-ACREB, CAG-RFP, shNKCC1, and shCTR retroviruses, or 1.5 μl of 1:1 mixtures of CAG-GFP/CAG-RFP, CAG-ACREB/CAG-RFP, shNKCC1/CAG-TagRFP, shCTR/CAG-TagRFP, shNKCC1/CAG-RFP, shNKCC1/CAG-CREBFY, and CAG-GFP IRES CRE/CAG-RFP retroviruses into the left and right dentate gyrus [coordinates from bregma were (in mm): −1.9 anterior/posterior, ±1.6 medial/lateral, −1.9 dorsal/ventral from dura]. Animals were transcardially perfused with PBS, pH 7.4, followed by 4% paraformaldehyde (PFA) at the respective time points (group sizes, n = 3–6).
For bromodeoxyuridine (BrdU) pulse-chase studies, 8-week-old female C57BL/6 mice were intraperitoneally injected with a single dose of BrdU (100 mg/kg body weight).
Immunohistochemistry.
Tissue and cells were fixed and processed for immunostaining as previously described (Lie et al., 2005). Primary antibodies and dilutions used for this study included rabbit anti-GFP (1:250, Invitrogen), chicken anti-GFP (1:1000, Aves), goat anti-doublecortin (DCX) (1:200, Santa Cruz Biotechnology), goat anti-NeuroD (1:200, Santa Cruz Biotechnology), rabbit anti-phosphorylated CREB (pCREB) (1:200, S133) (Cell Signaling Technology), rabbit and mouse anti-calbindin d28-K (1:1000, Swant), mouse anti-calretinin (1:250, Swant), and rabbit anti-Ki67 (1:200, Vision BioSystems). Secondary antibodies coupled to the flourophores Cy3, Cy5, FITC, or Alexa 488 were obtained from The Jackson Laboratory and were used at a dilution of 1:250 after resuspension in 200 μl of H2O and 200 μl of glycerol. Images were obtained using an Olympus FluoView 1000 or a Leica SP5 confocal microscope.
Phenotyping of transduced cells and analysis of coinjection assays.
To phenotype transduced cells and to determine the phosphorylation status of CREB, equidistant sections throughout the entire transduced region were selected and stained. Transduced cells were identified based on the expression of GFP and/or RFP. Transduced cells were analyzed by confocal microscopy for immunoreactivity for the respective marker (n = 20–50 cells per animal and marker). For evaluation of pCREB immunoreactivity, confocal microscope settings were adjusted to detect signal above background. Background was defined as signal, which was present in the hilus of the dentate gyrus and was not associated with a nucleus. Images were obtained using an Olympus, FluoView 1000, or Leica, SP5 confocal microscope.
To analyze the ratio of colabeled cells after injection of viral mixtures, virally labeled cells throughout the rostral-caudal extent of the transduced region were counted. The fraction of double-transduced (GFP and RFP double-positive cells) among all control-transduced cells (all RFP-positive cells) was determined. We performed statistical analysis using at least 10 sections derived from both hemispheres per mouse (n = 3–6 mice per experimental group). For survival analysis in the context of loss of CREB signaling, two-way ANOVA was performed followed by post hoc Student's t test. For all other data, we used Student's t test. Differences were considered statistically significant at *p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SD.
Morphological analysis.
To determine whether transduced newborn neurons had developed an apical dendrite spanning the granule cell layer toward molecular cell layer, projection images were made from Z-series stacks of confocal images of transduced cells. Transduced immature neurons, which were identified on the basis of GFP or RFP expression and DCX immunoreactivity, were analyzed for the direction of the apical dendrite with respect to the dentate granule cell layer, which was identified by 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) nuclear counterstain.
For analysis of the dendritic length, three-dimensional reconstruction of GFP- or ACREB-transduced DCX+ neurons were made from Z-series stacks of confocal images. The projection images were traced with NIH ImageJ (http://rsb.info.nih.gov/ij) using the NeuronJ plugin (http://www.imagescience.org/meijering/software/neuronj/), and total dendritic length was subsequently analyzed. A total of 15 neurons from three animals of each experimental group were analyzed.
Results
Time course of phosphorylation of CREB in adult hippocampal neurogenesis
The phosphorylated form of CREB (pCREB), which is the activated form of this transcription factor, has been shown to be present in the majority of immature neurons in the adult dentate gyrus (Nakagawa et al., 2002b). To confirm these results, we performed immunofluorescent stainings against pCREB and DCX, a protein specifically expressed in newborn immature neurons. As reported previously, pCREB in the adult dentate gyrus was predominantly detected in the lower third of the dentate granule cell layer within the nuclei of newborn immature neurons (Fig. 1 a). A small fraction of DCX-positive immature neurons, however, was devoid of pCREB immunoreactivity in their nuclei (Fig. 1 a). Similar to DCX, the calcium-binding protein calretinin is transiently expressed in immature dentate granule neurons. Its expression, however, is initiated only after the induction of DCX expression (Brandt et al., 2003). Triple immunostaining for pCREB, DCX, and calretinin revealed that the fraction of pCREB-positive cells was higher among DCX/calretinin double positive cells (∼98.4%, n = 3 animals) compared with DCX-positive, calretinin-negative immature cells (∼66.7%, n = 3 animals), indicating that pCREB was more highly enriched in “older” immature neurons.

CREB is phosphorylated at a specific development stage of adult hippocampal neurogenesis. a , The phosphorylated form of CREB (pCREB, red) is present in many but not all DCX-expressing (green) immature neurons in the subgranular zone of the hippocampal dentate gyrus. DAPI in blue. White arrowheads indicate DCX-positive immature neurons, which are pCREB negative. Scale bars (from left), 50 and 25 μm, respectively. b , Time course of phosphorylation of CREB (red) and DCX expression (blue) in newborn hippocampal neurons using a retroviral birth-dating paradigm. Confocal images of GFP-transduced cells at 3, 14, and 28 dpi. Top, Low magnification; lower rows are higher magnifications of indicated boxes. At 3 dpi, a fraction of GFP-positive cells becomes DCX positive. Most of these cells are pCREB negative (white arrowhead). Some transduced cells are negative for DCX and pCREB (yellow arrowhead). At 14 dpi the vast majority of GFP-transduced cells are DCX positive and pCREB positive (red arrowheads). At 28 dpi a large fraction of transduced cells display the morphology of mature granule neurons and are DCX and pCREB negative (yellow arrowheads). Scale bar, 25 μm. c , Analysis of GFP-transduced cells for pCREB expression and expression of markers corresponding to different stages of adult neuronal development at different time points after injection. Phosphorylation of CREB is preceded by DCX and NeuroD expression. Loss of CREB phosphorylation is paralleled by the loss of DCX and NeuroD expression and the initiation of calbindin expression.
To further characterize the timing of pCREB and to correlate CREB phosphorylation with the developmental stages of adult hippocampal neurogenesis, we combined birth-dating studies using a retrovirus encoding for GFP (Zhao et al., 2006) with immunostaining for pCREB and proteins, which are expressed at specific developmental stages. Mouse brains were processed at different time points after GFP transduction. Transduced cells were analyzed for the presence of pCREB, for the expression of the proliferation marker, Ki67, the immature dendritic growth-associated neuronal protein, DCX (Brown et al., 2003; Cohen et al., 2008), the proneural basic helix-loop-helix (bHLH) transcription factor, NeuroD (Steiner et al., 2006; Seki et al., 2007), and the mature dentate granule neuron protein, calbindin. CREB phosphorylation was initiated between 5 and 7 days postviral injection (dpi) and persisted in the vast majority of GFP-transduced newborn cells (7 dpi, 73.7 ± 6.1%;14 dpi, 91.7 ± 2.5%; and 21 dpi, 74.8 ± 14.7%, n = 4 animals for each time point) until ∼21 dpi (Fig. 1 b,c). pCREB immunoreactivity of newborn cells declined in GFP-labeled cells at 28 dpi and was virtually absent at 42 dpi (28 dpi, 30.3 ± 10.4%; 48 dpi 2.1 ± 0.7%, n = 3–4 animals for each time point). From 2 to 5 dpi, GFP-transduced cells were found in clusters of Ki67+ cells, as previously described for the adult rat dentate gyrus (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) (Seki et al., 2007). These GFP-positive clusters were almost exclusively negative for pCREB (data not shown). Based on this observation, the timing of Ki67 expression (2 dpi, 21.3 ± 5.5%; 3 dpi, 21.8 ± 5.8%; 5 dpi, 7.5 ± 2.9%; 7 dpi, 4.5 ± 2.7%, n = 3 for each time point) and its relation to the onset of CREB phosphorylation, we conclude that CREB phosphorylation is predominantly induced after newly generated cells have exited cell cycle. This conclusion is strongly supported by the finding that only a minor fraction of pCREB-positive DCX-expressing neurons incorporate the thymidine analog BrdU after a 2 h BrdU pulse (5.6%, n = 2 animals) (supplemental Fig. S2a, available at www.jneurosci.org as supplemental material).
CREB activity in newborn neurons is preceded by the initiation of expression of the immature neuronal protein, DCX, and of the proneural bHLH transcription factor NeuroD since at 2–5 dpi, the vast majority of GFP-transduced DCX-positive cells and NeuroD-expressing cells were devoid of pCREB (Fig. 1 b,c). Loss of CREB phosphorylation after 21 dpi was paralleled by a loss of DCX and NeuroD immunoreactivity and an increase in calbindin expression. At 28 dpi, pCREB activity among the transduced cells was mainly confined to immature neurons expressing DCX or NeuroD (supplemental Fig. S2b, available at www.jneurosci.org as supplemental material), whereas GFP-labeled calbindin-positive cells were largely devoid of pCREB (88.9%; n = 2) (supplemental Fig. S2c, available at www.jneurosci.org as supplemental material).
Together, these data indicate that CREB phosphorylation in newborn cells follows a stereotypic temporal pattern, i.e., initiation of CREB phosphorylation subsequent to both mitotic arrest and the onset of NeuroD and DCX expression, persistence of CREB phosphorylation for ∼3 weeks, and termination of CREB phosphorylation concomitantly with the loss of the immature neuronal proteins, DCX and NeuroD, and the induction of the mature granule neuron protein, calbindin.
Consequences of loss of CREB signaling in adult hippocampal neurogenesis
The presence of phosphorylated CREB in newborn cells during a distinct developmental period suggests that CREB signaling fulfills important regulatory functions in the development of new granule cells. To specifically investigate the cell-autonomous role of CREB signaling in new dentate granule neurons, we applied retrovirus-mediated genetic manipulation of individual newborn cells in the adult mouse dentate gyrus (S. Ge et al., 2006; Tashiro et al., 2006a). Transcription factors within the CREB family [including CREB, cAMP-responsive element modulator (CREM), and activating transcription factor 1] display a remarkable degree of functional redundancy. This redundancy is clearly evident from the phenotype of single knock-out mice, which display only subtle defects in most brain tissues (Hummler et al., 1994; Lonze and Ginty, 2002). The dominant-negative CREB polypeptide ACREB heterodimerizes with and blocks the binding of all three endogenous CREB family members (but not of other bZIP proteins) to DNA (Ahn et al., 1998). Expression of ACREB has been successfully applied to analyze the function of CREB signaling in vivo (Herzig et al., 2001; Berdeaux et al., 2007). We generated a recombinant retroviral vector, pCAG-ACREB-IRES-GFP that expresses ACREB and GFP bicistronically under the control of the CAG promoter to allow both transgene expression and the GFP-based identification of transduced cells. Such an approach allows characterization of the cell-autonomous roles of CREB specifically in adult neurogenesis, without the potential developmental defects and/or compensations from other CREB family members.
To determine whether inhibition of CREB signaling affected the maturation and differentiation of newborn granule neurons, we compared both the morphology and the expression of immature neuronal proteins of ACREB-GFP-transduced cells with wild-type GFP-transduced cells at various time points after injection (2 d to 2 months). Until 5 dpi, the percentage of transduced cells expressing DCX was comparable between the experimental groups (Fig. 2 c), indicating that inhibition of CREB signaling did not affect neuronal fate determination and the onset of expression of immature neuronal proteins. Starting from 5 dpi, the percentage of cells expressing DCX was significantly reduced among the ACREB-transduced cells (42.1 ± 5.6%) compared with control cells (62.8 ± 7.9%; p < 0.05 at 5 dpi) (Fig. 2 a,c). Similarly, in an independent experiment, a significant reduction in the fraction of NeuroD-expressing cells among the transduced cells was observed in ACREB-injected animals starting from 7 dpi (GFP, 96.5 ± 1.6%; ACREB, 57.5 ± 5.3%; p < 0.01 at 7 dpi) (Fig. 2 b,d).
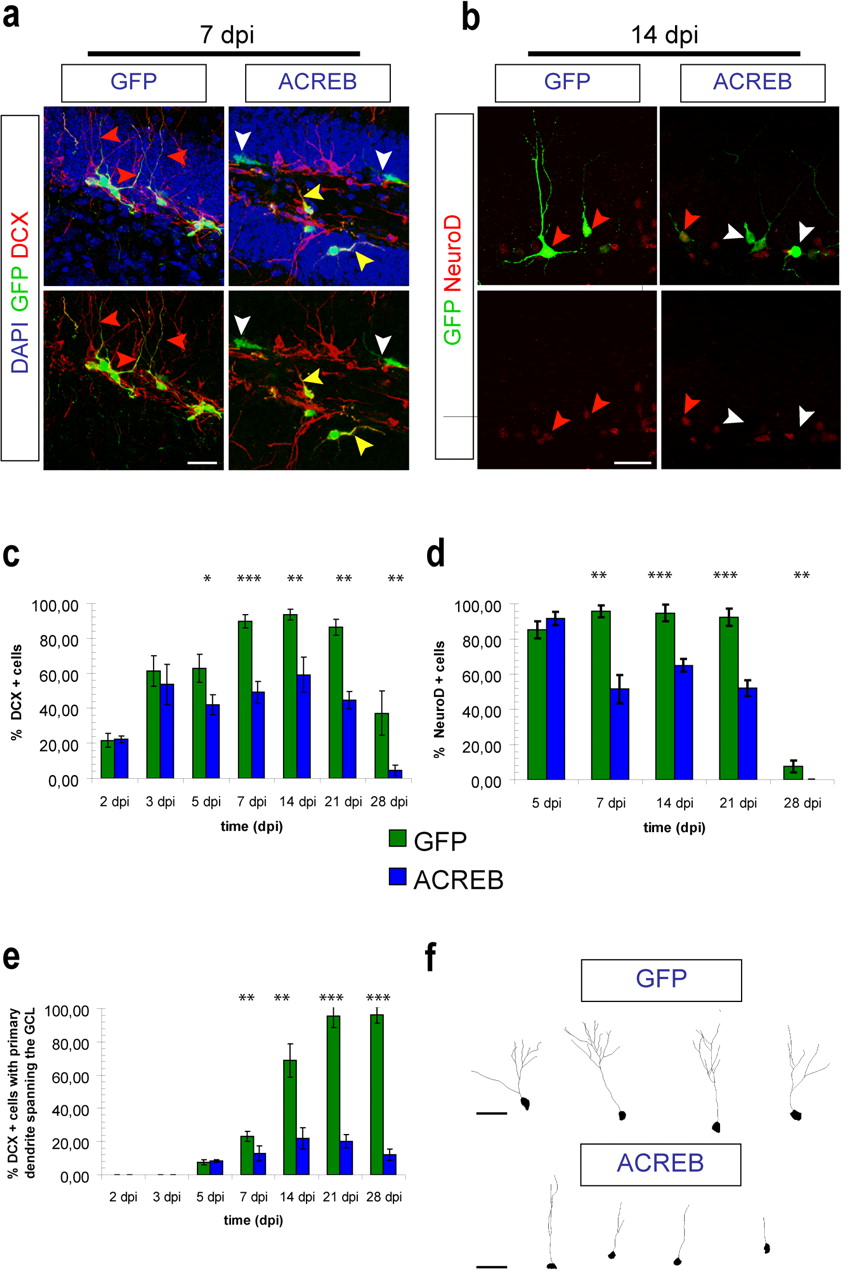
Loss of CREB function affects the differentiation and morphological maturation of newborn dentate granule cells. a , Analysis of DCX expression and morphology of newborn cells transduced with GFP-retrovirus (GFP; green) or ACREB-retrovirus (ACREB; green) at 7 dpi. GFP-transduced cells express DCX (red) and have extended an apical dendrite toward the molecular layer (red arrowhead). In contrast, a large number of ACREB-transduced cells are negative for DCX (white arrowhead). ACREB-transduced DCX+ neurons frequently failed to extend an apical dendrite toward the molecular layer (yellow arrowhead). DAPI in blue. Scale bar, 25 μm. b , NeuroD expression in newborn cells transduced with GFP-retrovirus (GFP; green) or ACREB-retrovirus (ACREB; green) at 14 dpi. The majority of GFP-transduced cells are NeuroD positive (red) (red arrowheads). In contrast, ACREB-transduced cells are frequently negative for NeuroD expression (white arrowheads). Scale bar, 25 μm. c , Quantification of the percentage of retrovirally transduced cells in the granule cell layer expressing DCX. Expression of DCX is significantly reduced among ACREB-transduced cells starting at 5 dpi (*p < 0.05,**p < 0.01, ***p < 0.001). d , Quantification of the percentage of retrovirally transduced cells in the granule cell layer expressing NeuroD. Expression of NeuroD is significantly reduced among ACREB-transduced cells after 5 dpi (**p < 0.01, ***p < 0.001). e , Morphological analysis of retrovirally transduced immature neurons. The percentage of ACREB-transduced DCX+ immature neurons, which extend an apical dendrite through the granule cell layer toward the molecular layer, is significantly reduced (**p < 0.01, ***p < 0.001). f , Confocal three-dimensional reconstruction of dendrites of GFP- and ACREB-transduced immature neurons, which had developed an apical dendrite spanning the granule cell layer at 14 dpi. Scale bar, 25 μm.
Next, we analyzed the morphological development of transduced immature neurons at different time points. Transduced immature neurons were identified on the basis of GFP expression and expression of DCX. As previously reported, GFP-transduced neurons consistently showed a highly polarized morphology with a single apical dendrite branching into the outer granule cell layer and extending into the molecular layer between 1 and 4 weeks after viral injection (Zhao et al., 2006). In striking contrast, the vast majority of ACREB-GFP-transduced immature neurons had lost the typical polarity of dentate granule cells and extended dendrites primarily parallel to the granule cell layer or even toward the hilus. These differences were already apparent at 7 dpi (GFP, 26.5 ± 3.0%; ACREB, 7.0 ± 0.7%; p < 0.01) (Fig. 2 a,e) and became even more pronounced at later developmental time points, i.e., 14, 21, and 28 dpi, when the majority of control cells but only a minor proportion of ACREB-transduced DCX-expressing neurons had sent dendritic processes through the granule cell layer into the molecular layer (Fig. 2 e). Further analysis of the small fraction of ACREB-transduced DCX-expressing neurons, which extended dendritic processes toward the molecular cell layer at 14 dpi, revealed that loss of CREB signaling also led to reduced dendritic growth (CTR, 257 ± 30 μm; ACREB, 91 ± 16 μm; p < 0.001) (Fig. 2 f). Thus, we conclude that intact CREB signaling is required for the proper morphological development of newborn neurons in adult neurogenesis.
In addition to the significant loss of DCX and NeuroD expression and the dramatic morphological changes after CREB signaling inhibition, we observed that there were consistently fewer transduced cells at 7–28 dpi in animals injected with the CREB mutant isoform, although retroviruses of comparable titer were injected. Moreover, we observed that ACREB-transduced cells frequently displayed hallmarks of programmed cell death such as pyknotic nuclei, membrane blebbing, and cell shrinkage (Hossain et al., 1997) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), suggesting that inhibition of CREB signaling affected survival of newborn neurons. To determine whether inhibition of CREB signaling affected proliferation or survival of transduced cells, animals (n = 5 at 7, 14, 21, and 28 dpi, n = 3, at 2, 3, and 5 dpi) were coinjected with a mixture of two retroviruses, i.e., ACREB-GFP-encoding retrovirus or GFP-encoding control virus together with another control retrovirus encoding for RFP. In this paradigm, the fraction of double-transduced cells among the RFP-transduced cells at different time points allows the evaluation of the influence of the CREB mutant transduction on the number of newborn cells, largely regardless of variations in injection sites and titer (Tashiro et al., 2006b). Two-way ANOVA showed a significant interaction between activity of CREB signaling and time for the fraction of double-transduced cells among RFP-transduced cells (p < 0.001). Post hoc comparisons revealed that the fraction of double-transduced, ACREB/RFP cells among the RFP-transduced cells was strongly diminished from 7 dpi onwards (7 dpi, 17 ± 6%) compared with animals injected with a mixture of GFP/RFP (control) (7 dpi, 50 ± 6%, p < 0.001) but not at earlier time points. Remarkably, by 28 dpi, ACREB/RFP-transduced cells were almost completely absent (ACREB/RFP, 5 ± 3%; GFP/RFP, 51 ± 4%, p < 0.001) (Fig. 3). To further characterize whether loss of CREB signaling in newborn cells affects their proliferation, we analyzed a set of animals at 2, 3, 5, and 7 dpi (n = 3 for each time point) and stained with the proliferation marker Ki67. As previously reported, numerous GFP and ACREB-GFP-transduced cells at 2 and 3 dpi were found in clusters of Ki67+ cells (Seki et al., 2007). Loss of CREB signaling did not alter the fraction of Ki67+ cells among the transduced cells at any time point (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Together, these results indicate that inhibition of CREB signaling reduces survival but not proliferation of newly generated neurons.

Loss of CREB function affects the survival rate of newborn dentate granule cells. a , Confocal images of transduced cells in control animals (GFP/RFP) and ACREB-injected animals (ACREB/RFP) at 5, 14, and 28 dpi. The percentage of double-transduced cells (yellow) in ACREB-injected animals is decreased compared with controls at 14 and 28 dpi. RFP-only transduced cells appear in red. DAPI in blue. Scale bar, 25 μm. b , Significant reduction in the survival rate of newborn cells, in which CREB signaling has been ablated starting at 7 dpi (***p < 0.001).
GABA-mediated depolarization but not NMDA activation controls CREB phosphorylation in immature neurons at early stages
A great body of work has demonstrated that the phosphorylation of CREB and CREB activity in neurons is influenced by neuronal activity (Lonze and Ginty, 2002). We sought to determine whether CREB phosphorylation in developing granule neurons of the adult dentate gyrus is also controlled by activity-dependent mechanisms. Previous work has identified an important role of GABAergic input (Tozuka et al., 2005; S. Ge et al., 2006; Overstreet-Wadiche et al., 2006) and NMDA-receptor-mediated synaptic input (Tashiro et al., 2006b) on the differentiation, morphological maturation, and integration of young neurons into the adult hippocampal network. GABA exerts initially tonic and then phasic excitatory actions on adult-generated granule neurons during the first 2–3 weeks of their development as a result of the expression of NKCC1 (S. Ge et al., 2006). Thereafter, GABA becomes inhibitory, presumably as a consequence of the downregulation of NKCC1 and the upregulation of a K+-Cl− cotransporter (KCC2) (S. Ge et al., 2006). In contrast, glutamatergic inputs and NMDA-receptor-mediated synaptic input are present on newborn granule neurons only from ∼14 to 16 d after their birth (Espósito et al., 2005; S. Ge et al., 2006). Based on the timing of CREB phosphorylation and GABA-induced excitation of newborn neurons, we hypothesized that GABA-induced depolarization but not NMDA-receptor-mediated signaling would be a key signal for regulating CREB activation at early time points after their birth.
To test the contribution of GABAergic and NMDA-receptor-mediated input to the activation of CREB signaling in newborn neurons, we performed genetic ablation of GABA-mediated depolarization and of NMDA-mediated synaptic input specifically in newborn neurons of the adult hippocampus. To ablate NMDA-receptor-mediated input we followed a previously described strategy, which disrupts functional NMDA receptor signaling in new cells through retroviral-mediated CRE-recombinase (CRE) expression in floxed NR1 (fNR1) transgenic mice (Tashiro et al., 2006b). fNR1 transgenic mice carry conditional alleles for the gene encoding for NMDA receptor subunit 1, which is a required subunit of functional NMDA receptors. We constructed a retroviral vector to bicistronically express GFP and CRE under the control of the CAG promoter and injected it together with an RFP-encoding retrovirus into the dentate gyrus of adult fNR1 mice. Animals were killed at 7 and 14 dpi, which correspond to time points at which we observed high levels of pCREB and a pronounced developmental phenotype after inhibition of CREB signaling. CRE-transduced cells were identified on the basis of GFP expression. Control cells were identified by the exclusive expression of RFP. These populations were analyzed for the phosphorylation status of CREB. Based on the time course of CREB phosphorylation, i.e., activation in the majority of newborn cells ∼5–7 d and the late onset of NMDA-mediated input on maturing granule cells, we expected that ablation of NMDA-mediated input would not have major effects on CREB phosphorylation. Indeed, ablation of the NR1 subunit did not significantly alter the percentage of pCREB-positive cells among the transduced immature neurons at 7 dpi (CRE-GFP, 73.3 ± 4.9%; RFP, 81.1 ± 5.6%) and 14 dpi (CRE-GFP, 96.6 ± 3.2%; RFP, 86.7 ± 0.9%) (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Furthermore, loss of NMDA activation did not affect the expression of DCX and NeuroD or the overall developmental morphology of the immature neurons at these time points (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
The ability of GABA to depolarize target cells is dependent on the expression of the Na+-K+-Cl− cotransporter NKCC1 (Ben-Ari, 2002; Owens and Kriegstein, 2002). To ablate GABA-mediated depolarization in newborn neurons we transduced proliferating cells in the dentate gyrus of adult mice with retroviruses encoding for an effective shRNA against NKCC1 (shNKCC1) or shRNA against dsRED (shCTR) as previously described (S. Ge et al., 2006). These retroviruses contain an additional expression cassette for GFP for the detection of transduced cells. Analysis of shNKCC1- and shCTR-transduced DCX-positive cells at 7 and 14 dpi (n = 5 animals per group for each time point) revealed that the fraction of pCREB-positive immature neurons was greatly reduced in shNKCC1-transduced cells compared with shCTR-transduced cells (shCTR, 92.1 ± 3.0%; shNKCC1, 39.6 ± 2.8%, p < 0.001, at 7 dpi; shCTR, 95.9 ± 3.2%; shNKCC1, 45.4 ± 10.3%, p < 0.001, at 14 dpi) (Fig. 4 a,b). These results demonstrate that GABA-mediated depolarization is involved in the phosphorylation of CREB during the first 2 weeks of development of new dentate granule neurons. Intriguingly, impaired GABA excitation in newborn neurons led to a developmental phenotype that was strikingly similar to the loss of CREB signaling phenotype: NKCC1 knockdown resulted in the loss of DCX expression (shCTR, 82.2 ± 3.1%; shNKCC1, 29.6 ± 4.2%, p < 0.001, at 7dpi; shCTR, 93.6 ± 2.4%; shNKCC1 64.2 ± 2.9% p < 0.001, at 14 dpi), the loss of NeuroD expression (shCTR, 86.3 ± 3.0%; shNKCC1, 42.3 ± 5.3%, p < 0.001, at 7dpi; shCTR, 92.5 ± 3.5%; shNKCC1 58.6 ± 3.7%, p < 0.001, at 14 dpi), and the impaired morphological development of newborn neurons (shCTR, 23.8 ± 2.7%; shNKCC1, 11.0 ± 1.2%, p < 0.01, at 7dpi; shCTR, 73.1 ± 7.1%, shNKCC1, 21.8 ± 3.0% p < 0.01, at 14 dpi) (Fig. 4 d–f). In addition, mice were coinjected with a mixture of shNKCC1- and TagRFP-encoding retrovirus (n = 4 for each time point) or a mixture of shCTR- and TagRFP-encoding retrovirus (n = 4 for each time point) to determine whether NKCC1 knockdown and the resulting impairment of GABA-mediated excitation would have an impact on the survival of newborn neurons. These experiments revealed that the fraction of shNKCC1-transduced cells among all TagRFP-transduced cells decreased significantly between 7 and 14 dpi (shNKCC1/TagRFP, 37.2 ± 2.3%, at 7 dpi; 20.6 ± 3.1%, at 14 dpi, p < 0.01), whereas the fraction of shCTR-transduced cells among all TagRFP-transduced cells remained stable (shCTR/TagRFP, 56.1 ± 3.1%, at 7 dpi; 48.6 ± 1.6%, at 14 dpi), indicating that impaired GABA-mediated excitation—similar to loss of CREB signaling—decreases the survival of newborn neurons (Fig. 5). Together, these results strongly suggest that GABA-mediated excitation controls the development of newborn granule neurons via activation of CREB signaling.

GABA-mediated depolarization regulates CREB phosphorylation, differentiation, and morphological maturation of new dentate granule cells during the first 2 weeks after their birth. a , Analysis of CREB phosphorylation and DCX expression after shRNA-mediated knockdown of NKCC1. Confocal images of control shRNA-transduced cells (shCTR) (green) and shNKCC1-transduced cells (green) at 14 dpi. Bottom, Magnifications of indicated boxes. A large fraction of shNKCC1-transduced DCX+ (blue) immature neurons is negative for pCREB (red) (white arrowhead). Moreover, many shNKCC1-transduced cells do not express DCX (yellow arrowhead). pCREB+-transduced immature neurons (red arrowheads). Scale bars, 25 μm. b , The percentage of pCREB-positive immature neurons (identified by DCX expression) among the shNKCC1-transduced immature neurons is significantly lowered (***p < 0.001). c , Expression of DCX is significantly reduced among shNKCC1-transduced cells (***p < 0.001). d , Analysis of NeuroD expression (red) in newborn cells after transduction with shCTR (green) or shNKCC1 (green) at 14 dpi. Scale bar, 25 μm. A large percentage of shNKCC1-transduced cells do not express NeuroD (white arrowheads). Red arrowheads indicate transduced cells, which express NeuroD. e , Quantification of NeuroD expression in transduced cells. Expression of NeuroD is significantly reduced among the shNKCC1-transduced cells (**p < 0.01). f , The percentage of shNKCC1-transduced immature neurons (DCX+), which extend an apical dendrite through the granule cell layer toward the molecular layer, is significantly reduced (**p < 0.01).

NKCC1 knockdown affects the survival rate of newborn dentate granule cells. a , Confocal images of transduced cells in control animals (shCTR/TagRFP injected) and shNKCC1/TagRFP-injected animals at 7 and 14 dpi. The percentage of double-transduced cells (yellow) in shNKCC1/TagRFP animals is decreased. shCTR- and shNKCC1-transduced cells in green, TagRFP in red. Scale bars, 25 μm. b , Quantification of the survival rate. There is a significant reduction in the fraction of double cells in the shNKCC1/TagRFP group between 7 and 14 dpi (**p < 0.01), indicating that NKCC1 knockdown affects survival of newborn neurons.
To further address this hypothesis, we investigated whether forced CREB signaling would compensate the NKCC1-knockdown phenotype in developing granule neurons. To this end, adult mice were stereotactically injected into the dentate gyrus with different mixtures of retroviruses. One group of mice was injected with a shNKCC1-encoding retrovirus together with a retrovirus encoding for RFP (shNKCC1/RFP group, n = 5). A second group was injected with a shNKCC1-encoding retrovirus together with a retrovirus encoding for RFP and the mutant CREB protein CREB-Y134F (shNKCC1/CREBYF group, n = 5), which displays increased affinity for putative kinases that have CREB as substrate, including protein kinase A and mitogen-activated protein (MAP) kinase (Du et al., 2000). Animals were analyzed at 7 dpi. As expected, analysis of the shNKCC1/RFP group showed that the percentage of cells expressing DCX and NeuroD was severely reduced in shNKCC1-transduced cells compared with single RFP-transduced cells (DCX: RFP+, 88.1 ± 9.1%; shNKCC1+, 41.7 ± 9.9%, p < 0.001; NeuroD: RFP+, 89.1 ± 11.7%; NKCC1+, 49.9 ± 7.0%, p < 0.01) and that shNKCC1-transduced immature neurons (i.e., DCX-positive cells) less frequently displayed processes spanning the dentate granule cell layer (RFP+, 29.6 ± 5.4%; shNKCC1+, 3.2 ± 2.6%, p < 0.001) (Fig. 6).

NKCC1-knockdown phenotype can be compensated by enhanced activation of CREB signaling. Analysis of neuronal protein expression and cellular morphology of animals injected with shNKCC1- and RFP-encoding retroviruses and animals injected with shNKCC1- and CREBYF-encoding retroviruses. a , Confocal images from animals at 7 dpi. Analysis of DCX expression and of the morphology of newborn cells. shNKCC1 (green)- and RFP (red)-injected control animals in left column. shNKCC1 (green)- and CREBYF (red)-injected animals in right column. Many shNKCC1/RFP-cotransduced cells are negative for DCX (blue) (white arrowhead). Yellow arrowheads indicate RFP-only transduced cells expressing DCX. Right column, shNKCC1/CREBFY-cotransduced cells expressing DCX and sending an apical dendrite toward the molecular cell layer (red arrowheads). Yellow arrowhead indicates CREBYF-only transduced immature neuron. Scale bar, 25 μm. b , Confocal images from animals at 7 dpi. Analysis of NeuroD expression and of the morphology of newborn cells. shNKCC1 (green)- and RFP (red)-injected control animals in left column. shNKCC1 (green)- and CREBYF (red)-injected animals in right column. The majority of shNKCC1/CREBFY double-transduced cells express NeuroD (blue) (red arrowheads). White arrowheads indicate shNKCC1-transduced cells which have lost NeuroD expression. Yellow arrowheads indicate RFP- or CREBYF-transduced cells which are NeuroD+. Scale bar, 25 μm. c , Quantification of single-transduced (shNKCC1, RFP, or CREBYF) and double-transduced (shNKCC1/RFP or shNKCC1/CREBYF) cells expressing DCX. CREBYF expression significantly increases the fraction of DCX+ immature neurons among shNKCC1-transduced cells (***p < 0.001). d , Quantification of single- and double-transduced cells expressing NeuroD. CREBYF expression significantly increases the fraction of NeuroD+ cells among shNKCC1-transduced cells (**p < 0.01) e , Quantification of single-transduced and double-transduced immature neurons, which extended an apical dendrite spanning the granule cell layer. CREBYF expression significantly increases the fraction of immature neurons, which have developed a dendrite spanning the granule cell layer among single-transduced cells and shNKCC1-transduced cells (***p < 0.001).
Similarly, in shNKCC1/CREBYF-injected animals, single shNKCC1-transduced cells again displayed a reduction in the fraction of DCX- and NeuroD-expressing cells and in the fraction of neurons that developed processes spanning the dentate granule cell layer (Fig. 6). In contrast, the fraction of cells expressing DCX or NeuroD was significantly greater among shNKCC1/CREBYF double-transduced cells than among shNKCC1/RFP double-transduced cells (DCX: shNKCC1+/CREBYF+, 95.2 ± 5.1%; shNKCC1+/RFP+, 33.9 ± 8.7%, p < 0.001; NeuroD: shNKCC1+/CREBYF+, 88.8 ± 6.2%; shNKCC1+/RFP+, 51.3 ± 14.6%, p < 0.01) and was comparable to single RFP-transduced cells in the shNKCC1/RFP group. Moreover, a large proportion of the shNKCC1/CREBYF-cotransduced cells had developed a primary dendritic processes spanning the dentate granule cell layer (shNKCC1+/CREBYF+, 55.3 ± 10.6%; shNKCC1+/RFP+, 6.0 ± 2.8%, p < 0.001) (Fig. 6 e). These data demonstrate that activation of the CREB pathway is sufficient to rescue the NKCC1-knockdown phenotype with regard to neuronal gene expression and defect in morphological development, and strongly support the notion that the effects of GABA excitation on the development of newborn granule neurons is, at least in part, mediated via activation of CREB signaling (supplemental Fig. S6, available at www.jneurosci.org as supplemental material).
It is important to point out that the proportion of cells that had developed a primary dendrite spanning the dentate granule cell layer was significantly higher among CREBYF-transduced cells (RFP+, 29.6 ± 5.4%; CREBYF, 66.6 ± 4.8%, p < 0.001) (Fig. 6 e) and shNKCC1/CREBYF-cotransduced cells than among control cells (RFP single-transduced cells) and that this proportion reached levels that were expected only at later time points (>7 dpi) during the development of adult generated dentate granule neurons, which suggests that unregulated or sustained CREB signaling may accelerate the morphological development of new dentate granule neurons.
Discussion
In this study, we have characterized the timing of CREB activation and the consequences of loss of CREB signaling in the development of new dentate granule neurons during adulthood. The phosphorylated form of CREB is first observed in newborn immature neurons ∼5–7 d after their birth, after neuronal fate commitment and very likely after neuroblasts underwent their final division. Subsequently, CREB phosphorylation is observed in the vast majority of immature newborn neurons for the subsequent 2–3 weeks, indicating that CREB phosphorylation is sustained during this developmental period.
Our study demonstrates that intact CREB signaling is essential for survival and morphological development of newborn neurons, and for the maintenance of expression of proteins involved in neuronal development and the control of neurogenic transcriptional programs. It has previously been noted that drug-induced systemic activation of cAMP signaling promotes the proliferation and morphological maturation of newborn cells (Nakagawa et al., 2002b; Fujioka et al., 2004), whereas global inhibition of CREB signaling in neurons results in impaired proliferation and morphological development of immature neurons (Nakagawa et al., 2002a; Fujioka et al., 2004). The present study extends these findings and demonstrates a specific cell-autonomous role of CREB signaling in morphological maturation and survival of newborn neurons. Moreover, we show that CREB is predominantly active in postmitotic cells and that inhibition of CREB signaling in newly generated cells does not affect proliferation. These findings indicate that cAMP signaling controls proliferation in adult hippocampal neurogenesis through a CREB-independent pathway or through activation of CREB signaling in other cell populations.
A recent study reported that CREB-deficient mice, i.e., CREB αΔ mice, have increased levels of hippocampal neurogenesis (Gur et al., 2007), which has raised the question of whether CREB may be dispensable for the regulation of hippocampal neurogenesis. However, in CREB αΔ mice, expression of other CREB family members, including CREM, is upregulated, which may compensate for the lack of CREB in newborn neurons (Blendy et al., 1996). Moreover, CREB αΔ mice lack CREB in all neurons starting from embryonic development. Given that CREB signaling controls synaptic plasticity and neural circuit modification (Marie et al., 2005; Aguado et al., 2009) and that adult hippocampal neurogenesis is tightly controlled by hippocampal network activity, it is possible that the neurogenesis phenotype of CREB αΔ mice is the consequence of alterations in the hippocampal network rather than of impaired CREB signaling in newborn neurons. Indeed, we provide strong evidence that intact CREB signaling is indispensable for hippocampal neurogenesis by demonstrating that acute inhibition of CREB signaling specifically in newborn neurons inhibits hippocampal neurogenesis.
Survival, maturation, and integration of new neurons into the hippocampal circuit are closely connected in time (van Praag et al., 2002; Schmidt-Hieber et al., 2004; Espósito et al., 2005; Zhao et al., 2006; Tashiro et al., 2007; Toni et al., 2008). Our observation, that loss of CREB signaling affects survival and the structural and molecular basis of maturation and integration, i.e., dendrite development and neuronal gene expression, suggests that CREB signaling may represent a universal and rate-limiting regulator for each of these developmental steps. CREB targets include genes involved in the regulation of survival and dendritic growth (Lonze and Ginty, 2002; Impey et al., 2004). Moreover, bioinformatic analysis has indicated that CREB may be a coactivator for the proneural transcription factor neurogenin 2 (Ngn2) in the regulation of neuronal gene expression programs during embryonic neurogenesis (Gohlke et al., 2008). Interestingly, DCX and NeuroD, which are downregulated in newborn granule neurons on loss of CREB signaling, are targets of Ngn2 in developmental neurogenesis (W. Ge et al., 2006; Guillemot et al., 2006). The putative interaction of CREB signaling with Ngn2 becomes even more interesting, in light of the recent finding that Ngn2 is expressed in proliferating precursors and postmitotic immature neurons of the adult hippocampal neurogenic lineage (Ozen et al., 2007; Hodge et al., 2008). These observations suggest that CREB signaling directly regulates survival, maturation and integration, and neuronal gene expression in new granule cells. Given, however, that survival and integration are likely to be interdependent processes in adult neurogenesis (Tashiro et al., 2006b, 2007), we cannot exclude the possibility that CREB signaling primarily regulates morphological development and neuronal gene expression and that cell death in the context of loss of CREB signaling is a secondary consequence of failed neuronal development and integration or vice versa. Thus, characterization of the downstream targets of CREB in newborn granule neurons is needed to determine which developmental processes are directly controlled by CREB signaling in adult hippocampal neurogenesis.
The peak of CREB phosphorylation is observed around the period during which GABAA receptor activation results in depolarization of newborn neurons, which is essential for proper dendritic morphogenesis of newly generated dentate granule neurons (S. Ge et al., 2006). The dependency of neuronal maturation on GABA-induced membrane depolarization is not confined to adult neurogenesis but is a recurring theme throughout CNS development (Ben-Ari, 2002; Cancedda et al., 2007). The signaling pathways, which mediate the developmental effects downstream of GABA, are poorly defined. Several lines of evidence from the present study indicate that CREB signaling confers the effects of GABA-induced depolarization on the development of new granule neurons. First, inhibition of GABA-mediated excitation results in the loss of CREB phosphorylation in newborn neurons. Second, the phenotype of newborn cells after loss of GABA-mediated excitation resembles the phenotype observed after loss of CREB signaling, including impaired dendritic development and loss of expression of the immature neuronal proteins, DCX and NeuroD. Third and most importantly, forced CREB signaling compensates the observed developmental defects in newborn neurons caused by loss of GABA-mediated excitation. One caveat to the present study is the fact that we examined the role of the GABA–CREB pathway in female mice. Several studies have described sex differences in the regulation of CREB (Kudo et al., 2004) and the effects of GABA in the adult hippocampus (Nuñez and McCarthy, 2007). It is therefore possible that the present data may not fully translate to the regulation of adult neurogenesis in the hippocampus of male mice.
Strikingly, forced CREB signaling repeatedly led to precocious morphological maturation, indicating that proper development of new granule neurons is not only dependent on the activation of CREB signaling but also dependent on the precise regulation of timing and strength of CREB signaling. CREB-signaling activity is limited not only by the phosphorylation of CREB but also by the activation of coactivators (Screaton et al., 2004; Ravnskjaer et al., 2007). This raises the possibility that although CREB phosphorylation is sustained in immature new neurons, CREB signaling itself may only be sporadically active. Future studies will need to investigate the temporal activity of CREB signaling as well as the potential role of coactivators during adult hippocampal neurogenesis.
Survival and integration of newborn neurons are considered crucial determinants of the rate of hippocampal neurogenesis. The survival of new neurons is determined during the first 3 weeks after neuronal birth—a period that has also been described as a critical period for survival—and is strongly influenced by hippocampal network activity. Neurons that have survived this period are incorporated for an extended time into the hippocampal network (Dayer et al., 2003; Kempermann et al., 2003; Kee et al., 2007; Tashiro et al., 2007). Here, we demonstrate that immature neurons display high levels of CREB phosphorylation during the critical period of survival and that survival is dependent on intact CREB signaling. NMDA receptor activation plays a role in the selection of newborn neurons for survival during the third week after neuronal birth (Tashiro et al., 2006b). A significant fraction of newborn neurons, however, is lost before the presence of synaptic glutamatergic inputs (Biebl et al., 2000; Kempermann et al., 2003; Tashiro et al., 2006b), suggesting that other signals control the survival of newborn neurons at earlier stages. Indeed, we did not detect any changes in CREB phosphorylation in newborn neurons after NMDA-receptor-signaling ablation during the first 2 weeks after their birth, demonstrating that CREB phosphorylation is independent of NMDA receptor signaling during this period. It has been hypothesized that the level of GABAergic signaling in the dentate gyrus relays information on general hippocampal network activity to newborn neurons (Ge et al., 2007). We found that GABA excitation-mediated signaling controls CREB phosphorylation during the first 2 weeks after neuronal birth and that CREB signaling is a major regulator of survival and morphological integration of newborn neurons during this developmental period (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Thus, our data provide evidence for the notion that the critical period for survival is divided into at least two phases, which are both activity dependent but are distinct with regard to the survival signal.
In summary, we have provided evidence for an essential pathway regulating survival and maturation of new hippocampal neurons involving GABA-mediated excitation and subsequent initiation of CREB signaling. Adult hippocampal neurogenesis may represent a candidate target process for the treatment of age-related cognitive decline (Drapeau and Nora Abrous, 2008), seizure-induced cognitive impairment (Jakubs et al., 2006; Jessberger et al., 2007), and mood disorders (Duman, 2004; Sahay and Hen, 2008). Hence, the characterization of the GABA–CREB pathway may set the stage for the development of novel strategies for the treatment of these neuropsychiatric diseases.
Footnotes
-
R.J. is supported by a fellowship of the Alexander von Humboldt Foundation and by the European Research Area in Ageing/Future Leaders in Aging Research Program of the European Union through the Bundesministerium für Bildung und Forschung (BMBF). Work in the Lie laboratory is supported by the European Young Investigator Award of the European Science Foundation (DFG 858/6-1), the Marie Curie Excellence Team Program and the Marie Curie Reintegration Program of the European Union, the Bavarian Research Network on Adult Neural Stem Cells “FORNEUROCELL,” the Helmholtz Alliance for Mental Health in an Ageing Society, the BMBF Network “Cell Based Regenerative Medicine,” and the European Commission Coordination Action ENINET (Network of European Neuroscience Institutes) (contract number LSHM-CT-2005-19063). We thank K. Wassmer and B. Eble-Müllerschön for excellent technical support, Dr. Marc Montminy for providing CREB constructs, and members of the Lie laboratory for helpful discussion and suggestions. We are grateful to Dr. Christian Stigloher for artwork in supplemental Figure 6 and to Drs. S. Jessberger, X. Duan, L. Bally-Cuif, B. Berninger, N. Abrous, and S. de Marchis for discussion and comments on this manuscript.
- Correspondence should be addressed to D. Chichung Lie, Research Group/Adult Neural Stem Cells and Neurogenesis, Institute of Developmental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, Ingolstaedter Landstrasse 1, 85764 Munich-Neuherberg, Germany. chichung.lie{at}helmholtz-muenchen.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}