

Abstract
Amyloid plaques are primarily composed of extracellular aggregates of amyloid-β (Aβ) peptide and are a pathological signature of Alzheimer's disease. However, the factors that influence the dynamics of amyloid plaque formation and growth in vivo are largely unknown. Using serial intravital multiphoton microscopy through a thinned-skull cranial window in APP/PS1 transgenic mice, we found that amyloid plaques appear and grow over a period of weeks before reaching a mature size. Growth was more prominent early after initial plaque formation: plaques grew faster in 6-month-old compared with 10-month-old mice. Plaque growth rate was also size-related, as smaller plaques exhibited more rapid growth relative to larger plaques. Alterations in interstitial Aβ concentrations were associated with changes in plaque growth. Parallel studies using multiphoton microscopy and in vivo microdialysis revealed that pharmacological reduction of soluble extracellular Aβ by as little as 20–25% was associated with a dramatic decrease in plaque formation and growth. Furthermore, this small reduction in Aβ synthesis was sufficient to reduce amyloid plaque load in 6-month-old but not 10-month-old mice, suggesting that treatment early in disease pathogenesis may be more effective than later treatment. In contrast to thinned-skull windows, no significant plaque growth was observed under open-skull windows, which demonstrated extensive microglial and astrocytic activation. Together, these findings indicate that individual amyloid plaque growth in vivo occurs over a period of weeks and may be influenced by interstitial Aβ concentration as well as reactive gliosis.
Introduction
Amyloid-β (Aβ) is a 38-43 aa peptide that is produced in neurons by the sequential proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase and γ-secretase (Selkoe, 2001). Under normal conditions, Aβ is secreted into the extracellular space [interstitial fluid (ISF)] of the brain, where it is found in soluble form throughout life (Cirrito et al., 2003). Aggregation of Aβ into compact amyloid plaques is associated with local neuritic dystrophy (Knowles et al., 1999; D'Amore et al., 2003; Lombardo et al., 2003; Brendza et al., 2005; Garcia-Alloza et al., 2006; Meyer-Luehmann et al., 2008), neuronal and astrocytic calcium dysregulation (Busche et al., 2008; Kuchibhotla et al., 2008, 2009) and disruption of cortical synaptic integration (Stern et al., 2004) and is widely considered to represent a necessary factor in Alzheimer's disease (AD) pathogenesis (Hardy and Selkoe, 2002). In vitro studies of Aβ aggregation kinetics demonstrate that Aβ monomers polymerize to form higher order aggregates in a concentration-dependent manner (Burdick et al., 1992). Although similar aggregation steps are believed to occur during plaque pathogenesis in the AD brain, the dynamics of this process in vivo are not well understood. Since amyloid deposits are primarily present in the extracellular space of the brain, the concentration of soluble Aβ in the ISF is likely a key determinant of Aβ aggregation. This hypothesis is supported by transplantation studies in which wild-type neural grafts in APP transgenic hosts developed amyloidosis, suggesting that host-derived soluble Aβ diffused through the ISF and seeded amyloid formation in the graft (Meyer-Luehmann et al., 2003). However, a direct relationship between soluble, extracellular Aβ and amyloid plaque growth dynamics in vivo has not been demonstrated.
Since β-secretase and γ-secretase activity is necessary for Aβ generation, drug discovery efforts have aimed to inhibit these enzymes to halt or reverse the progression of amyloid pathology in AD. However, γ-secretase is also required for the proteolysis of a multitude of other type 1 transmembrane proteins including Notch (De Strooper et al., 1999), ErbB4 (Ni et al., 2001) and CD44 (Lammich et al., 2002). Thus, complete inhibition of this enzyme is associated with significant mechanism-based toxicity, including atrophy of the thymus and altered intestinal cell differentiation (Wong et al., 2004). Interestingly, genetic reduction of γ-secretase activity throughout life by as little as 30% is sufficient to attenuate amyloid pathology in a mouse model of AD while limiting toxicity (Li et al., 2007). However, it is not known whether, and to what extent, transient pharmacological reduction of extracellular Aβ is associated with altered amyloid plaque growth dynamics in adult animals.
Given the overwhelming evidence that Aβ accumulation is necessary for AD pathogenesis, it is critical to understand the mechanisms that govern Aβ plaque formation and growth in vivo. To this end, we used serial in vivo multiphoton microscopy to monitor the growth of individual plaques over time and in vivo microdialysis to determine the relationship between ISF Aβ concentration and amyloid plaque growth in APPswe/PS1dE9 (APP/PS1) transgenic mice.
Materials and Methods
Animals.
Male and female APPswe/PS1dE9 mice (Savonenko et al., 2005); (APP/PS1; The Jackson Laboratory) were aged to 6 ± 0.5 or 10 ± 0.5 months for multiphoton microscopy experiments and 3 ± 0.5 months for in vivo microdialysis experiments. All experimental protocols were approved by the Animal Studies Committee at Washington University.
Open-skull cranial window surgery.
Open-skull cranial windows were prepared on the day of the first imaging session as previously described (Bolmont et al., 2008; Meyer-Luehmann et al., 2008). Briefly, mice were anesthetized under volatile isoflurane (2% induction, 1.5% maintenance), and the skin and periosteum were removed to expose the skull. Lines forming a square (6 mm) were drilled into the skull surface using a high-speed drill (Fine Science Tools), and a pair of angled forceps was used to remove the circumscribed region of skull without damaging the dura mater. A glass coverslip (8 mm in diameter) was placed over the open-skull region and was sealed to the skull with dental cement. A 2-mm-high wall of dental cement was constructed around the coverslip to create a water-immersion chamber for subsequent imaging (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Thinned-skull cranial window surgery.
Thinned-skull cranial windows were prepared on the day of the first imaging session as previously described (Christie et al., 2001; Tsai et al., 2004). Briefly, mice were anesthetized (as described above), and the skin and periosteum were removed to expose the skull. A high-speed drill and microsurgical blade (Surgistar) were used to thin the skull until the skull window was transparent and displayed flexibility. Pial vasculature was clearly visualized through the window and served as a guide to repeatedly locate the same sites in the brain at subsequent imaging sessions (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Two thinned-skull windows (each 0.8–1.0 mm in diameter) were prepared on each animal.
Multiphoton microscopy.
To examine plaque growth, individual plaques were observed longitudinally. Twenty-four hours before each imaging session, animals were injected intraperitoneally with methoxy-X04 (5 mg/ml in 10% DMSO, 45% propylene glycol, and 45% saline), a fluorescent compound that crosses the blood–brain barrier and binds to amyloid plaques (Klunk et al., 2002). The animal was anesthetized and mounted on a custom-built stereotaxic apparatus (Brendza et al., 2005). For thinned-skull window preparations, a small ring of molten bone wax was applied to the skull surrounding the perimeter of the window to create a chamber for water immersion. The skull window was placed directly under the objective lens on a two-photon microscope [LSM 510 META NLO system (Carl Zeiss Inc.) with a Chameleon Ti: Sapphire laser (Coherent Inc.)]. To image methoxy-X04-labeled plaques, two-photon fluorescence was generated with 750 nm excitation, and fluorescent emission was detected in the range of 435–485 nm. A 10× water-immersion objective [numerical aperture (NA) = 0.33, Zeiss] was used to create a site map during initial imaging and a 40× water-immersion objective (NA = 0.75, Zeiss) was used for detailed analysis of individual plaques. A z-stack image series was acquired from the skull surface to a depth of ∼200 μm into cortex. Incremental z-step distance was 10 μm under the 10× objective and 5 μm under the 40× objective. At the conclusion of each imaging session, the animal was removed from the stage and the scalp was sutured. To examine plaque growth over time, the same sites for each animal were imaged on day 0 and day 7, 28, or 90. To determine whether laser intensity might impact plaque size determination, a visual field containing multiple plaques was repeatedly imaged using increasing laser power (14–20%) while detection gain and amplification parameters were held constant (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). No significant differences were found between the different laser intensities.
Image analysis.
Collapsed z-stack images for each individual plaque were measured by cross-sectional area and intensity using SigmaScan Pro Image Analysis Software (Systat Software) with a preset threshold (threshold = mean + 4 * SD) (supplemental Fig. 2C,D, available at www.jneurosci.org as supplemental material). Plaques were excluded from analysis if they were located on the edge of the thinned-skull window, if they exhibited a fluorescence intensity less than the mean intensity of an adjacent background region, or if the images were affected by motion artifacts (from heartbeat or respiration).
Double immunofluorescence.
To compare microglial and astrocytic activation in cortex under open- and thinned-skull window preparations, animals were transcardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.01 m PBS immediately after the second multiphoton imaging session. Brains were removed and postfixed in 4% paraformaldehyde for an additional 4 h. Coronal free-floating sections (50 μm) were cut on a cryostat. Sections were incubated overnight at 4°C in a combination of rabbit anti-Iba1 (1:1000; Wako Chemicals) and mouse anti-GFAP monoclonal antibody (1:1000; Sigma). A secondary antibody mixture of Cy3-conjugated donkey anti-rabbit IgG (1:800; Jackson ImmunoResearch Laboratories) and Alexa Fluor 488-conjugated donkey anti-mouse IgG (1:400; Invitrogen) was applied. Sections were washed, mounted, and examined using a confocal microscope (Zeiss LSM).
Compound E treatment.
To examine the effect of reduced Aβ concentration on plaque growth dynamics, mice were treated with either vehicle or the potent γ-secretase inhibitor, Compound E (3 mg/kg; Axxora) daily for 7 or 28 d (Grimwood et al., 2005). To examine the effect of Compound E on ISF Aβ levels, mice were treated with either a single dose of 3 mg/kg Compound E (i.p.) or treated daily with vehicle or 3 mg/kg Compound E for 7 d.
Amyloid plaque load quantification.
To determine the effect of Compound E treatment on overall cortical amyloid plaque load, 6- and 10-month-old animals were treated with vehicle or Compound E (3 mg/kg, i.p.) daily for 28 d. Mice were subsequently transcardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.01 m PBS. Brains were dissected, fixed with 4% paraformaldehyde, cryosectioned (50-μm-thick slices), and stained with 100 μm X-34 in staining buffer (40% ethanol/60% PBS, pH 10) for 20 min and differentiated with staining buffer three times, each, for 2 min to visualize compact plaques (Styren et al., 2000). Cortical plaque load was expressed as percentage of cortical area covered by X-34-positive staining.
In vivo microdialysis.
In vivo microdialysis used to measure Aβ in the brain ISF of awake, freely behaving mice was performed as previously described (Cirrito et al., 2003, 2005, 2008; Kang et al., 2007). One day before drug treatment, guide cannulae (BR-style, Bioanalytical Systems) were stereotaxically implanted into the left barrel cortex (at bregma, 2.0 mm lateral to midline, and 1.0 mm below the dura mater at a 32° angle) of APP/PS1 mice under isoflurane volatile anesthesia. After guide cannula implantation, a 2 mm microdialysis probe [BR-2, 38 kDa MWCO (molecular weight cutoff membrane), Bioanalytical Systems] was inserted through the guide cannula so the microdialysis membrane was contained entirely within the barrel cortex. Mice were allowed to recover from anesthesia and were housed in a Raturn Cage system (Bioanalytical Systems) with ad libitum access to food and water for the remainder of the experiment. The microdialysis probe was connected to a syringe pump (Stoelting Co.) and artificial CSF (aCSF), containing (in mm) 1.3 CaCl2, 1.2 MgSO4, 3 KCl, 0.4 KH2PO4, 25 NaHCO3, and 122 NaCl, pH 7.35, was continuously perfused through the microdialysis probe at a constant flow rate of 0.7 μl/min. Artificial CSF contained 0.15% bovine serum albumin to limit nonspecific loss of Aβ. Dialysis samples were collected using a refrigerated fraction collector (SciPro Inc.) into polypropylene tubes for subsequent measurements of Aβ1-x, Aβx-40, or Aβx-42 by ELISA, as described below. Mice were treated intraperitoneally with either a single dose of Compound E (3 mg/kg) or daily with vehicle or Compound E (3 mg/kg) for 7 d. In single-dose experiments, baseline levels of ISF Aβ were defined as the mean concentration of Aβ 8 h before drug treatment. Because artifactual fluctuations in ISF Aβ levels are occasionally observed 3–4 d after insertion of the microdialysis probe (our unpublished observations), we chose to monitor ISF Aβ levels during days 5, 6, and 7 of treatment in multiple-dose experiments. Based on our finding that ISF Aβ levels collected 16–24 h after a single dose of Compound E did not differ from pretreatment baseline levels, baseline levels of ISF Aβ in multiple-dose experiments were defined as the mean concentration of Aβ from hours 16–24 after each injection of Compound E. After each experiment, animals were killed.
Aβ ELISA assay.
Microdialysis samples were analyzed for Aβ1-x, Aβx-40, or Aβx-42 using species-specific sandwich ELISAs. Briefly, Aβ1-x, Aβx-40, and Aβx-42 were captured using monoclonal antibodies targeted against amino acids 13-28 (m266), 35-40 (HJ2), or 33-42 (21F12) of Aβ, respectively. The antibodies m266, 21F12, and 3D6 were gifts from Eli Lilly. For Aβ1-x assays, a biotinylated N-terminal domain monoclonal antibody (3D6B) followed by streptavidin-poly-HRP-20 was used to detect (Fitzgerald). For Aβx-40 and Aβx-42 assays, a biotinylated central domain monoclonal antibody (HJ5.1B) followed by streptavidin-poly-HRP-40 was used to detect (Fitzgerald). All assays were developed using Super Slow ELISA TMB (Sigma) and read on a Bio-Tek FL-600 plate reader at 650 nm.
Statistical analysis.
Longitudinal changes in cross-sectional area of individual plaques were analyzed using paired t tests. Differences in average fold increase of amyloid plaques at multiple time points as a function of imaging interval were analyzed using a one-way ANOVA followed by Dunn's post hoc test. To assess differences in individual plaque growth as a function of drug treatment, the fold increase of mean plaque size was analyzed using a paired t test (SigmaStat statistical software v 2.0, Systat Software). Group differences in percentage area covered by X-34-positive staining were analyzed by Student's t tests. In microdialysis experiments, time point comparisons of ISF Aβ levels were performed using one- and two-way ANOVAs followed by Bonferroni post hoc t tests. Differences in mean ISF Aβ levels between groups were compared using a paired t test (GraphPad Prism v 4.0). All data in figures represent mean ± SEM. In all tests, the level of significance was set at p ≤ 0.05.
Results
Amyloid plaques exhibit age- and size-related growth under thinned-skull windows
Using serial intravital multiphoton microscopy, individual amyloid plaques were imaged through thinned-skull cranial windows over 7, 28, and 90 d intervals in 6- and 10-month-old APP/PS1 transgenic mice. In 6-month-old mice, plaque size increased significantly over 7, 28, and 90 d intervals (Figs. 1, 2A–C; supplemental Fig. 4A,C, available at www.jneurosci.org as supplemental material). Over a 7 d interval, 21% of plaques grew by at least twofold, with a maximum of fourfold growth (Fig. 2A; supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Over a 28 d interval, 62% of plaques increased by twofold or more, with a maximum of sixfold growth (Fig. 2B; supplemental Fig. 4A,C, available at www.jneurosci.org as supplemental material). Notably, the average fold increase in plaque size over a 90 d period was no greater than that observed over the 28 d period, suggesting that plaques adopted a stable size before these two imaging time points (Fig. 2C,D; supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Although the net effect of plaque dynamics indicated average plaque growth, there was a great deal of variability. Indeed, many plaques remained stable during the observed time intervals and rare examples of plaque regression were also evident (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). In contrast to plaque growth observed in 6-month-old animals, plaques imaged in 10-month-old mice did not exhibit significant growth over 7 or 90 d periods (Fig. 2D; supplemental Figs. 3A,B, 4B, available at www.jneurosci.org as supplemental material). Plaque growth was also related to plaque size, as smaller plaques exhibited a greater growth rate compared with larger plaques regardless of animal age (Fig. 2E). Consistent with these findings, plaques sampled in 6-month-old mice exhibited a smaller mean size than 10-month-old mice and showed greater mean growth (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material). Moreover, newly formed plaques were significantly smaller than preexisting plaques (supplemental Fig. 3D, available at www.jneurosci.org as supplemental material). Thus, individual amyloid plaque growth in vivo is related to animal age and plaque size.
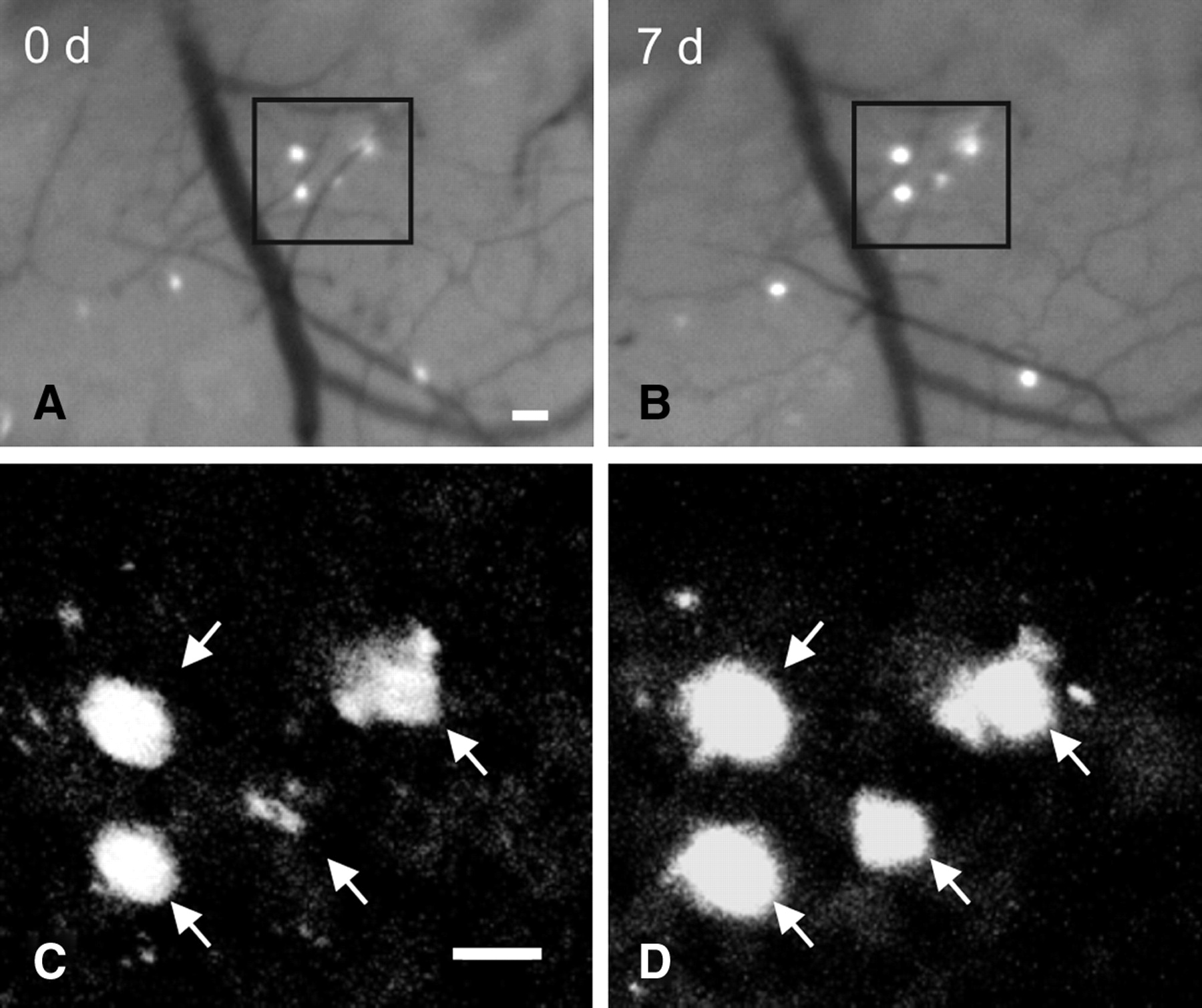
Serial in vivo multiphoton microscopy demonstrates growth of individual amyloid plaques. A, B, Epifluorescence micrographs of 4 plaques labeled with methoxy-X04 and imaged over a 7 d interval in the brain of a 6-month-old APP/PS1 mouse. C, D, Multiphoton micrographs of those same 4 plaques illustrate growth of individual plaques over time. Scale bars, 20 μm.

Amyloid plaques exhibit age- and size-related growth under thinned-skull window preparations. Using serial intravital multiphoton microscopy, individual plaques were labeled with methoxy-X04 and imaged at multiple time points. Plaque growth was expressed as fold increase in cross-sectional area relative to initial plaque size. A–C, In 6-month-old APP/PS1 mice, plaques exhibited significant growth over 7, 28, and 90 d intervals (paired t test, *p ≤ 0.05). D, Time course of plaque growth in 6- and 10-month-old mice. In 6-month-old animals, the average fold increase in plaque size over a 28 d period was significantly greater than that observed over a 7 d period. There was no difference between average fold increase over a 28 d period compared with that over a 90 d period (one-way ANOVA followed by Dunn's post hoc test). Plaques imaged in 10-month-old animals did not exhibit significant plaque growth at either the 7 or 90 d interval (paired t test). E, Average plaque growth is plotted as a function of initial plaque size. Regardless of animal age, smaller plaques grew at a greater rate compared with larger plaques. F, Plaques imaged in 6-month-old animals under open-skull window preparations did not exhibit significant growth over a 28 d interval.
Open-skull cranial window preparations do not show plaque growth and are associated with extensive gliosis
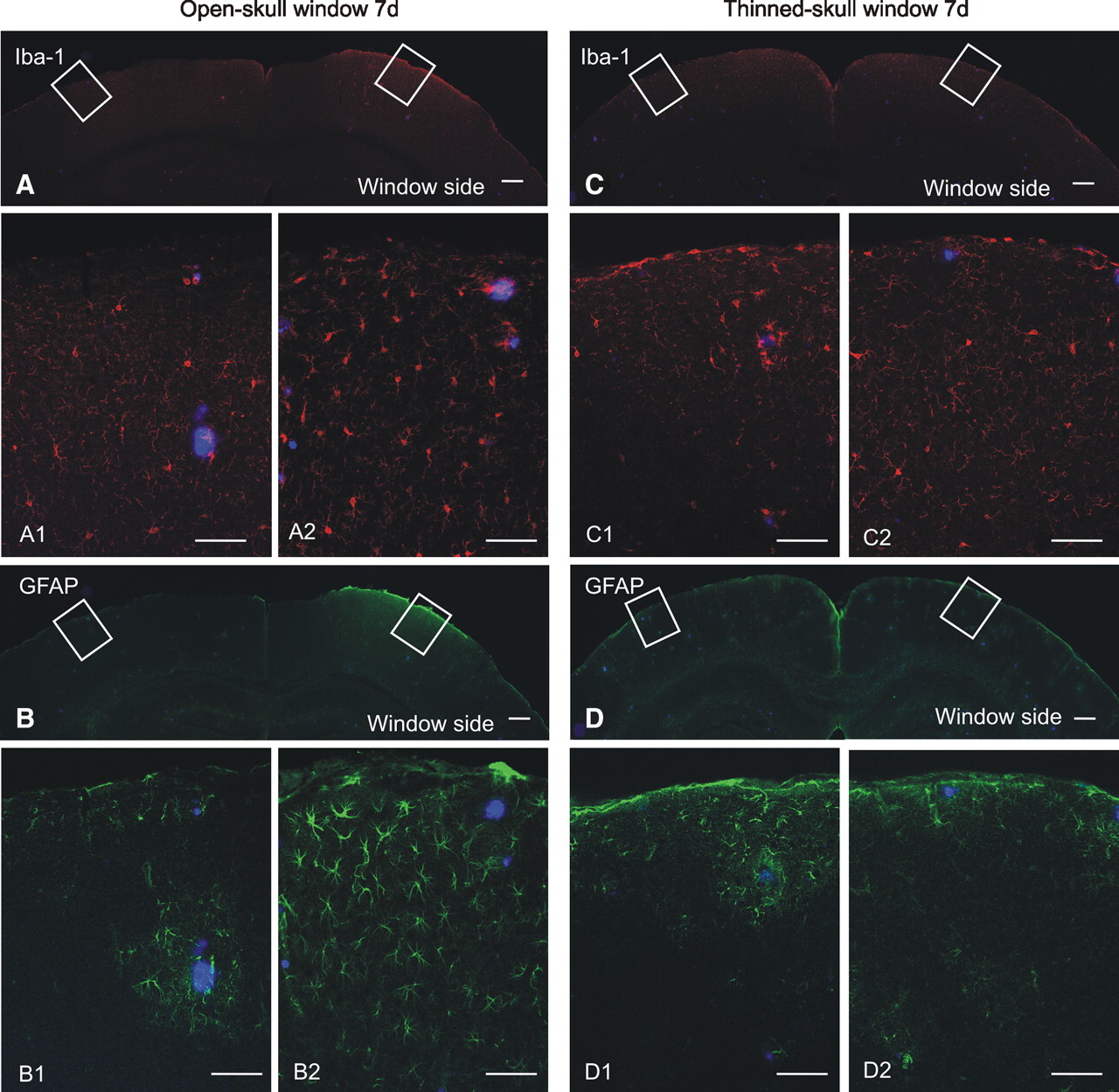
In contrast to the robust plaque growth observed under thinned-skull window preparations, plaques imaged in 6-month-old APP/PS1 mice under open-skull cranial window preparations did not exhibit significant growth but did show some evidence of regression over a 28 d interval (Fig. 2F; supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). These findings are consistent with two recent two-photon studies using open-skull windows (Bolmont et al., 2008; Meyer-Luehmann et al., 2008). Technical differences between the two window types are shown in supplemental Figure 1, available at www.jneurosci.org as supplemental material, and include open (craniotomy)- versus closed-skull preparations, size differences (6 mm diameter in open-skull vs <0.8 mm in closed-skull windows), and the presence of a foreign body (coverslip) in open-skull windows versus none in closed-skull windows. To examine functional consequences of the different cranial window types, we performed a comparative analysis of microglial and astrocytic activation under thinned- and open-skull window preparations. Seven days after open-skull surgery, extensive microglial activation (Iba-1 immunostain, Fig. 3A) was present in cortex under the open-skull window, whereas activated microglia were largely absent in the contralateral control hemisphere. Astrocytic activation (GFAP immunostain, Fig. 3B) was also abundant in cortex under the open-skull window but rare in contralateral control cortex. Even at low power, extensive immunostaining is visible in the region immediately below the cranial window. In contrast, microglial and astrocytic activation occurred only immediately surrounding plaques under thinned-skull window preparations and was indistinguishable from the respective contralateral control hemispheres (Fig. 3C,D). These results indicate that open-skull, but not thinned-skull, window preparations are associated with extensive cortical gliosis and suggest that reactive glial activation may underlie the suppressed plaque growth dynamics observed under open-skull window preparations. Our findings are consistent with those of Dr. W. G. Bao, who has reported increased gliosis under open-skull windows associated with altered dynamics in synaptic spine behavior (Xu et al., 2007). Thus, for the remaining multiphoton studies, all experiments were performed using thinned-skull windows.

Open-skull cranial window preparations are associated with extensive gliosis. A, Low-magnification image of Iba-1 immunofluorescence (red) illustrates extensive microglial activation in cortex under the open-skull cranial window but not in contralateral control cortex. High-power micrographs colabeled for methoxy-X04-positive plaques (blue) reveal little microglial activation in control hemisphere (A1), but robust microglial activation under the open-skull window (A2). B, Low-magnification image of GFAP immunofluorescence (green) demonstrates extensive activation of astrocytes in cortex under the open-skull cranial window. High-power micrographs illustrate astrocytic activation only in the immediate vicinity of plaques in control cortex (B1), whereas robust astrocytic activation is present throughout cortex under the open-skull window (B2). C, Iba-1-positive microglia are not abundantly visible at low magnification under a thinned-skull cranial window preparation. High-power micrographs illustrate rare microglial activation in control cortex (C1) and under the thinned-skull window (C2). D, Astrocytic activation is not robust at low magnification under a thinned-skull window preparation. Astrocytic activation is rare in control cortex (D1) and under the thinned-skull window (D2). Scale bars: A–D, 200 μm; A1–D2, 50 μm.
γ-Secretase inhibition suppresses plaque growth and new plaque appearance in vivo
To assess the effects of reduced Aβ production on longitudinal growth of individual plaques, we measured plaque growth in 6- and 10-month-old APP/PS1 mice treated with either the potent γ-secretase inhibitor, Compound E (3 mg/kg), or vehicle daily for 7 or 28 d (n = 4/group). Compound E suppressed plaque growth in 6-month-old mice over 7 and 28 d intervals (Fig. 4). Treatment for 28 d in 6-month-old animals also dramatically attenuated the appearance of new plaques: 0.4390 ± 0.1391 new plaques per mm3 in vehicle-treated versus 0.0506 ± 0.0339 in Compound E-treated mice (Student's t test, *p ≤ 0.05). In contrast, plaques imaged in 10-month-old mice did not exhibit significant growth over the 7 or 90 d interval regardless of drug treatment (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Importantly, although Compound E treatment was sufficient to dramatically decrease plaque growth, it did not induce regression in average plaque size.

γ-Secretase inhibition suppresses plaque growth and new plaque formation. A, B, Representative multiphoton micrographs of plaques imaged at a 28 d interval in a vehicle-treated 6-month-old APP/PS1 mouse illustrate growth of existing plaques (arrows) and appearance of new plaques (arrowheads). C, D, Plaque growth was suppressed in mice treated with the γ-secretase inhibitor, Compound E (3 mg/kg, i.p). All images are collapsed z-stack images (100–120 μm thick). E, Plaques imaged in vehicle-treated mice exhibited significant growth over 7 and 28 d intervals, whereas plaque growth was attenuated in Compound E-treated mice. (n = 4 mice/group; Student's t test, *p ≤ 0.05). The dotted line indicates no growth. Scale bars, 20 μm.
To determine whether suppressed growth of individual plaques was reflective of changes in overall plaque burden, 6- and 10-month-old APP/PS1 mice were treated with Compound E (3 mg/kg) or vehicle daily for 28 d (n = 8/group); mice were killed, brains were sectioned and immunostained, and compact plaque load was determined. Compound E reduced X-34-positive cortical plaque burden by 23% in 6-month-old mice compared with vehicle-treated controls (Fig. 5). No difference was detected in 10-month-old mice, similar to findings recently reported (Garcia-Alloza et al., 2009). These results indicate that modest γ-secretase inhibition was sufficient to decrease amyloid plaque load and size when administered during the plaque growth phase and suggest that longitudinal growth of individual amyloid plaques is a fundamental mechanism by which plaque load increases in AD. Of note, individual plaque growth measured by multiphoton microscopy (n = 4) is a much more sensitive measure than average grouped plaque burden measures (n = 8).

γ-Secretase inhibition reduces plaque load in 6-month-old APP/PS1 mice. Six- and 10-month-old APP/PS1 mice were treated daily with Compound E (3 mg/kg, i.p.) or vehicle for 28 d. Compound E reduced X-34-positive cortical plaque load by 23% in 6-month-old animals compared with vehicle-treated controls. No difference was present between groups in 10-month-old animals (n = 8 mice/group; Student's t test, *p ≤ 0.05).
Modest reduction in ISF Aβ concentration is associated with a dramatic reduction in amyloid plaque growth in vivo
We next performed in vivo microdialysis experiments to determine the extent to which this dose of Compound E affects ISF Aβ concentrations. Three-month-old APP/PS1 mice were treated with a single dose of Compound E (3 mg/kg) while cortical in vivo microdialysis was performed to directly measure ISF Aβ levels in living mice. Compound E treatment decreased ISF Aβ1-x by 27% during the first 8 h after treatment and had no effect during the remaining 16 h of sample collection (n = 6; supplemental Fig. 6A,B, available at www.jneurosci.org as supplemental material). To determine the effect of 7 d treatment with Compound E on ISF Aβ, we treated 3-month-old APP/PS1 mice with Compound E (3 mg/kg) or vehicle for 7 d and performed microdialysis during the final 3 d of drug treatment. Compound E treatment decreased ISF Aβx-40 and Aβx-42 levels by 41% (n = 4/group) and 44% (n = 4/group), respectively, during the first 8 h after injection compared with controls (Fig. 6A,B). Aβx-40 and Aβx-42 levels exhibited more modest reductions during hours 8–16 after Compound E treatment and were no different from controls during the final 8 h after injection (Fig. 6A,B). Expressed as relative levels over a 24 h period, Compound E reduced ISF Aβx-40 levels by 22% (n = 4/group) and Aβx-42 by 25% (n = 4/group) compared with controls (Fig. 6C). Together, these results suggest that a modest decrease in ISF Aβ levels may be sufficient to arrest amyloid plaque growth in vivo.

γ-Secretase inhibition results in modest decreases in ISF Aβ levels in cortex. Three-month-old APP/PS1 mice were treated daily with Compound E (3 mg/kg, i.p.) or vehicle for 7 d. In vivo microdialysis was performed to measure ISF Aβx-40 and Aβx-42 in cortex throughout the final 3 d of drug treatment. A, B, Compound E reduced ISF Aβx-40 and Aβx-42 levels by 42% and 44%, respectively, during the first 8 h after each treatment compared with vehicle-treated controls. ISF Aβx-40 and Aβx-42 exhibited more modest decreases during hours 8–16 after Compound E treatment and were no different from controls during hours 16–24 (n = 4/group, two-way ANOVA with post hoc Bonferroni tests; *p ≤ 0.05). C, Expressed as relative levels over 24 h, mice treated with Compound E for 7 d exhibited a 22% decrease in ISF Aβx-40 and a 25% decrease in Aβx-42 compared with controls (n = 4/group; paired t test; *p ≤ 0.05).
Discussion
Amyloid plaques, primarily composed of aggregated Aβ, exist in the extracellular space of the brain and are a pathological hallmark of AD. However, the factors that govern the formation and growth of plaques in the living brain are unknown. In the present study, we used serial in vivo multiphoton microscopy in APP/PS1 mice to directly quantify amyloid plaque formation and growth in vivo. We found that although many plaques remained stable in size at both ages examined, plaques imaged in 6-month-old APP/PS1 mice exhibited robust growth relative to plaques found in 10-month-old mice, suggesting that plaque growth is more prominent early in disease pathogenesis. This finding is consistent with reports of biphasic development of plaque load in APP transgenic mice; compact plaque load is reported to increase with age (Sturchler-Pierrat et al., 1997; Wengenack et al., 2000; Jack et al., 2005; Braakman et al., 2006; Harigaya et al., 2006) before stabilizing at later disease stages (Gordon et al., 2002). Plaque load stabilization has also been hypothesized to occur in human AD patients, as amyloid burden does not correlate with disease duration (Hyman et al., 1993; Engler et al., 2006) and plaque burden in a subset of patients with mild cognitive impairment is indistinguishable from AD patients (Lopresti et al., 2005; Price et al., 2005; Mintun et al., 2006). Our study also indicated that plaque growth was related to plaque size, as smaller plaques exhibited greater rates of growth compared with larger plaques, regardless of age.
We also found that treatment with the γ-secretase inhibitor, Compound E, markedly decreased the appearance of new plaques and growth of preexisting plaques in APP/PS1 mice. This suppression in plaque appearance and growth was reflected in a decrease in total plaque burden in parallel cross-sectional studies. To determine the extent to which Compound E treatment reduced soluble extracellular Aβ levels in vivo, we used in vivo microdialysis to measure ISF Aβ levels in APP/PS1 mice treated with Compound E. Chronic dosing of Compound E over a 7 d period decreased ISF Aβx-40 and Aβx-42 levels by only 20–25% over a 24 h period. That reduced extracellular Aβ concentration is associated with inhibition of amyloid plaque growth but not plaque regression is consistent with previous data obtained using transgenic mice that overexpress mutant APP under the regulation of a tetracycline-responsive promoter. Inhibition of mutant APP expression for 6 months after plaque formation arrested the progression of amyloid pathology but did not reduce overall plaque burden (Jankowsky et al., 2005). A more recent report demonstrated that 3 weeks of treatment with an orally active γ-secretase inhibitor did not reduce size of existing plaques in APP/PS1 mice (Garcia-Alloza et al., 2009). Together, these studies suggest that a kinetic disequilibrium between Aβ plaque aggregation and dissociation may exist in vivo.
The hypothesis that soluble extracellular Aβ concentration is a key determinant of Aβ aggregation in vivo is supported by data demonstrating that areas of the brain that ultimately develop plaque pathology have higher basal ISF Aβ levels early in life relative to brain regions that do not develop pathology (Cirrito et al., 2003). Moreover, intracerebral injection of Aβ-containing brain extract from human AD patients or APP transgenic mice induces cerebral amyloidosis in APP transgenic mice in a concentration-dependent manner (Meyer-Luehmann et al., 2006). Although correlative, the present study is the first demonstration of the relationship between soluble ISF Aβ and amyloid plaque growth in vivo. Moreover, it is the first report that this relationship can be modulated with pharmacological intervention. That a modest reduction of ISF Aβ levels is associated with inhibition of amyloid plaque growth and attenuation of new plaque formation is consistent with recent data demonstrating that a 30% reduction in γ-secretase activity, as seen with knocking out γ-secretase components throughout life, can attenuate plaque burden in a mouse model of AD (Li et al., 2007). Thus, partial γ-secretase inhibition may be sufficient to arrest amyloid plaque progression in AD.
A number of potential therapeutic implications follow from the results of the present study. First, the observation that γ-secretase inhibition can prevent growth of existing plaques and attenuate new plaque formation, but not induce plaque regression suggests that anti-Aβ treatments may be most efficacious if administered early in disease pathogenesis. This hypothesis is supported by our cross-sectional study, showing that 28 d Compound E treatment reduced amyloid plaque load in 6-month-old APP/PS1 mice, but not in 10-month-old mice. Moreover, our finding that a modest reduction in soluble, extracellular Aβ is associated with a dramatic reduction in plaque growth and formation may have important implications for drug dosing and pharmacodynamic effects of anti-Aβ therapeutics to be used in clinical trials.
Our present observations of individual plaque growth over a period of weeks are in contrast to a recent report which showed that plaques reach a mature size within 24 h after appearance (Meyer-Luehmann et al., 2008). We hypothesized that these differences might be due to different techniques used to create transcranial windows. We used a small (<0.8 mm diameter) closed thinned-skull window in our experiments, in contrast to the larger (6 mm diameter) open-skull (craniotomy) window used by Meyer-Luehmann et al. (2008) (see supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). A direct comparison of plaque growth using these two techniques revealed robust growth under thinned-skull window preparations, but no significant growth under open-skull preparations. In addition, the open-skull preparation was associated with extensive cortical microglial and astrocytic activation, which was largely absent under the thinned-skull window. These findings are consistent with a previous comparison of these two techniques, which demonstrated that the open-skull window resulted in significant reactive gliosis and an alteration of dendritic spine dynamics compared with the closed thinned-skull window (Xu et al., 2007). Coupled with previous reports implicating reactive gliosis in plaque size maintenance and regression (Gordon et al., 2002; Wyss-Coray et al., 2003; El Khoury et al., 2007; Takata et al., 2007; Bolmont et al., 2008), this may explain the discrepancy between the findings using the two different techniques.
In summary, our results suggest that individual amyloid plaques grow over a period of weeks and that the rate of plaque growth is related to disease stage, plaque size, gliosis, and soluble extracellular Aβ concentration. Thus, growth of individual plaques may be a fundamental mechanism by which plaque load increases in AD. Furthermore, the present results suggest that a decrease in ISF Aβ levels by as little as 20–25% at key time points in plaque development may be sufficient to prevent the progression of amyloid pathology.
Footnotes
-
This work was supported by grants from the National Institutes of Health (NIH) (R01 NS048283, P01 NS032636), the American Health Assistance Foundation, and the Hope Center for Neurological Disorders (J.-M.L.), and the NIH (P30 NS057105, R37 AG13956), Eli Lilly, and Cure Alzheimer's Fund (D.M.H.). We thank Dr. Robert H. Mach for generously providing methoxy-X04 and X-34. P.Y., A.W.B., J.R.C., D.M.H., and J.-M.L. designed the study. P.Y., A.W.B., Q.X., X.H., Y.W., and E.G. performed the experiments. P.Y., A.W.B., J.R.C., D.M.H., and J.-M.L. interpreted the results. P.Y., A.W.B., J.R.C., D.M.H., and J.-M.L. wrote this manuscript.
- Correspondence should be addressed to Dr. Jin-Moo Lee, Department of Neurology and the Hope Center for Neurological Disorders, Washington University School of Medicine, 660 South Euclid Avenue, Campus Box: 8111, St. Louis, MO 63110. leejm{at}neuro.wustl.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}