

Abstract
Alzheimer's disease (AD) is characterized by synaptic dysfunction and cardinal neuropathological features including amyloid plaques and neurofibrillary tangles. Soluble amyloid-β (Aβ) can suppress synaptic activities by interacting with α7 nicotinic acetylcholine receptors (α7nAChRs). Here, we show that α7nAChR and NMDA glutamatergic receptor (NMDAR) activities are severely compromised in synaptosomes prepared from AD and Aβ1-42 (Aβ42)-exposed control frontal cortex slices from postmortem tissue. Whereas Aβ12-28 prevents Aβ42 from binding to α7nAChRs, 2-[2-(4-bromophenyl)-2-oxoethyl]-1-methyl pyridinium (S 24795), a novel α7nAChR partial agonist, facilitates release of Aβ42 from Aβ42–α7nAChR and –Aβ42 complexes. S 24795 interacts with α7nAChR and Aβ15-20 region of the Aβ42 to enable partial recovery of the α7nAChR and NMDAR channel function. These findings suggest that the Aβ–α7nAChR interaction may be an upstream pathogenic event in AD and demonstrate that some recovery of neuronal channel activities may be achieved in AD brains by removing Aβ from α7nAChRs.
Introduction
Transgenic animals with increased amyloid-β (Aβ) can model early-onset familial Alzheimer's disease (AD) and help elucidate the mechanisms underlying memory and cognitive deficits (Eriksen and Janus, 2007). Exceedingly high brain levels of Aβ in these transgenic animals or in subjects with familial AD appear to exacerbate neuronal dysfunction, leading to the neuropathological characteristics of AD and to memory and cognitive defects (Tanzi and Bertram, 2005).
In postmortem AD brains, the magnitude of synaptic and neurotransmission deficits, neurofibrillary lesions, dementia, and cognitive impairment have a higher correlation with soluble Aβ levels than with Aβ-rich plaque counts (Näslund et al., 2000). Furthermore, antisense targeting Aβ and removal of Aβ by immunization in AD transgenic mice can induce partial recovery of cognitive function. Together, these findings suggest that soluble Aβ is intimately involved in the AD pathogenesis and that preventing Aβ from interacting with its brain targets may slow AD progression (Tanzi and Bertram, 2005; Shankar et al., 2007).
Aβ binds with high affinity to neuronal α7 nicotinic acetylcholine receptors (α7nAChRs) (Wang et al., 2000a,b). This interaction leads to intraneuronal accumulation of Aβ1-42 (Aβ42)–α7nAChR complexes (Nagele et al., 2002), rapid tau phosphorylation (Wang et al., 2003), severe impairment of α7nAChR channels (Liu et al., 2001; Pettit et al., 2001), cholinergic neurotransmission defects (Lee and Wang, 2003), and neuronal cell death (Wang et al., 2000a). Additionally, Aβ suppresses NMDA glutamatergic receptor (NMDAR)-mediated long-term potentiation (LTP), a synaptic mechanism underlying memory and cognitive processing (Shankar et al., 2007). The specific α7nAChR antagonist α-bungarotoxin (α-BTX) reduces Aβ-induced NMDAR internalization, which indicates that the Aβ42–α7nAChR interaction may negatively impact NMDAR function (Snyder et al., 2005). Therefore, chronic perturbation of the α7nAChRs by Aβ42 in AD brains could cause neuronal dysfunctions and neurodegeneration resulting in the accumulation of Aβ-rich amyloid plaques and phosphorylated tau-containing neurofibrillary tangles (NFTs). Hence, disrupting Aβ42–α7nAChR interaction may represent a novel approach to reduce Aβ42-mediated functional deficits, neurodegeneration, and possibly the clinicopathological features of AD.
Recent data have shown that the α7nAChR-specific agonists and partial agonists enhance learning and memory in animal models of cognitive deficits (Pichat et al., 2007; Beracochea et al., 2008; Marighetto et al., 2008). However, it is not clear how the existing Aβ42–α7nAChR complexes are affected by the α7nAChR agents and whether removal of Aβ42 affords functional recovery of the α7nAChRs as well as downstream NMDARs and neuronal activities regulated by these channels.
2-[2-(4-Bromophenyl)-2-oxoethyl]-1-methyl pyridinium (S 24795) is a novel selective α7nAChR partial agonist (Lopez-Hernandez et al., 2007). The observations that S 24795 improves contextual memory of aged mice (Beracochea et al., 2008; Marighetto et al., 2008) and that aged brains have a higher Aβ load (Piccini et al., 2005; Lindner et al., 2006) led us to explore whether this α7nAChR partial agonist may alleviate Aβ-mediated neuronal dysfunction in AD brain by disrupting Aβ42–α7nAChR interaction. We offer insights into mechanism through which S 24795 added in vitro removes Aβ42 from α7nAChRs and normalizes α7nAChR and NMDAR function in synaptosomes from postmortem AD and Aβ-exposed control brain tissues.
Materials and Methods
Materials and chemicals.
Leupeptin, aprotinin, phenylmethylsulfonyl fluoride (PMSF), pepstatin A, soybean trypsin inhibitor, NaF, sodium vanadate, β-glycerophosphate, 2-mercaptoethanol, NMDA, glycine, Tween 20, NP-40, anti-α7nAChR (M-220), ATP, choline kinase, physostigmine sulfate, acetylcholine chloride, AgNO3, tetraphenylboran, n-butyronitrile, and digitonin were all purchased from Sigma-Aldrich. 45Ca2+ (10 Ci/mmol) and [methyl-3H]choline chloride (85.1 Ci/mmol) were purchased from PerkinElmer. N-(3R)-1-Azabicyclo[2.2.2]oct-3-yl-4-chlorobenzamide (PNU 282987), galantamine, memantine, and methyllycaconitine (MLA) were purchased from Tocris Bioscience. Aβ1-42 was obtained from Invitrogen. Aβ1-11, Aβ10-20, Aβ15-20, Aβ12-28, Aβ25-35, Aβ37-43, Aβ29-40, biotinated Aβ1-42, and FITC-conjugated Aβ1-42 were obtained from AnaSpec. Anti-Postsynaptic density-95 (PSD-95) (05494), -Aβ42 (AB5739), and -NR3A (07-356) were from Millipore Bioscience Research Reagents. Anti-phosphotyrosine (sc-508), -neuronal nitric oxide synthase (nNOS) (sc-5302), -phospholipase C-γ1 (sc-7290), -NR1 (sc-9058), -NR2A (sc-9056), -NR2B (sc-9057), -actin (sc-7210), and -β-actin (sc-47778) were all purchased from Santa Cruz Biotechnology. γPKC (P82820) was from BD Biosciences Transduction Laboratories; pY402PyK2 (A00498) and pY416Src (A00396) were from GenScript. Reacti-Bind NeutrAvidin high binding capacity coated 96-well plates, covalently conjugated protein A-agarose beads, biotinylation kit, antigen elution buffer, and chemiluminescent reagents were purchased from Pierce Endogen. All Aβ-derived peptides were dissolved in 50 mm Tris, pH 9.0, containing 10% DMSO and stored at −80°C. All test agents were made freshly according to the manufacturer's recommendation. If DMSO was used as the solvent, the highest DMSO concentration in the incubation medium was 1%.
Postmortem tissue.
This study protocol conformed to the Declaration of Helsinki: Ethical Principles for Biomedical Research Involving Human Beings (the fourth amendment) as reflected in a previous approval by the City College of New York and City University of New York Medical School human research committee. The participants had a uniform clinical evaluation that included a medical history, complete neurological examination, cognitive testing including Mini-Mental state examination and other cognitive tests on episodic memory, semantic memory and language, working memory, perceptual speed, and visuospatial ability as well as psychiatric rating. Based on this information, subjects received AD diagnoses based on National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria (McKhann et al., 1984). Postmortem brain tissues [frontal cortex (FCX)] from patients with clinically diagnosed sporadic AD (n = 11; age range, 66–92; mean, 78.5 ± 2.3) and control tissues from normal, age-matched, neurologically normal (n = 11; age range, 67–90; mean, 78.1 ± 2.3) individuals were obtained from the Harvard Brain Tissue Resource Center (HBTRC) (Belmont, MA) and University of California, Los Angeles, Brain Tissue Resource Center (UBTRC) (Los Angeles, CA) (Table 1). Both HBTRC and UBTRC are supported in part by Public Health Service grants from the National Institutes of Health. The postmortem time intervals for collecting these brains were ≤13 h (mean postmortem intervals for collection of AD and control brain samples were 6.0 ± 0.9 and 5.8 ± 0.8 h, respectively). Diagnostic neuropathological examination was conducted on fixed sections stained with hematoxylin and eosin stain and with modified Bielschowsky silver staining (Yamamoto and Hirano, 1986) to establish any disease diagnosis according to the criteria defined by the National Institute on Aging and the Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of AD (Hyman and Trojanowski, 1997), and brain tissue from age-matched controls was similarly screened. The presence of both neuritic (amyloid) plaques and neurofibrillary tangles in all AD brains was confirmed by Nissl and Bielschowsky staining as well as characterized immunohistochemically with anti-Aβ42 and -NFT staining in frontal and entorhinal cortex as well as hippocampus as described previously (Wang et al., 2000a). Control tissues exhibited no gross and minimal, localized microscopic neuropathology of AD (0–3 neuritic plaques/10× field and 0–6 NFTs/10× field in hippocampus).
Demographic information on the subjects of postmortem brains
One gram blocks of FCX were dissected using a band saw from fresh frozen coronal brain sections maintained at −80°C. These blocks were derived from Brodmann areas 10 and/or 46.
All postmortem tissues were identified by an anonymous identification number, and experiments were performed as a best matched pair without knowledge of clinical information.
Animal treatment for specificity and simulating postmortem study.
Pathogen-free, 10-week-old male Sprague Dawley rats weighing ∼200–215 g (Taconic Farms) were housed individually in a 12 h light/dark cycle with ad libitum access to food and water. All animal procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the City College of New York Animal Care and Use Committee. To determine how postmortem changes may influence the function of α7nAChR, NMDAR, and voltage-gated calcium channels, frontal cortices were either processed immediately after CO2 asphyxiation of rats to make synaptosomes (“fresh, 0 h”), placed on dry ice (“frozen, 0 h”) or retained in their bodies and kept at 25°C for 4 h (“25°C, 4 h”), or refrigerated for 4, 8, or 16 h and then dissected (“4°C, 4 h,” “4°C, 8 h,” and “4°C, 16 h,” respectively). After freezing at −80°C for 7 d before the experimentation, frontal cortices were slowly thawed (from −80 to −20 to 4°C), and synaptosomes were prepared as described below. In addition, synaptosomes were prepared from fresh rat frontal cortices to determine the selectivity of interaction between Aβ42 and the α7nAChRs.
In vitro treatment of brain slices for the assessment of Aβ42–α7nAChR association, Ca2+ influx, and NMDAR signaling.
Postmortem FCX tissues were gradually thawed (from −80 to −20°C) and sliced using a chilled McIlwain tissue chopper (200 μm × 200 μm × 3 mm). Approximately 20 mg of the brain slices were suspended in 1 ml of ice-cold oxygenated Krebs'–Ringer's solution (K-R), containing 25 mm HEPES, pH 7.4, 118 mm NaCl, 4.8 mm KCl, 1.3 mm CaCl2, 1.2 mm KH2PO4, 1.2 mm MgSO4, 25 mm NaHCO3, 10 mm glucose, 100 μm ascorbic acid, 50 μg/ml leupeptin, 0.2 mm PMSF, 25 μg/ml pepstatin A, and 0.01 U/ml soybean trypsin inhibitor, and centrifuged briefly. After two additional washes with 1 ml of ice-cold K-R, brain slices were suspended in 1 ml of K-R.
To determine whether exposure to exogenous Aβ42 increases Aβ42–α7nAChR association and causes Aβ42-induced α7nAChR and NMDAR dysfunction, ∼20 mg of FCX slices from either control subjects or AD individuals were incubated with 0.1 μm Aβ42 at 37°C for 1 h. To test their effects, the following drugs were added immediately after Aβ42: S 24795 (1–100 μm), Aβ12-28 (10 μm), memantine (30 μm), galantamine (30 μm), PNU 282987 (30 μm), MLA (10 μm), or MLA (10 μm) plus S 24795 (10 μm). Incubation continued for 1 h in the dark to minimize light destruction of the test agents such as S 24795. The incubation mixture in a total incubation volume of 0.5 ml was aerated with 95% O2/5% CO2 every 15 min for 1 min during the incubation. Reaction was terminated by the addition of 1.5 ml of ice-cold Ca2+-free K-R. Tissue slices were harvested by brief centrifugation and used as the tissue sources for various assays.
Synaptosome preparation for Aβ42–α7nAChR association, Ca2+influx assays, and determination of high-affinity choline uptake and choline acetyltransferase.
Synaptosomes were prepared from postmortem FCX slices after in vitro incubation by the method described previously (Hahn et al., 2006; Battaglia et al., 2007). Briefly, brain tissues were homogenized in 10 vol of ice-cold homogenization buffer (10 mm HEPES, pH 7.4, 0.32 m sucrose, 0.1 mm EDTA homogenization solution containing 50 μg/ml leupeptin, 10 μg/ml aprotinin, 2 μg/ml soybean trypsin inhibitor, 0.04 mm PMSF, a mixture of protein phosphatase inhibitors, and 0.2% 2-mercaptomethanol) using a Teflon/glass homogenizer (10 strokes). The homogenates were cleared by a centrifugation (1000 × g for 10 min) and the supernatants were centrifuged at 15,000 × g at 4°C for 30 min to pellet the synaptosomes (P2 fraction). The synaptosomes were washed twice at 4°C in 1 ml of ice-cold K-R solution (25 mm HEPES, pH 7.4, 118 mm NaCl, 4.8 mm KCl, 25 mm NaHCO3, 1.3 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, 10 mm glucose, 100 μm ascorbic acid, 50 μg/ml leupeptin, 10 μg/ml aprotinin, 2 μg/ml soybean trypsin inhibitor, 0.04 mm PMSF, and a mixture of protein phosphatase inhibitors). The synaptosomes were then resuspended in 1 ml of K-R solution, and the protein concentrations were determined by the Bradford method (Bio-Rad) according to the manufacturer's instruction.
In vitro treatment of brain synaptosomes for the assessment of Aβ fragments on Aβ42–α7nAChR association and S 24795-mediated changes in Aβ42–α7nAChR association.
To determine the effect of Aβ fragments on Aβ42–α7nAChR interaction, 200 μg of synaptosomes prepared from control subjects were incubated with 0.1 μm Aβ42, 0.1 μm Aβ42 with 10 μm Aβ1-11, Aβ15-20, Aβ25-35, or Aβ37-43 in 250 μl of oxygenated Krebs'–Ringer's solution at 37°C for 30 min. Reactions were stopped by adding 750 μl of ice-cold 0.5 mm EGTA containing Ca2+-free Krebs'–Ringer's solution and synaptosomes were pelleted by centrifugation. The level of Aβ42–α7nAChR association in the obtained synaptosomes was measured by coimmunoprecipitation.
To determine the effect of various Aβ fragments on S 24795-mediated changes in Aβ42–α7nAChR interaction, 200 μg of FCX synaptosomes prepared from control subjects were first incubated with 0.1 μm Aβ42 in 250 μl of oxygenated Krebs'–Ringer's solution at 37°C for 30 min. Reactions were stopped and synaptosomes pelleted by centrifugation. After two more washes with 1 ml of Ca2+-free Krebs'–Ringer's solution, synaptosomes were resuspended in 250 μl of oxygenated Krebs'–Ringer's solution containing 10 μm S 24795 or 10 μm S 24795 with 10 μm Aβ1-11, Aβ15-20, or Aβ29-40, and the incubation was performed at 37°C for 30 min in the dark (to reduce destruction of S 24795). Reactions were stopped and synaptosomes were pelleted by centrifugation. The level of Aβ42–α7nAChR association in the obtained synaptosomes was measured by coimmunoprecipitation as described below.
Assessment of Aβ42–α7nAChR association by coimmunoprecipitation.
Two hundred micrograms of synaptosomes were pelleted by centrifugation and then solubilized by brief sonication in 250 μl of immunoprecipitation buffer (25 mm HEPES, pH 7.5, 200 mm NaCl, 1 mm EDTA, 50 μg/ml leupeptin, 10 μg/ml aprotinin, 2 μg/ml soybean trypsin inhibitor, 0.04 mm PMSF, 5 mm NaF, 1 mm sodium vanadate, 0.5 mm β-glycerophosphate, and 0.02% 2-mercaptoethanol containing 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40) and incubated at 4°C with end-to-end shaking for 1 h. After dilution with 750 μl of ice-cold immunoprecipitation buffer and centrifugation (4°C) to remove insoluble debris, the Aβ42–α7nAChR complexes were isolated by immunoprecipitation with 16 h incubation at 4°C with immobilized rabbit anti-Aβ42 antibodies (1 μg)–protein A-conjugated agarose beads. The resultant immunocomplexes were pelleted by centrifugation at 4°C. After three washes with 1 ml of ice-cold PBS, pH 7.2, and centrifugation, the isolated Aβ42–α7nAChR complexes were solubilized by boiling for 5 min in 100 μl of SDS-PAGE sample preparation buffer (62.5 mm Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.1% bromophenol blue). The content of α7nAChRs in 50% of the obtained anti-Aβ42 immunoprecipitate was determined by Western blotting with monoclonal anti-α7nAChR antibodies. Immobilized rabbit anti-actin (0.5 μg)–protein A-conjugated agarose were added together with anti-Aβ42 in the coimmunoprecipitation process, and the content of β-actin in resultant immunoprecipitates was analyzed by immunoblotting with monoclonal anti-β-actin to illustrate even immunoprecipitation efficiency and loading.
To determine whether Aβ42 interacts selectively with the α7nAChRs, we compared immunoprecipitates of immobilized anti-receptor, -Aβ42, and preimmune rabbit IgG from lysate of Krebs'–Ringer's solution and Aβ42-incubated rat FCX synaptosomes. Rat FCX synaptosomes (5 mg) were incubated with Krebs'–Ringer's solution or 0.1 μm Aβ42 peptides for 30 min at 37°C. Synaptosomes were collected by centrifugation, resuspended by sonicating in 0.5 ml of immunoprecipitation buffer, and solubilized with 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40 for 1 h at 4°C. After centrifugation, the resultant lysate was diluted fivefold and the protein concentration was measured by the Bradford method. Extracts (200 μg) from Krebs'–Ringer's solution incubated synaptosomes were immunoprecipitated with either the indicated receptor antibody or preimmune rabbit IgG (1 μg each), whereas extracts (200 μg) from Aβ42-incubated synaptosomes were immunoprecipitated with anti-Aβ42. The presence of the indicated receptors and Aβ42 in the immnoprecipitates was determined by Western blotting with preimmune rabbit IgG as the negative control.
Assessment of Ca2+ influx in synaptosomes.
NMDAR-, α7nAChR-, and voltage-gated calcium channel-mediated 45Ca2+ influx was studied using synaptosomes prepared from postmortem FCX slices after in vitro treatments. In brief, brain synaptosomes (50 μg for simulating postmortem study and 100 μg for postmortem study) were incubated at 37°C for 5 min in oxygenated 0.3 mm Mg2+ K-R [low Mg2+ K-R (LMKR)] containing 5 μm 45Ca2+ (10 Ci/mmol) followed by incubation with vehicle, 0.1–10 μm PNU 282987, a specific α7nAChR agonist, or 0.1–10 μm NMDA plus 1 μm glycine for 5 min or 65 mm K+ (made with isomolar replacement of Na+) for 1 min in a total incubation volume of 0.5 ml. The reaction was terminated by addition of 0.5 ml of ice-cold Ca2+-free K-R containing 0.5 mm EGTA and centrifugation at 4°C. After two additional washes, 45Ca2+ contents in synaptosomes were assessed using scintillation spectrometry (Beckman). The background 45Ca2+ was estimated using hypotonically lysed synaptosomes. Typically, the background 45Ca2+ was ∼10% of basal levels. The absolute Ca2+ influx was calculated by subtracting background 45Ca2+ count. The percentage increase in Ca2+ influx was calculated as follows: % [(drug-treated − vehicle)/vehicle].
NMDAR activation, signaling, and assemblies.
NMDAR activation, signaling, assemblies, and their interaction with synaptic anchoring protein, PSD-95, were compared in rat brain slices from simulating postmortem study as well as K-R, 0.1 μm Aβ42, or 0.1 μm Aβ42 plus 10 μm S 24795-exposed control versus K-R and S 24795-exposed AD brain slices. NMDAR activation and signaling was initiated by incubation of ∼10 mg of in vitro treated brain slices with either LMKR (basal) or LMKR containing 10 μm NMDA and 1 μm glycine at 37°C for 30 min. The incubation mixture was aerated with 95% O2/5% CO2 every 10 min for 1 min during the stimulation. Ligand stimulation was terminated by the addition of 1 ml of ice-cold Ca2+-free K-R containing 0.5 mm EGTA and 0.1 mm EDTA. Brain slices were harvested by a brief centrifugation and were homogenized in 0.25 ml of ice-cold immunoprecipitation buffer. The homogenates were centrifuged at 1000 × g for 5 min (4°C), and the supernatant (postmitochondrial fraction) was sonicated for 10 s on ice. The proteins were solubilized in 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40 for 60 min at 4°C with end-to-end rotation. The resultant lysates were then cleared by centrifugation at 50,000 × g for 5 min and diluted with 0.75 ml of immunoprecipitation buffer. Protein concentrations were measured by the Bradford method (Bio-Rad).
To measure the magnitude of NMDA activation, the abundance of tyrosine-phosphorylated NR2A and NR2B subunits (pY-NR2A/2B) in response to NMDA (10 μm)/glycine (1 μm) was determined in anti-NR2A/2B immunoprecipitates. To determine the subunit compositions of the NMDAR complexes and their association with PSD-95 as well as NMDAR signaling, the levels of NMDAR subunits, PSD-95, and NMDAR-associated signaling molecules were measured in anti-NR1 immunoprecipitates. In these experiments, brain slice lysates (200 μg) were immunoprecipitated overnight at 4°C with 2 μg of immobilized anti-NR1 or -NR2A/-NR2B onto covalently conjugated protein A-agarose beads (Pierce Endogen). Anti-NR1 or -NR2A/-NR2B immunoprecipitates were incubated with 75 μl of antigen elution buffer and 2% SDS for 2 min on ice, centrifuged to remove antibody–protein A-agarose complexes, and neutralized immediately with 10 μl of 1.5 m Tris buffer, pH 8.8, followed by addition of 65 μl of 2× PAGE sample buffer and boiled for 5 min. Seventy-five microliters of the obtained eluates (50%) were then size fractionated on 7.5% SDS-PAGE. Proteins were transferred to nitrocellulose membrane and the levels of various NMDA receptor subunits, PSD-95, and signaling proteins were measured using Western blotting with antibodies for NR2A, NR2B, NR1, NR3A, PSD-95, nNOS, phospholipase C-γ1, γPKC, pY402PyK2, pY416Src, or phosphotyrosine. The blots were stripped and reprobed with anti-NR1 or -NR2A/-NR2B to assess the loading as appropriate.
Western blot analysis.
Solubilized immunoprecipitates derived from coimmunoprecipitation assays were separated by either 7.5 or 10% SDS-PAGE and then electrophoretically transferred to nitrocellulose membranes. The membranes were washed with PBS and blocked overnight at 4°C with 10% milk in PBS with 0.1% Tween 20 (PBST). After three 5 min washes with 0.1% PBST, the membranes were incubated at room temperature for 2 h with the selected antibody at 1:500–1:1000 dilutions. After three 2 min washes in 0.1% PBST, membranes were incubated for 1 h with anti-species IgG-HRP (1:5000 dilution) and washed with 0.1% PBST three times, 2 min each. Immunoreactivity was visualized by reacting with chemiluminescent reagent for exactly 5 min and immediately exposing to x-ray film. Specific bands were quantified by densitometric scanning (GS-800 calibrated densitometer; Bio-Rad).
Determination of high-affinity choline uptake and choline acetyltransferase activity.
To assess the viability of the postmortem tissues, high-affinity choline uptake and choline acetyltransferase (ChAT) activity were measured in synaptosomes of the postmortem FCX from all 11 control/AD pairs in duplication. Briefly, high-affinity [3H]choline uptake was measured in 100 μg synaptosomes that were incubated in 250 μl of oxygenated Krebs'–Ringer's solution containing 0.1 μm [3H]choline at 37°C for 5 min. The reaction was terminated by fivefold dilution with ice-cold Ca2+-free Krebs'–Ringer's solution and chilled on ice. Nonspecific choline uptake for each sample was measured with 0.1 μm [3H]choline at 4°C for 5 min. [3H]Choline-labeled synaptosomes were harvested by rapid vacuum filtration onto Whatman GF/B filter. After two washes with 5 ml of ice-cold Ca2+-free Krebs'–Ringer's solution, filters were dried and [3H]choline levels were determined by liquid scintillation spectrometry. [3H]Choline uptake was defined by subtracting [3H]choline derived at 4°C from those incubated at 37°C.
Separately, ChAT activity was also estimated in FCX synaptosomes from all 11 control/AD pairs by conversion of [3H]choline to [3H]ACh. Synaptosomes (100 μg) were incubated in 250 μl of oxygenated Krebs'–Ringer's solution containing 0.1 μm [3H]choline at 37°C for 30 min in the presence of 10 μm physostigmine, a cholinesterase inhibitor. The reaction was terminated by fivefold dilution with ice-cold Ca2+-free Krebs'–Ringer's solution, chilling on ice, and centrifugation. The pelleted synaptosomes were washed twice with 1 ml of ice-cold Ca2+-free Krebs'–Ringer's solution and chilled on ice and sonicated 10 s in 1 ml of ice-cold 25 mm HEPES, pH 7.5, containing protease inhibitors. After centrifugation to remove cell debris, [3H]ACh and [3H]choline were extracted from synaptosomal lysate into equal volume of n-butyronitrile containing 10 mg/ml tetraphenylboron as described in our previous work (Wang et al., 1994). [3H]ACh and [3H]choline were recovered from the organic phase with 250 μl of AgNO3 in H2O (20 mg/ml), and the organic phase was discarded. Excessive Ag+ was precipitated by addition of 12 μl of 200 μg/ml MgCl2 solution, and samples were centrifuged for 5 min. The supernatant was recovered and subsequently lyophilized in a vacuumed centrifuge. [3H]ACh and [3H]choline in the lyophilized samples were separated by organic extraction after phosphorylation of the choline. [3H]ACh was extracted into 0.5 ml of n-butyronitrile containing 10 mg/ml tetraphenylboron and determined using liquid scintillation spectrometry.
In vitro assessment of S 24795 on Aβ42–α7nAChR interaction and Aβ42–α7nAChR/Aβ42–Aβ42 complexes.
To determine the effect of S 24795 on Aβ42–α7nAChR interaction, 2 nm immunopurified biotinated α7nAChR derived from SK-N-MC cells using a biotinylation kit (Pierce Endogen) was coated onto streptavidin-coated plates (Reacti-Bind NeutrAvidin high binding capacity coated 96-well plate. Plates were washed three times with ice-cold K-R (200 μl) and incubated at 37°C with K-R or varying concentrations of S 24795 either together with 20 nm FITC-tagged Aβ42 or sequentially (10 min for K-R or S 24795 followed by 60 min FITC-Aβ42). Plates were washed twice with 200 μl of ice-cold K-R and the residual FITC-Aβ42 signals were determined by multimode plate reader (DTX880; Beckman).
To assess whether S 24795 is effective in removing Aβ42 from Aβ42–Aβ42 and/or Aβ42–α7nAChR complexes and to determine the S 24795 interacting site on Aβ42, 2 nm biotinated Aβ42 peptides or α7nAChRs were coated onto streptavidin-coated 96-well plate for 60 min at 25°C. Plates were washed twice with ice-cold K-R (200 μl each) and then incubated with 20 nm FITC-tagged Aβ42 for 60 min at 37°C to form Aβ42–Aβ42 or –α7nAChR complexes. After washing, varying concentrations of S 24795, 10 μm indicated Aβ fragments, or combination of S 24795 and indicated Aβ fragment (10 μm each) were added, and the incubation was continued for 60 min at 37°C. After two washes with 200 μl of ice-cold K-R, the residual FITC-Aβ42 signals were detected using a multimode plate reader, DTX-880. Negligible FITC-Aβ42 was noted when biotinated Aβ42 peptides or α7nAChRs were omitted.
Data analysis.
All data are presented as mean ± SEM. Treatment effects were evaluated by ANOVA as appropriate. Individual differences in the dose–response curve were evaluated by the Newman–Keuls multiple comparisons. The threshold for significance was p < 0.05. Between-group comparisons were also conducted for all parameters using linear mixed effects models as the general framework to account for the cluster structure attributable to pair matching and to include the impact of covariates (age, sex, and postmortem interval).
Results
Effects of S 24795, Aβ12-28, memantine, galantamine, and PNU 282987 on Aβ42–α7nAChR association
To validate that Aβ42 interacts selectively with the α7nAChRs, lysates from Aβ42-incubated synaptosomes prepared from rat FCX were immunoprecipitated with anti-Aβ42 and tested for the presence of various surface receptors. Our data (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) indicate that Aβ42 binds to the α7nAChRs with relative selectivity. We then determined whether S 24795 added in vitro affects Aβ42–α7nAChR interaction in human postmortem FCX slices. Accordingly, the effect of S 24795 on Aβ42–α7nAChR complexes preexisting in postmortem AD and newly formed by adding exogenous Aβ42 was measured in synaptosomes prepared from postmortem human FCX slices of 11 well matched normal control and AD pairs (Table 1) after in vitro incubation with K-R, 0.1 μm Aβ42, 1–100 μm S 24795, or 0.1 μm Aβ42 plus 1–100 μm S 24795. In synaptosomes prepared from postmortem human FCX slices of control individuals, incubation with 0.1 μm Aβ42 in vitro markedly increased the abundance of Aβ42–α7nAChR complexes to the level of AD (10- to 18-fold higher than that of matching controls) as demonstrated by a higher α7nAChR level in the anti-Aβ42 immunoprecipitate (Fig. 1A). Similar incubation of the AD FCX slices with 0.1 μm Aβ42, however, did not further increase Aβ42–α7nAChR complexes. Densitometric quantification of the α7nAChRs coimmunoprecipitated with Aβ42 reveals that S 24795 reduced Aβ42–α7nAChR complexes in a concentration-dependent manner in both AD and Aβ42-exposed control and AD FCX synaptosomes. Although 1 μm S 24795 limited Aβ42–α7nAChR interaction in in vitro Aβ42-exposed control FCX slices by 47.6 ± 5.4%, a 10-fold higher S 24795 concentration was required to reduce Aβ42–α7nAChR complexes in AD FCX slices without and with in vitro addition of Aβ42 by 49.9 ± 6.4 and 47.3 ± 6.9%, respectively (Fig. 1B). Although the maximal S 24795-mediated inhibition was observed at 100 μm (57.8 ± 4.0 and 66.0 ± 4.6% for AD and Aβ42-exposed control slices, respectively), the magnitudes of maximal inhibition was not statistically different from that achieved at 10 or 30 μm (Fig. 1B). The comparable β-actin levels in anti-Aβ42/-β-actin immunoprecipitates demonstrated equal immunoprecipitation efficiencies and loading.
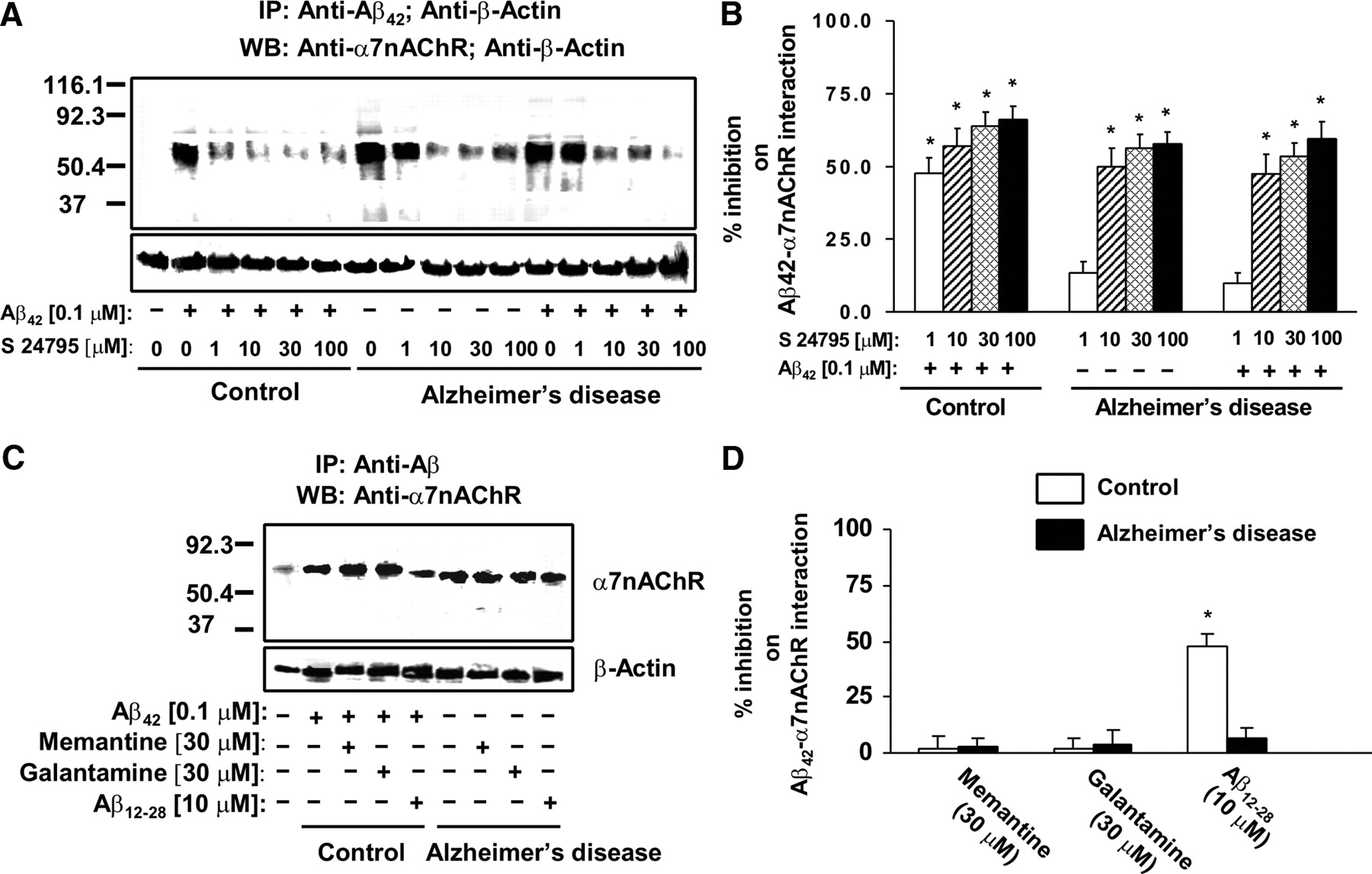
S 24795 but not Aβ12-28, memantine, or galantamine removes Aβ42 from existing Aβ42–α7nAChR complexes in a concentration-dependent manner. A, A representative blot showing the level of Aβ42–α7nAChR complexes in the anti-Aβ42/-actin immunoprecipitates of synaptosomal lysates prepared from 1 h vehicle- or 1–100 μm S 24795-treated postmortem FCX slices of K-R or Aβ42-exposed control and AD subjects. Synaptosomes were lysed using nonionic detergents and the resultant synaptosomal lysates were immunoprecipitated with anti-Aβ42 and anti-actin. The levels of Aβ42-associated α7nAChRs as well as β-actin (immunoprecipitation and loading control) were then measured in the anti-Aβ42/-actin immunoprecipitates by Western blotting with antibodies to α7nAChR and β-actin. B, Densitometric data derived from FCX slices of 11 matched control/AD pairs showing the effect of varying in vitro concentrations of S 24795 on Aβ42-associated α7nAChRs levels. Data are means ± SEM of percentage inhibition comparing the optical intensity of the α7nAChR band derived from independent determinations from an individual postmortem brain with in vitro S 24795 treatments to Aβ42-treated control and vehicle- and Aβ42-treated AD groups, respectively. *p < 0.01 compared with respective Aβ42-treated control and vehicle- and Aβ42-treated AD level. C, A representative blots showing the level of Aβ42–α7nAChR complexes in the anti-Aβ42/-actin immunoprecipitates of synaptosomal lysates prepared from FCX slices of controls exposed to 10 μm Aβ12-28, 30 μm memantine, or 30 μm galantamine alone or with addition of 0.1 μm Aβ42 as well as AD FCX slices incubated with 10 μm Aβ12-28, 30 μm memantine, or 30 μm galantamine. D, Densitometric data derived from FCX slices of six matched control/AD pairs showing the effect of Aβ12-28, memantine, and galantamine on Aβ42-associated α7nAChRs levels. Data are means ± SEM of percentage inhibition comparing the optical intensity of the α7nAChR band derived from independent determinations from an individual postmortem brain with specific in vitro treatments to Aβ42-treated control and vehicle-treated AD groups, respectively. *p < 0.01 compared with respective Aβ42-treated control and vehicle-treated AD level.
Next, we determined whether dissociation of Aβ42 from α7nAChR is a unique property of S 24795. To this end, we compared the in vitro effect of Aβ12-28, an Aβ42 fragment that blocks Aβ42–α7nAChR interaction (Wang et al., 2000a), and galantamine and memantine, two clinically used anti-AD drugs, on Aβ42–α7nAChR levels by incubating these agents alone in AD FCX slices or with 0.1 μm Aβ42 in control FCX slices. Both galantamine and memantine, which bind α7nAChRs, only minimally affected Aβ42–α7nAChR association at 30 μm concentrations (Fig. 1C,D). Although Aβ12-28, at 10 μm, reduced Aβ42–α7nAChR interaction by 48.1 ± 5.4% in control FCX slices that were simultaneously exposed to Aβ12-28 and Aβ42, Aβ12-28 did not lessen Aβ42–α7nAChR levels in AD FCX slices (Fig. 1C,D).
Because S 24795 is an α7nAChR partial agonist, we assessed whether α7nAChR agonist activity is necessary for dissociating Aβ42–α7nAChR complexes. Accordingly, we compared (1) the effect of PNU 282987, a full α7nAChR agonist, to S 24795 on Aβ42–α7nAChR linkage, and (2) the effect of S 24795 on Aβ42–α7nAChR interaction when its agonist activity was blocked by a selective α7nAChR antagonist, MLA. PNU 282987 inhibited Aβ42–α7nAChR interaction with only 20% efficacy of S 24795 (Fig. 2A,B). The inhibition of S 24795 agonist activity by MLA did not affect the magnitude of S 24795-mediated Aβ42–α7nAChR dissociation (Fig. 2A,B). Collectively, the data presented in Figure 2 indicate that agonist activity for the α7nAChR agents is not essential for their ability to disrupt Aβ42–α7nAChR association.

The α7nAChR agonist activity is not a critical determinant of S 24795-induced reduction in Aβ42–α7nAChR complexes. A, A representative blot showing the level of Aβ42–α7nAChR complexes in the anti-Aβ42/-actin immunoprecipitates of synaptosomal lysates prepared from postmortem FCX slices from control and AD subjects that were incubated with the indicated test agents. Control slices were incubated simultaneously for 1 h with 0.1 μm Aβ42 and S 24795; PNU 282987; or MLA alone or with addition of S 24795 (each at 10 μm). AD FCX slices were incubated with 10 μm S 24795; PNU 282987; or MLA alone or with addition of 10 μm S 24795. Synaptosomes were prepared from these FCX slices and lysed using nonionic detergents. The resultant synaptosomal lysates were immunoprecipitated with anti-Aβ42/-actin and the levels of Aβ42-associated α7nAChRs and β-actin in the anti-Aβ42/-actin immunoprecipitates were measured by Western blotting with antibodies against α7nAChR and β-actin. B, Densitometric quantification of Aβ42-associated α7nAChRs levels of four matched control/AD pairs showing the effect of PNU 282987, MLA, S 24795, or MLA plus S 24795 on Aβ42-associated α7nAChRs levels. Data are expressed as means ± SEM of percentage inhibition by PNU 282987, MLA, S 24795, or MLA plus S 24795 comparing optical intensity of the α7nAChR band derived from four independent determinations from an individual postmortem brain of control and AD groups with indicated in vitro treatments to Aβ42 alone control and AD vehicle group, respectively. *p < 0.01 compared with respective Aβ42-treated control and vehicle-treated AD level.
Functional consequence of interfering with Aβ42–α7nAChR association
We further determined the functional consequences of altering Aβ42–α7nAChR interaction using FCX slices from postmortem AD and control brains. First, the viability of the synaptosomes prepared from postmortem tissues was validated using a simulating postmortem study in rat. In this study, synaptosomes prepared from FCX of rats subjected to 0–16 h postmortem conditions were used to examine Ca2+ influx and NMDAR signaling. Although postmortem delays progressively reduce basal (nonstimulated), tissues remained responsive to stimuli that selectively activate the α7nAChRs, NMDARs, and voltage-gated Ca2+ channels. In accord, Ca2+ influx through α7nAChRs, NMDARs, and voltage-gated Ca2+ channels was increased in response to PNU 282987 (a selective α7nAChR agonist), NMDA/glycine, and K+ depolarization, respectively, in a concentration-dependent manner (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The fact that increasing postmortem delays did not affect NMDAR signaling and assembly further reinforces the notion that postmortem tissues remain viable for the assessing Ca2+ influx and NMDAR signaling at least 16 h after death (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
In addition, the viability and integrity of the synaptosomes prepared from postmortem FCX of the 11 pairs of matched AD and controls brains used in the present study was directly examined by a study that assesses high-affinity choline uptake and ChAT activity, two energy-dependent processes that are known to be altered in AD and highly sensitive to postmortem delays. Although the magnitude of choline uptake was inversely related to postmortem intervals and to age (data not shown), high-affinity choline uptake was similar in FCX synaptosomes from control and AD postmortem brains (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). The conversion of choline into acetylcholine by ChAT was clearly detectable in all synaptosomes with a 40.6% reduction found in AD FCX (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). Using the ratio of high-affinity choline uptake to choline acetyltransferase activity as an index of uptake per viable nerve terminal, a twofold increase in choline uptake was noted in synaptosomes from AD FCX (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). Our data are qualitatively similar to those reported previously by Slotkin et al. (1990), and together they indicate that choline uptake in AD is adaptively increased per viable nerve terminal to compensate for the destruction of cholinergic neurons. Together with the data from simulating postmortem study in rat (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material), our data suggest that postmortem FCX used in this study retain sufficient viability for functional assays such as Ca2+ influx and NMDAR signaling.
Next, synaptosomes from FCX slices from postmortem AD and control brains were used to assess the Ca2+ influx through α7nACh and NMDA receptors as well as voltage-gated Ca2+ channels. The basal 45Ca2+ influx was 1422 ± 120 and 1548 ± 261 cpm for control and AD, respectively. Incubation with 0.1 μm Aβ42, 10 μm S 24795, or 10 μm Aβ12-28 in vitro did not affect the basal 45Ca2+ influx in either control or AD synaptosomes. By comparison, the basal 45Ca2+ influx in 100 μg synaptosomes prepared from fresh rat FCX slices that had been exposed to vehicle and Aβ42 was 11,416 ± 309 and 10,076 ± 560 cpm, respectively.
Furthermore, we detected that Aβ42 limited the α7nAChR- and NMDAR-mediated Ca2+ influx and reduced the potency of PNU 282987 and NMDA (as indicated by the rightward shifted dose–response curves of PNU 282987 and NMDA) without affecting K+-induced Ca2+ flux in synaptosomes prepared from control FCX slices that have been exposed to 0.1 μm Aβ42 (Fig. 3A,B). In AD FCX synaptosomes, Ca2+ influx through α7nAChR, NMDAR, and voltage-gated Ca2+ channels in response to PNU 282987, NMDA/glycine, and K+ depolarization, all reduced markedly (51.4–68.7%) compared with vehicle-exposed FCX synaptosomes from matched control subjects (Fig. 3A,B). Parallel to the reduced Aβ42–α7nAChR complexes shown in Figure 1, A and B, 1 h incubation with 10 μm S 24795 nearly doubled the level of agonist-induced Ca2+ influx through α7nAChRs and NMDARs in both AD and Aβ42-exposed control brains without affecting the K+-induced Ca2+ influx in AD brains (Fig. 3A). Similar to the data showing Aβ12-28 reduces Aβ42–α7nAChR interaction in synaptosomes prepared from Aβ42-incubated control but not native AD FCX slices, Aβ12-28 blocked Aβ42-inhibited α7nAChR and NMDAR Ca2+ fluxes in Aβ42-exposed control FCX but did not reverse Ca2+ influx deficits through α7nAChR, NMDAR, or voltage-gated Ca2+ channels in AD brain synaptosomes (Fig. 3B).

S 24795 reduces Aβ42-induced suppression of α7nAChR and NMDAR function as indicated by a higher agonist-induced Ca2+ influx. Synaptosomes prepared from AD FCX slices treated 1 h with vehicle or 10 μm S 24795 or Aβ12-28 as well as from control FCX slices treated 1 h with 10 μm S 24795 or Aβ12-28 plus 0.1 μm Aβ42 were used to assess α7nAChR, NMDAR, and voltage-gated Ca2+ channel function, respectively, by Ca2+ influx in response to 5 min 0.1–10 μm PNU 282987, 5 min 0.1–10 μm NMDA/1 μm glycine, and 1 min 65 mm K+ depolarization, respectively. A, Effect of S 24795 on Ca2+ influx through α7nAChRs, NMDARs, and voltage-gated Ca2+ channels in AD and Aβ42-exposed control brains compared with vehicle-incubated 11 pairs of control and AD brains. B, Effect of Aβ12-28 on Ca2+ influx through α7nAChRs, NMDARs, and voltage-gated Ca2+ channels in six pairs of AD and Aβ42-exposed control brains compared with vehicle-incubated control and AD brains. Data are expressed as means ± SEM of the percentage stimulation calculated based on stimulated/basal 45Ca2+ levels in cpm of 11 (control and AD) or 6 (Aβ12-28 and Aβ42-exposed control) independent determinations from an individual postmortem brain. *p < 0.01, **p < 0.05 compared with vehicle-exposed control brains; #p < 0.01 compared with vehicle-incubated AD and Aβ42-exposed control brains; p̂ < 0.01 compared with Aβ42 alone; +p < 0.01 compared with control and Aβ12-28-exposed control brains.
Since removal of Aβ42 from Aβ42–α7nAChR complexes appears to correlate with partial restoration of the NMDAR-mediated Ca2+ influx to control values (Figs. 1, 3), we further compared NMDAR activation by the level of pY-NR2A/2B and signaling in control FCX slices that have been exposed to vehicle, Aβ42, and S 24795 plus Aβ42 in vitro as well as AD FCX slices that have been exposed to vehicle and S 24795.
Incubation with NMDA plus glycine resulted in a 5.8-fold increase in pY-NR2A/2B level compared with the baseline in control FCX slices. This NMDA/glycine-mediated effect was reduced by 58.2 ± 4.0 and 64.6 ± 7.6%, respectively, in AD and after Aβ42 exposure to control FCX slices when compared with K-R incubated control FCX slices (Fig. 4A,B). There were no appreciable changes in basal pY-NR2A/2B level in AD and after Aβ42 exposure. Compared with controls, AD brains also showed a 29.9–30.8% reduction in total NR2A and NR2B densities (Fig. 4A,B). Parallel to reducing Aβ42–α7nAChR association and reversing Aβ42-inhibited NMDAR Ca2+ fluxes by S 24795, in vitro incubation with S 24795 dramatically improved NMDA/glycine-induced pY-NR2A/2B levels in AD and Aβ42-exposed FCX slices to 80.7 ± 1.4 and 81.3 ± 2.6%, respectively, of the levels obtained in K-R-exposed control FCX slices (Fig. 4A,B).

S 24795 normalizes tyrosine phosphorylation of NMDAR-NR2A/2B subunits and NMDAR signaling in AD and Aβ42-exposed control brains. Brain slices were stimulated with 10 μm NMDA/1 μm glycine and then lysed. NMDA receptor activation was measured in lysates prepared from FCX slices of vehicle-treated control–AD pairs (n = 11), 10 μm S 24795-treated AD (n = 11), and 0.1 μm Aβ42-incubated controls with or without 1 h 10 μm S 24795 treatment (n = 6 each) by the magnitude of pY-NR2A and -NR2B levels (A, B) and NMDAR signaling (C, D). A, NR2A/NR2B in brain slice lysates were immunoprecipitated with anti-NR2A/-NR2B and the level of the pY-NR2A/-NR2B was determined by Western blotting using phosphotyrosine antibody. B, Densitometric quantifications of the pY-NR2A/-NR2B optical intensity levels (means ± SEM) in A. C, NMDAR signaling defined by the levels of nNOS, PLC-γ1, and γPKC recruited into NMDARs and NMDAR-associated pY402-PyK2 and pY416-Src in anti-NR1 immunoprecipitates of FCX slice lysates of vehicle- and NMDA/glycine-stimulated by Western blotting using specific antibodies. D, Bar plots showing the optical intensity levels (means ± SEM) of nNOS, PLC-γ1, γPKC, pY402-PyK2, and pY416-Src as well as NR1 bands in response to 10 μm NMDA/1 μm glycine in controls, vehicle, and S 24795 treated AD and Aβ42-exposed controls. *p < 0.01; **p < 0.05 compared with respective response in vehicle-exposed control group; #p < 0.01, +p < 0.01, compared with vehicle-exposed tissues in the same group.
Next, we determined whether 1 h S 24795 incubation with AD FCX slices and Aβ42-incubated control FCX slices also have favorable effect on NMDAR signaling by measuring the abundance of NMDAR-associated nNOS, PLC-γ1, and PKCγ recruited to the NMDAR complexes and the level of NMDAR-coupled activated (tyrosine-phosphorylated) protein tyrosine kinase-2 (PyK2) and Src tyrosine kinases elicited by an incubation with NMDA/glycine. In response to NMDA/glycine stimulation, NMDAR-associated nNOS, PLC-γ1, γPKC, pY402-PyK2, and pY416-Src levels were increased by at least fivefold in K-R-exposed control FCX slices (Fig. 4C,D). These NMDA/glycine-induced responses were reduced in AD by 45.7–71.4% and in Aβ42-exposed control FCX slices by 41.6–75.5% (Fig. 4C,D). Although Aβ42 exposure did not affect the NR1 level in lysate of the control FCX slices, a 30.5–34.9% reduction in NR1 was noted in AD FCX (Fig. 4C,D). Neither AD nor Aβ42 exposure altered basal pY402-PyK2 and pY416-Src levels, suggesting that basal PyK2 or Src activities are intact in AD (Fig. 4C,D). Remarkably, in vitro incubation with 10 μm S24795 for 1 h reinvigorated NMDAR signaling in AD and Aβ42-exposed control FCX slices as indicated by improving the pY402-PyK2, pY416-Src, PKCγ, PLC-γ1, and nNOS levels in the NMDAR complexes by at least 47.2–83.9% of control values (Fig. 4C,D).
To determine whether changes in NMDAR activation and signaling elicited by in vitro Aβ42 and S 24795 were mediated by their influence on the NMDAR assemblies, we compared the subunit compositions of the NMDARs in control and AD FCX slices after exposure to K-R, Aβ42, or S 24795. The effect of Aβ42 and S 24795 on NMDAR signaling appears to occur without changes in NMDAR subunit compositions or numbers in synapses as those parameters were comparable between K-R and Aβ42- or S 24795-exposed groups (Fig. 5A–C). Compared with controls, AD frontal cortices have lower abundance of the NMDAR assemblies with 41.9 ± 3.0, 26.5 ± 2.4, 44.2 ± 3.4, and 58.9 ± 4.6% reduction in NR1, NR2A, NR2B, and NR3A, respectively (Fig. 5A,B). Although Aβ42 or S 24795 did not affect the ratios of NR2A/NR1, NR2B/NR1, NR3A/NR1, and PSD-95/NR1 in control FCX slices, the NR2A/NR1 ratio was increased by 32.6 ± 3.5% and NR3A/NR1 ratio was decreased by 21.7 ± 2.6% in AD FCX compared with controls that were not affected by S 24795 (Fig. 5C). Collectively, the data summarized in Figure 5 suggest that Aβ42-mediated reduction and S 24795-induced restoration of NMDAR activation and signaling are not mediated by altering NMDAR assemblies. Moreover, our data indicate a reduced NMDAR density in AD FCX with altered NR2A/NR2B ratio and additional negative effect on abundance of NR3A.

S 24795 restores NMDA/glycine-responsive NMDAR–PSD-95 linkage without affecting NMDAR assemblies in AD and Aβ42-treated control brains. NMDAR assembly and interaction with PSD-95 was determined in anti-NR1 immunoprecipitate derived from solubilized postmitochondrial lysate of FCX slices of 11 matched control and AD pairs as well as 6 Aβ42-exposed control with or without 1 h in vitro incubation with S 24795. A, The effect of S 24795 on NMDAR subunit compositions and NMDAR–PSD-95 interaction after 10 μm NMDA/1 μm glycine in 11 control-AD pairs and 6 Aβ42-exposed controls. B, Densitometric quantifications of the NMDAR subunit assemblies and NMDAR-associated PSD-95 levels shown in A. C, Bar plots showing ratios of NMDAR subunits and PSD-95 to NR1 levels. Data are expressed as means ± SEM of optical intensity or ratios of NMDAR subunits and PSD-95 to NR1 levels derived from 6 to 11 independent determinations from an individual postmortem brain of the indicated diagnostic groups with indicated in vitro treatments. *p < 0.01, **p < 0.05 compared with respective response in vehicle-exposed control group; #p < 0.01 compared with vehicle-exposed tissues in the same group.
Since PSD-95 anchors the NMDARs to PSDs within the synaptic membranes and thereby regulate NMDAR activation and signaling, we probed whether Aβ42, S 24795, or AD affect NMDAR–PSD-95 linkage. Although NMDA/glycine increased NMDAR–PSD-95 association by 73.5 ± 7.7% in control brains, this NMDA/glycine effect was reduced by 63.7 ± 12.1 and 79.4 ± 11.8% in AD and Aβ42-exposed control FCX, respectively (Fig. 5A,B). Incubation with S 24795 markedly improved NMDA/glycine-induced NMDAR–PSD-95 coupling in AD and Aβ42-exposed control FCX slices to 87.4 and 76.9% of control response, respectively (Fig. 5A–C). These data suggest that in vitro S 24795 exposure normalizes NMDAR–PSD-95 coupling to improve NMDAR activation and signaling.
Effects of S 24795 on Aβ42–α7nAChR and Aβ42–Aβ42 association in a cell-free system
To more precisely unravel the site of action for S 24795-mediated Aβ42–α7nAChR uncoupling effect, we tested the effect of S 24795 on Aβ42–α7nAChR interaction as well as on preformed Aβ42–α7nAChR and Aβ42–Aβ42 complexes using a cell-free system. We first validate the effect of S 24795 on Aβ42–α7nAChR interaction using FITC-conjugated Aβ42 peptides and biotinated α7nAChRs that were bound to a streptavidin-coated plate. The data summarized in Figure 6A indicate that S 24795 reduced α7nAChR–Aβ42 interaction, although S 24795 preexposure was more effective than a simultaneous exposure. In light of S 24795 being capable of removing Aβ42 from preexisting Aβ42–α7nAChR complexes in AD FCX, we examined whether S 24795 also dissociates Aβ42 from preformed Aβ42–α7nAChR and –Aβ42 complexes. Furthermore, we elucidate the potential interacting site on Aβ42 for S 24795 by measuring the effect of S 24795 on preexisting Aβ42–α7nAChR and Aβ42–Aβ42 complexes in the absence or presence of Aβ fragments including those surrounding Aβ15-20, the proposed epicenter of Aβ oligomerization (Austen et al., 2008). Our data summarized in Figure 6, B and C, indicate that, although Aβ12-28, Aβ10-20, Aβ1-11, and Aβ15-20 themselves did not affect Aβ42 contents in the preformed Aβ42–α7nAChR and Aβ42–Aβ42 complexes, Aβ15-20-containing fragments (i.e., Aβ12-28, Aβ10-20, and Aβ15-20) prevented S 24795 from removing Aβ42 from Aβ42–α7nAChR and Aβ42–Aβ42 complexes. The abundance of Aβ42 was also not affected by C terminus of Aβ: Aβ25-35, Aβ37-43, or Aβ29-40 themselves, although these Aβ fragments modestly but significantly increased the magnitude of S 24795-mediated Aβ42 release from Aβ42–α7nAChR and Aβ42–Aβ42 complexes (Fig. 6B,C).

S 24795 reduces Aβ42–α7nAChR association and removes Aβ42 from preformed Aβ42–Aβ42 and Aβ42–α7nAChR complexes by binding to Aβ15-20. Biotin-tagged α7nAChRs or Aβ42 peptides were trapped on streptavidin-coated 96-well plate by incubating with 2 nm α7nAChRs or Aβ42 at 25°C for 1 h. A, S 24795 added either simultaneously with or 10 min before 20 nm FITC-conjugated Aβ42 dose-dependently reduced FITC-conjugated Aβ42 interaction with biotinated α7nAChRs trapped on streptavidin-coated 96-well plate as measured by the residual FITC signals after washing. The data are expressed as mean ± SEM of percentage inhibition comparing S 24795- to vehicle-treated wells (n = 6–8). B, Aβ15-20 containing Aβ fragments (i.e., Aβ12-28, Aβ10-20, and Aβ15-20) reduced or prevented, whereas C terminus Aβ peptides (i.e., Aβ25-35, Aβ29-40, and Aβ37-43) enhanced S 24795-induced Aβ42 release from preformed Aβ42–α7nAChR complexes resulting from FITC-Aβ42 incubation with biotinated α7nAChRs trapped on streptavidin-coated 96-well plates. The data are mean ± SEM of percentage dissociation by S 24795, the indicated Aβ fragments alone or plus S 24795 (10 μm each) (n = 4–8). C, Aβ15-20-containing peptides (i.e., Aβ12-28, Aβ10-20, and Aβ15-20) blocked, whereas C terminus Aβ peptides (i.e., Aβ25-35, Aβ29-40, and Aβ37-43) increased S 24795-mediated release of Aβ42 from preformed Aβ42–Aβ42 complexes deriving from FITC-Aβ42 incubation with biotinated Aβ42 trapped on streptavidin-coated 96-well plates. The data expressed are mean ± SEM of percentage dissociation by S 24795, the indicated Aβ fragments alone or plus S 24795 (10 μm each) (n = 4–8). *p < 0.01, **p < 0.05 compared with vehicle control; #p < 0.01, ##p < 0.05 compared with 10 μm S 24795 alone.
In light of the possibility that the conformation of the FITC- and/or biotin-tagged α7nAChRs and Aβ42 may be different from the native proteins and thereby influence the pattern of interaction, we tested the ability for selective Aβ fragments to block Aβ42–α7nAChR interaction using synaptosomes containing native α7nAChRs prepared from postmortem FCX of control subjects. Similar to that observed using cell-free system, Aβ42–α7nAChR interaction was blocked by simultaneous incubation of FCX synaptosomes with Aβ42 and Aβ15-20 but not Aβ1-11, Aβ25-35, or Aβ37-43 (Fig. 7A). To validate the putative S 24795 action site on Aβ42-associated α7nAChR, the ability for specific Aβ42 fragments to block S 24795 induced dissociation of Aβ42–α7nAChR complexes was tested. In this case, Aβ42–α7nAChR complexes were first established by incubating synaptosomes prepared from FCX of control subjects with Aβ42. The synaptosomes containing Aβ42–α7nAChR complexes were then incubated with S 24795 in the absence or presence of Aβ1-11, Aβ15-20, or Aβ29-40. In accord with the data obtained using cell-free system, Aβ15-20 but not Aβ1-11, Aβ29-40 blocked S 24795-induced reduction in Aβ42–α7nAChR complexes (Fig. 7B). Collectively, the data summarized in Figure 7 confirm that Aβ15-20 is both the interaction site on Aβ42 for α7nAChRs and the action site for S 24795.

Aβ15-20 is the primary Aβ42 binding domain to α7nAChRs and the action site of S 24795 on dissociating Aβ42 from Aβ42–α7nAChR complexes. A, Top, A representative blot depicting the level of Aβ42–α7nAChR complexes in the anti-Aβ42/-actin immunoprecipitates of lysates preparing from synaptosomes of postmortem FCX of control subjects incubated with Aβ42 and Aβ15-20, Aβ1-11, Aβ25-35, or Aβ37-43. The levels of Aβ42-associated α7nAChRs and β-actin (immunoprecipitation and loading control) were detected in the anti-Aβ42/-actin immunoprecipitates by Western blotting with antibodies to α7nAChR and β-actin and quantified by densitometric scanning (bottom). Data are means ± SEM of the percentage inhibition by comparing optical intensity of the α7nAChR band of Aβ42 plus the indicated Aβ peptides to Aβ42 alone group derived from four independent determinations from an individual postmortem control brain. There were no discernible changes in β-actin level by any of the in vitro treatments. *p < 0.01 compared with Aβ42 alone. B, A representative blot (top) depicting the level of Aβ42–α7nAChR complexes in the anti-Aβ42/-actin immunoprecipitates of lysates preparing from synaptosomes of postmortem FCX of control subjects that were incubated with Aβ42 and then exposed to S 24795, S 24795 plus Aβ15-20, Aβ1-11, or Aβ29-40. The levels of Aβ42-associated α7nAChRs and β-actin (immunoprecipitation and loading control) were detected in the anti-Aβ42/-actin immunoprecipitates by Western blotting with antibodies to α7nAChR and β-actin and quantified by densitometric scanning (bottom). Data are means ± SEM of the percentage inhibition by S 24795 or S 24795 plus the indicated Aβ peptides comparing optical intensity of the α7nAChR band of S 24795 and S 24795 plus the indicated Aβ peptides groups with Aβ42 alone. The data were derived from four independent determinations from an individual postmortem brain of control subject. There were no discernible changes in β-actin level by any of the in vitro treatments. *p < 0.01 compared with S 24795 group.
Discussion
The present study shows that in vitro exposure to S 24795 reduced the existing Aβ42–α7nAChR complexes in AD FCX synaptosomes and slices to normalize α7nAChR- and NMDAR-mediated Ca2+ influx and improves NMDAR signaling in AD and Aβ42-exposed control brain synaptosomes. S 24795 facilitates Aβ42 release from Aβ42–α7nAChR and –Aβ42 complexes by interacting with the Aβ15-20 region of the Aβ42, the pivotal Aβ42 binding domain to the α7nAChR. In contrast, neither galantamine nor memantine, two clinically used anti-AD agents, nor PNU 282987, a selective α7nAChR full agonist, affect Aβ42–α7nAChR complexes under the same experimental conditions.
These findings support the notion that AD pathogenesis involves multiple pathways including those mediated through Aβ–α7nAChR interaction (Wang et al., 2000a, 2003), NMDAR defects (Battaglia et al., 2007), cholinergic degeneration resulting from NGF deficiency (Capsoni et al., 2002), and excitotoxicity (Yamada and Nabeshima, 2000), which are differentially targeted by S 24795, memantine, and galantamine.
We examined the α7nAChR and NMDAR function using their Ca2+ permeability and signaling in the postmortem tissues after incubation with selective test agents. These systems have been used extensively in live cells and fresh animal tissues but not in postmortem tissues. We validated these protocols in postmortem tissues and have successfully used them to study other signaling mechanisms (Wang and Friedman, 2001; Hahn et al., 2006). The viability of the postmortem FCX used in this study was illustrated by the presence of high-affinity choline uptake and ChAT activity, which resonates with a previous demonstration that postmortem brain slices (up to 8 h in ambient temperature) remain alive for several weeks in culture (Verwer et al., 2002). Although the degree to which the result of this protocol corresponds to in vivo measurements remains unclear, these postmortem brain function protocols can provide valuable information on functional status of the disease brains and effects of drugs on specific brain activities.
Soluble Aβ oligomers dampen LTP (Walsh et al., 2002; Shankar et al., 2007), impair α7nAChR-dependent LTP induction (Chen et al., 2006), and disrupt cognitive function (Yamada et al., 2005). Since Aβ42 dramatically reduces α7nAChR- and NMDAR-mediated Ca2+ influx and NMDAR signaling, a higher diffusible Aβ in AD CSF and brain (Georganopoulou et al., 2005) likely causes α7nAChR and NMDAR deficiencies and elevates Aβ42–α7nAChR complex levels in AD observed here and previously (Wang et al., 2000a). Since reducing Aβ42–α7nAChR complexes (S 24795) or blocking Aβ42–α7nAChR interaction (Aβ12-28) attenuates Aβ42 deleterious effects on α7nAChRs and NMDARs, removing Aβ42 from α7nAChRs may improve synaptic function regulated by α7nAChRs and NMDARs. Nevertheless, protracted Aβ exposure in AD eventually causes synaptic loss (Hsieh et al., 2006; Shankar et al., 2007) and reduces α7nAChR and NMDAR function and density (Scheuer et al., 1996; Wevers et al., 1999). This synaptic loss is reflected by a weaker reversal of α7nAChR and NMDAR function in AD versus Aβ42-exposed controls and the failure of S 24795 to rescue the reduced K+ depolarization-induced Ca2+ influx in AD brain tissues. The improved NMDAR function we show after removal of Aβ42 from α7nAChRs, along with the prevention by α-BTX of Aβ-induced NMDAR-NR1 subunit internalization (Snyder et al., 2005) and the milder cognitive deficit induced by Aβ after eliminating α7nAChRs (Dziewczapolski et al., 2009), all indicate that α7nAChR is a pivotal upstream NMDAR regulator and mediator of Aβ-induced pathologies and cognitive deficit.
Aβ42 and Aβ40 bind to α7nAChRs with high affinity but not to NMDARs (Wang et al., 2000a,b) (our unpublished data). Aβ40 is 1000-fold less potent than Aβ42 and its effects are reversible (Lee and Wang, 2003), whereas Aβ42 effects are irreversible and it internalizes with α7nAChRs (Nagele et al., 2002). Collectively, these data suggest that Aβ40 is less pathogenic than Aβ42 and that S 24795 should be more efficacious in disrupting Aβ40–α7nAChR interaction. Since both Aβ29-40 and Aβ37-43 failed to influence the Aβ42–α7nAChR interaction but increased the S 24795-induced Aβ42 dissociation from Aβ42–α7nAChR complexes, the C terminus of these two Aβ species does not account for their differential reversibility and is not the critical Aβ42–α7nAChR interaction site. With a higher propensity to aggregate (Masuda et al., 2008; Shen et al., 2008), the C terminus of Aβ42 may bind to Aβ42 released by S 24795 and thereby prevent their reassociation with α7nAChRs and Aβ42.
Although S 24795 is a α7nAChR partial agonist, the agonist-binding site is not essential for its Aβ42 releasing effects. Specifically, (1) MLA did not block S 24795-mediated reduction in Aβ42–α7nAChR association, (2) PNU 282987 only marginally reduced Aβ42–α7nAChR interaction, and (3) S 24795 removed Aβ42 from Aβ42–α7nAChR complexes with concentrations lower than its apparent affinity for rat brain α7nAChRs (4.6 μm) (Lopez-Hernandez et al., 2007).
Both S 24795 and Aβ12-28 reduce Aβ42–α7nAChR interaction and Aβ42-mediated α7nAChR and NMDAR inhibition in Aβ42-incubated control tissues. However, S 24795 but not Aβ12-28 facilitates Aβ42 release from preformed Aβ42–α7nAChR complexes and enables partial recovery of α7nAChR and NMDAR function in AD. Thus, Aβ12-28 might limit Aβ42 effects by competing for the Aβ42 interaction sites on α7nAChRs. Although the differential abilities for S 24795 versus Aβ12-28 on AD tissues may be related to their cell permeability, our data collected from a cell-free assay showing S 24795 but not Aβ12-28 releases Aβ42 from preformed Aβ42–α7nAChR (Aβ42) argue against this possibility. Most importantly, removal of Aβ with S 24795 can significantly reverse functional deficits in AD brain, although the Aβ aggregation status and localization of the Aβ/α7nAChR aggregates remain ambiguous. Interestingly, a site-directed Aβ42 antibody, tetracyclines, and nordihydroguaiaretic acid also disaggregate preformed Aβ (Solomon et al., 1997; Forloni et al., 2001; Ono et al., 2002).
Accumulating data suggest that intraneuronal nonfibrillar Aβ42 oligomers damage neurons. In accord, the magnitude of NMDAR-dependent LTP and paired-pulse facilitation deficiencies and neurodegeneration correlate temporally with the appearance and levels of intraneuronal Aβ42 deposits without significant amyloid plaques and NFT (Chui et al., 1999; Oddo et al., 2003). Aβ42 internalization is also accompanied by nuclear DNA fragmentation (LaFerla et al., 1995), heightened tau phosphorylation (Wang et al., 2003), surface α7nAChRs internalization, and neuronal lysis (Nagele et al., 2002), all of which contribute to the destruction of α7nAChR- and NMDAR-expressing pyramidal neurons in AD. Hence, by removing Aβ42 from Aβ42–α7nAChR complexes, S 24795 should decrease intraneuronal Aβ42 aggregates and, thus, neurodegeneration and amyloid plaque formation resulting from lysis of neurons with excessive intraneuronal Aβ aggregates (Gouras et al., 2000; D'Andrea et al., 2001). The present study, however, cannot ascertain whether S 24795 also extracts Aβ42 from intraneuronal Aβ42–α7nAChR (Aβ42) aggregates.
The reduced NMDAR activation and signaling in Aβ42-incubated control brain slices and the report showing Aβ oligomers induce NMDAR-dependent long-term depression (LTD) in cultured hippocampal neurons (Shankar et al., 2007) illustrates that Aβ causes NMDAR-dependent synaptic defects in AD individuals and transgenic AD mice with heightened Aβ (Di Lazzaro et al., 2003; Inghilleri et al., 2006; Battaglia et al., 2007). NMDARs clustered in PSD through interaction with synaptic anchoring proteins like PSD-95 (Malenka and Nicoll, 1993). NMDAR stimulation activates PyK2 and Src family tyrosine kinases, increases NMDAR–PSD-95 coupling leading to augmented NMDAR signaling by providing the anchoring sites for signaling molecules such as PLCγ and PKCγ (Hahn et al., 2006), and increases coupling of NMDARs to signaling proteins including nNOS and CaMKIIα through PSD-95 (Williams et al., 2003; Hahn et al., 2006; Battaglia et al., 2007). Our data indicate that, through α7nAChRs, Aβ negatively impacts NMDAR–PSD-95 interaction leading to reduced NMDAR activity and signaling. By releasing Aβ42 from α7nAChRs, S 24795 improves NMDAR–PSD-95 coupling and hence NMDAR signaling and activity. The partial restoration of NMDAR and α7nAChR function by S 24795 in AD brains again suggests that some of the brain dysfunction in AD derives from reversible soluble Aβ42 oligomer-mediated suppression of the synaptic processes. The reduced NMDAR density we observed in AD brains likely contributes to reduce NMDAR activity that consequently triggers a compensatory increase in NR2A and decrease NR3A in NMDARs. This could result in a higher Mg2+ blockade with an improved Ca2+ permeability and current of NMDARs in AD brain to offset synaptic impairments (Das et al., 1998).
In summary, our present and previous results suggest that binding of soluble Aβ (oligomers) to α7nAChRs reduces α7nAChR and NMDAR activities, resulting in attenuated synaptic processes, increased intraneuronal Aβ42 aggregation, and tau phosphorylation (Nagele et al., 2002; Wang et al., 2003) (Fig. 8). We show that by removing Aβ42 from existing Aβ42–α7nAChR complexes, S 24795 reinvigorates α7nAChRs and NMDARs, two prominent synaptic modulators. Hence, S 24795 may achieve a therapeutic effect in AD by combating Aβ42-mediated α7nAChR and NMDAR suppression (Beracochea et al., 2008; Marighetto et al., 2008). Additionally, S 24795 may promote synaptic LTP by directly activating Aβ-free α7nAChRs to augment NMDAR function (Lagostena et al., 2008). Since aged brains also harbor a higher Aβ load (Piccini et al., 2005; Lindner et al., 2006), S 24795 could potentially lessen age-associated memory deficits in addition to the profound cognitive impairments of AD. Finally, our data also indicate that, although reversal of some AD dysfunction may be achievable, early detection of this disease clearly improves the “window of opportunity” for more effective treatments.

Hypothetical pathways through which S 24795 normalizes Aβ42-induced synaptic dysfunction. Based on the results of this study and our previous work (Wang et al., 2000a,b, 2003; Nagele et al., 2002; Battaglia et al., 2007), soluble Aβ42 or Aβ42 oligomers bind to α7nAChRs in AD brains to induce synaptic dysfunction by first markedly activating α7nAChRs (Dougherty et al., 2003) followed immediately by rapid desensitizing and restricting Ca2+ influx through α7nAChRs and downstream NMDAR activity and NMDAR signaling. In addition, Aβ42 activates α7nAChR-associated kinases leading to rapid tau phosphorylation and neurofibrillary lesions (Wang et al., 2003). Similar to partial pharmacological blockade of NMDARs, Aβ42-induced hypofunctioning NMDARs is expected to cause a reduced NMDAR-dependent LTP with increased LTD probability leading to dendritic spine shrinkage and retraction (Shankar et al., 2007) that further dampens excitatory neurotransmission. Treatment with S 24795 removes Aβ42 monomers and/or oligomers from Aβ42–α7nAChR complexes. This relieves Aβ42-inhibited α7nAChR activity leading to an improved NMDAR–PSD-95 linkage and consequently the NMDAR signaling and activity. The augmented NMDAR signal and activation favor LTP induction and healthier dendritic spines resulting in more normal excitatory neurotransmission and cognitive processing. The red triangles indicate the effect of Aβ42, and the blue lines and triangles reflect the impact of S 24795.
Footnotes
-
↵†Deceased, Jan. 6, 2008.
-
This work was supported by grants from Institut de Recherches Internationales Servier in France, who also supplied S 24795. We express our gratitude to the patients and their families for their generous participation. We thank Dr. Pierre-Alain Boyer, Dr. Maria Felice Ghilardi, and Dr. Lindsay Burns for helpful discussion and editorial suggestions. This paper is dedicated to the late Dr. Philippe Morain, whose knowledge of the field and enthusiasm for research were, and will continue to be, an inspiration to all of his coauthors and colleagues.
- Correspondence should be addressed to Hoau-Yan Wang, Department of Physiology and Pharmacology, Sophie Davis School of Biomedical Education, City University of New York Medical School, H-210F, Harris Hall, 160 Convent Avenue, New York, NY 10031. hywang{at}sci.ccny.cuny.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}