

Abstract
Macrophages dominate sites of CNS injury in which they promote both injury and repair. These divergent effects may be caused by distinct macrophage subsets, i.e., “classically activated” proinflammatory (M1) or “alternatively activated” anti-inflammatory (M2) cells. Here, we show that an M1 macrophage response is rapidly induced and then maintained at sites of traumatic spinal cord injury and that this response overwhelms a comparatively smaller and transient M2 macrophage response. The high M1/M2 macrophage ratio has significant implications for CNS repair. Indeed, we present novel data showing that only M1 macrophages are neurotoxic and M2 macrophages promote a regenerative growth response in adult sensory axons, even in the context of inhibitory substrates that dominate sites of CNS injury (e.g., proteoglycans and myelin). Together, these data suggest that polarizing the differentiation of resident microglia and infiltrating blood monocytes toward an M2 or “alternatively” activated macrophage phenotype could promote CNS repair while limiting secondary inflammatory-mediated injury.
Introduction
Clinical and experimental CNS trauma elicits an inflammatory response that comprises mostly macrophages (Soares et al., 1995; Holmin et al., 1998; Sroga et al., 2003; Fleming et al., 2006; Nagamoto-Combs et al., 2007). These cells exist in a state of dynamic equilibrium within the lesion environment; whether they differentiate into cells that exacerbate tissue injury or promote CNS repair likely depends on signals in the lesion microenvironment.
In vitro, unique stimuli endow macrophages with distinct molecular phenotypes and effector functions (for review, see Gordon, 2003; Mantovani et al., 2004). Lipopolysaccharide (LPS) and the proinflammatory cytokine interferon-γ (IFNγ) promote the differentiation of “classically activated” M1 macrophages. These cells produce high levels of oxidative metabolites (e.g., nitric oxide and superoxide) and proinflammatory cytokines that are essential for host defense and tumor cell killing but that also cause collateral damage to healthy cells/tissue (Ding et al., 1988). Conversely, activating macrophages in the presence of cytokines such as interleukin-4 (IL-4) or IL-13 promotes an “alternatively activated” M2 phenotype. M2 macrophages promote angiogenesis and matrix remodeling while suppressing destructive immunity (Sica et al., 2006). It is appreciated that polarized macrophage populations can contribute to systemic diseases (Gordon, 2007); however, whether distinct macrophage subsets predominate in the traumatically injured CNS is not known.
After spinal cord injury (SCI), CNS macrophages promote secondary injury and repair (Giulian and Robertson, 1990; Blight, 1994; Rapalino et al., 1998; Popovich et al., 1999; Saville et al., 2004; Vallières et al., 2006; Barrette et al., 2008). These divergent effects might be attributed to distinct macrophage subsets and the activation of specific intracellular signaling cascades. Using controlled nontraumatic models of intraspinal macrophage activation in which defined ligand/receptor pathways are engaged, we have shown that CNS macrophages can promote axonal regeneration and cell killing or enhance the turnover and maturation of oligodendrocyte lineage cells (Schonberg et al., 2007; Gensel et al., 2009). It is infinitely more complex to define similar cause/effect relationships at sites of SCI because there are multiple lesion-associated factors that can independently shape the phenotype and function of CNS macrophages (for review, see Popovich and Longbrake, 2008). Still, we and others have shown that acute depletion or functional inhibition of macrophages is neuroprotective and promotes recovery of function after SCI (Giulian and Robertson, 1990; Blight, 1994; Popovich et al., 1999; Gris et al., 2004). In this study, we show that this is likely because microglia and newly recruited monocytes differentiate into proinflammatory M1 macrophages at sites of SCI. M1 cells are maintained in the lesion sites for weeks after injury, in part because proinflammatory signaling mechanisms persist indefinitely in the lesion. In vitro, we show that M1 macrophages are neurotoxic and possess only moderate axon growth-promoting effects. In contrast, M2 macrophages are not neurotoxic and can promote long-distance axon growth, even in the face of potent growth inhibitory substrates [e.g., chondroitin sulfate proteoglycans (CSPG) or myelin].
Materials and Methods
Animals and histology.
C57BL/6 mice (The Jackson Laboratory) received a moderate midthoracic (T9–10) SCI using the Ohio State University electromechanical contusion (0.5 mm over 30 ms) device as described previously (Jakeman et al., 2000). Sham mice received a laminectomy without SCI. Naive mice (i.e., uninjured) were unmanipulated. For histological evaluation, tissue was collected 1, 3, 7, 14, or 28 d after SCI and stained with eriochrome cyanine plus cresyl violet as described previously (Kigerl et al., 2006). For immunohistochemical labeling, tissue sections were sequentially treated with blocking solution (1–2 h), incubated in primary antibodies overnight (4°C), and incubated with appropriate Alexa fluor-conjugated secondary antibodies (1–2 h) the following day as described previously (Kigerl et al., 2006). Antibody concentrations are listed in Table 1. Draq5 (Biostatus Limited) at 1:3000 dilution was used as a nuclear counterstain. Immunofluorescently labeled M1 and M2 cells were quantified under high-power magnification on a Carl Zeiss Axioplan II Imaging Microscope equipped with wide field-fluorescence. Proportional area of cells expressing either M1 or M2 antigens were quantified at the injury epicenter and in the dorsal columns (n = 4 mice per/time were considered). Macrophage specificity was confirmed by double labeling with macrophage markers (tomato lectin) and M1 or M2 markers. For phenotypic assessment of intraspinally injected macrophages, the total number enhanced green fluorescent protein-positive (EGFP+) cells were manually counted from 1-μm-thick confocal slices taken from areas of highest EGFP expression (one slice per subject; n = 3 subjects per group). Only cells with a Draq5-labeled nucleus in the confocal plane were included. The number of EGFP+ and CD206 or arginase double-labeled cells was quantified and expressed as a total of EFGP+ cells.
Antibodies for immunohistochemistry
Gene expression.
RNA for microarray was prepared and processed as described previously (Kigerl et al., 2007). Individual RNA samples, each derived from a single animal, were compared with pooled (uninjured and control spinal cord) references on a two-color microarray. Microarrays were printed on poly-l-lysine-coated glass slides using an OmniGrid microarrayer (GeneMachines) and quill-type printing pins (Telechem). Oligonucleotides (Mouse Oligonucleotide Library by Sigma-Genosys/Compugen) were resuspended to 40 μm in 3× SSC and printed at 24°C with a relative humidity of ∼50%. After printing, arrays were stored overnight and post-processed by standard procedures. Slides were stored at room temperature in a desiccator flushed with nitrogen and were used between 3 weeks and 3 months after printing. Each individual sample was analyzed on a single array with n = 4 per time point [1, 3, 7, 14, or 28 d post-injury (dpi)]. Post-SCI intraspinal gene expression changes were revealed through hierarchical clustering of genes showing greater than twofold change from sham spinal cords as detected using GeneTraffic (Iobion/Stratagene). RNA for quantitative real-time (QRT)-PCR was isolated from SCI animals as described previously (Kigerl et al., 2007). Gene primer pairs (Table 2) were used to detect mRNA expression in uninjured and SCI samples (n = 3–4 per group). Expression was normalized to 18S rRNA for each sample. Gene expression is expressed relative to control (tissue collected 6 h after sham surgery). For in vitro RT-PCR, RNA was isolated from cultured bone marrow-derived macrophages (BMDMs) 6 h after stimulation via Trizol extraction and reverse transcribed as described previously (Longbrake et al., 2007). All genes were run in triplicate for each individual condition.
Primer sequences for QRT-PCR analysis
Cell culture.
Bone marrow-derived macrophage cultures were generated as described previously from adult C57BL/6 mice (Longbrake et al., 2007). Briefly, BMDMs were obtained from bilateral femurs and tibias using aseptic techniques. Marrow cores were flushed into sterile tubes using syringes fit with 26 gauge needles and filled with RPMI 1640/10% FBS. Cells were triturated three to five times, and red blood cells were lysed in lysis buffer (0.15 m NH4Cl, 10 mm KHCO3, and 0.1 mm Na2EDTA, pH 7.4). Cells were washed once in media then plated and cultured in RPMI 1640 supplemented with 1% penicillin/streptomycin, 1% HEPES, 0.001% β-mercaptoethanol, 10% FBS, and 20% supernatant from sL929 cells. The sL929 (which contains macrophage colony-stimulating factor) is needed to promote differentiation of bone marrow cells into macrophages (7–10 d) (Burgess et al., 1985). To promote differentiation into M1 or M2 macrophages, cells were treated with LPS (100 ng/ml; Sigma-Aldrich) plus IFNγ (20 ng/ml; eBioscience) or IL-4 (20 ng/ml; eBioscience), respectively, for 12–24 h. Macrophage conditioned media (MCM) was prepared from supernatant of stimulated macrophages. Supernatant was collected and centrifuged (1200 rpm, 5 min).
Primary cortical neurons were from 1–2 d postnatal mouse pups. Using aseptic technique, the entire forebrain and brainstem were removed and placed in L-15/10% BSA. Bilateral cortices were separated from cerebellum, brainstem, and olfactory lobes. Meninges were removed using fine forceps before pieces were minced (using sterile razor blade) into ∼20 pieces per hemisphere. These were collected into a sterile tube and then incubated in a 1.0 mg/ml papain/L-15 solution at 37°C for 15 min. After enzymatic digestion, pieces were washed (Neurobasal A; Invitrogen) and then triturated through polished glass pipettes with progressively smaller tip diameters. Debris was removed by allowing the pieces to settle between trituration steps. After sufficient trituration, cells were suspended in fresh media [Neurobasal A with 2% B27 supplement (Invitrogen), 1% Glutamax, and 1% penicillin/streptomycin], and live cells were counted on a hemocytometer using the trypan blue exclusion method. Cells were grown in poly-d-lysine (25 μg/ml; Sigma) coated plastic culture plates at 37°C/5% CO2. Cultures were used 7–14 d after initial plating.
Cortical neuron viability was assessed using quantitative MAP2 ELISA. Neurons were fixed in 96-well cell culture plate using 2% paraformaldehyde for 20 min, followed by washing in 0.1 m PBS. Blocking solution (100 μl of 0.1 m PBS/10% FBS/0.1% Triton X-100) was applied for 1 h at room temperature, followed by primary antibody [MAP2 (SMI-52); Covance] diluted 1:1000 in blocking solution (above) overnight at 4°C. Primary antibody was aspirated, and cells were washed in 0.1 m PBS/0.1% Triton X-100. Alkaline-phosphatase-labeled secondary antibody was then applied (rat anti-mouse IgG1; eBioscience) was then applied (1:1000) for 2 h at room temperature. Positive MAP2 labeling was detected using para-nitrophenyl phosphate (PNPP) substrate (Southern Biotechnology Associates), and resulting development was read using a SpectraMax microplate reader at absorbance 405 nm. After development with PNPP, MAP2 staining was visualized using 5-bromo-4-chlor-indolyl-phosphate/nitroblue-tetrazolium-chloride substrate (KPL).
Adult wild-type C57BL/6 mice were terminally anesthetized, and DRG cultures were prepared on glass coverslips as described previously (Steinmetz et al., 2005). After 3 d in vitro, media from DRG cultures was removed, and an equivalent volume of macrophage-conditioned media (see below) was added for 24 h. To assess DRG neurite outgrowth, digital images from β-tubulin III-labeled DRGs were captured with a Carl Zeiss Axioplan 2 Imaging microscope from cells chosen randomly using the MCID Elite Image Analysis station (Imaging Research Corporation) stereology module. Using the MetaMorph (Molecular Devices) image analysis system, an automated Scholl ring analysis was performed using concentric circles at 50 μm intervals, and the number of crossings at each interval was recorded (Sholl, 1953).
DRG CSPG spot assay was done as described previously (Tom et al., 2004; Steinmetz et al., 2005). Glass coverslips were coated with poly-d-lysine (25 μg/ml) and nitrocellulose and air dried. Then a mixture of aggrecan (0.7 mg/ml) and laminin (5 μg/ml) in Ca2+/Mg2+-free (CMF) HBSS was spotted in 2 μl spots onto the coverslips. After aggrecan/laminin mixture had completely dried, coverslips were coated with laminin (5 μg/ml) for 3 h at 37°C. After laminin, coverslips were treated with 0, 0.25, or 0.5 U/ml chondroitinase ABC (chABC) HBSS for 3 h at 37°C. After chABC treatment, coverslips were washed 1× with CMF HBSS. DRG neurons were isolated and plated onto coverslips at a density of 5000 cells per coverslip. M1 or M2 MCM was added to the DRG neurons at the time of plating. Cells were fixed after 5 d in vitro with 2% paraformaldehyde and then stained with β-tubulin III. Axon crossings were quantified by manually counting the number of axon crossing around the entire circumference of the CSPG gradient.
Myelin-associated glycoprotein (MAG) neurite outgrowth assay was done using CHO cells engineered to express MAG (kindly provided by Dr. Marie Filbin, Hunter College, City University of New York, New York, NY) (Domeniconi et al., 2002). Briefly, a monolayer of MAG CHO cells was grown on glass coverslips until the cells were ∼80% confluent. DRG neurons were then plated on this monolayer with M1 or M2 MCM for 48 h. Cells were fixed with 2% paraformaldehyde after 48 h and stained with β-tubulin III. Analysis was done as described above.
Statistical analysis.
Immunohistochemical, morphometric, and gene expression levels were analyzed using one- or two-way ANOVA, followed by Bonferroni's, Tukey's, or Dunnett's post hoc comparisons. If appropriate, t tests were performed to assess differences between experimental conditions. Results were considered statistically significant at p < 0.05. All statistical analyses were conducted using GraphPad Prism 5.0 (GraphPad Software). All data are expressed as group mean ± SEM unless otherwise noted.
Results
Polarization of the CNS macrophage response after SCI
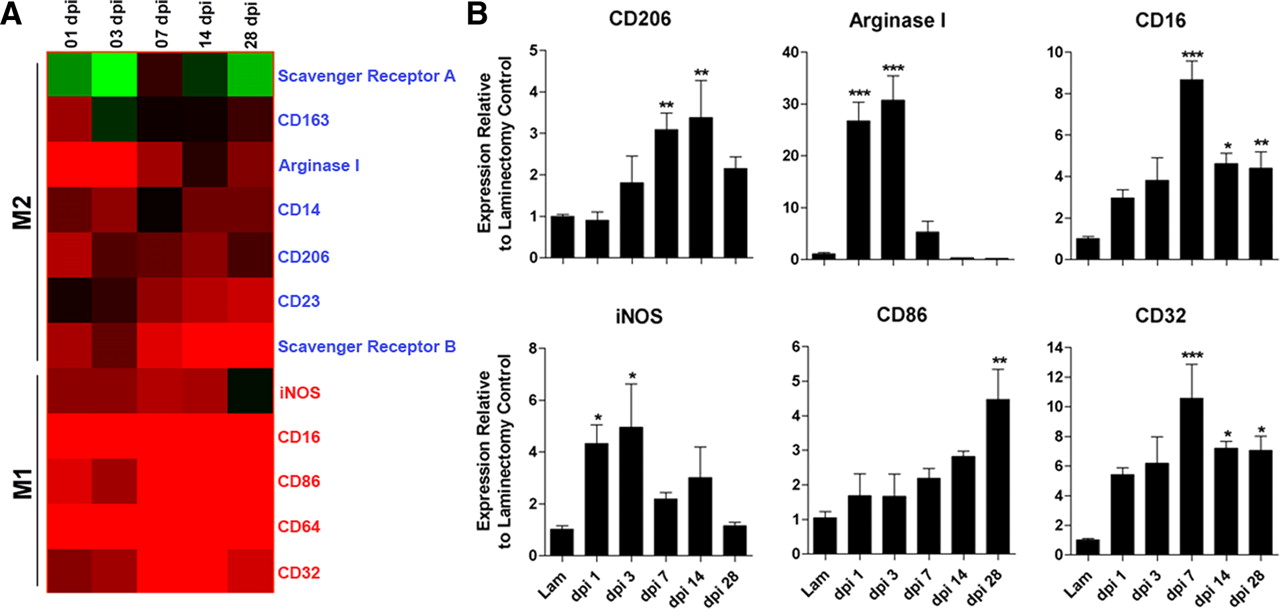
Using cDNA microarrays, we analyzed temporal changes in the expression profiles for genes associated with M1 or M2 macrophages between 1 and 28 d after injury (Mantovani et al., 2002; Gordon, 2003). In the injured spinal cord, M1 and M2 genes were rapidly induced. M2 gene expression, however, was transient and in most cases returned to preinjury levels by 7 dpi (Fig. 1 A). In contrast, M1 gene expression was maintained for up to 1 month after injury (longest time evaluated in this study).

Spinal cord injury induces changes in the expression of genes associated with M1 and M2 macrophages. A , cDNA microarray “heat map” showing clustering of M1 and M2 genes as a function of time after injury. All genes shown are upregulated or downregulated more than twofold compared with uninjured controls. Black indicates no change in gene expression, whereas green or red indicate a decrease or increase in expression, respectively, relative to uninjured control samples. Note transient induction of M2 genes in contrast with early and sustained induction of M1 genes. B , Quantitative real-time PCR confirms select M1 and M2 gene expression changes noted via microarray (n = 4 per time for microarray; n = 3–5 per time for PCR). (ANOVA, p < 0.01 for all genes; *p < 0.05, **p < 0.01, ***p < 0.001 vs laminectomy control.).
For a subset of genes, changes in expression were confirmed using real-time QRT-PCR (Fig. 1 B). Of those genes that define M2 macrophages in vitro and in vivo, arginase 1 (Arg1) and mannose receptor (CD206) may be most relevant for CNS repair (Nauta et al., 2003; Ghassabeh et al., 2006). Both Arg1 and CD206 increased early after SCI and then returned to baseline by 14 dpi (Fig. 1 B). Similarly, inducible nitric oxide synthase (iNOS), an enzyme that is preferentially induced in M1 macrophages (Dalton et al., 1993; Gordon, 2003), was maximally increased 1–3 dpi (Fig. 1 B). In contrast, expression of CD86, CD16, and CD32, surface receptors associated with M1 macrophages, increased as a function of time after SCI (Fig. 1 B).
These data suggest that cues in the dynamic lesion environment control macrophage phenotype and favor differentiation of M1 macrophages. Additional molecular analyses confirmed this hypothesis. Specifically, we found a delayed but persistent induction of IFNγ and IFNγ receptor mRNA in injured spinal cord (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Conversely, expression of IL-4 receptor (IL-4R) mRNA, a receptor needed for M2 macrophage development, was increased more than twofold during the first 72 h after injury (p < 0.01 vs sham-injured controls); however, IL-4 was below the level of detection (data not shown). Other macrophage receptors necessary for inducing M2 cells or for suppressing macrophage function, i.e., IL-13R and IL-10Rα, respectively, also were unchanged relative to uninjured control spinal cord (data not shown).
Post-SCI changes in M1 and M2 gene expression should predict the phenotype of CNS macrophages that occupy the lesion and lesion penumbra of the injured spinal cord. We tested this hypothesis using immunofluorescent double labeling and confocal microscopy. Specifically, we documented the distribution and magnitude of macrophages expressing M1 (CD86 and CD16/32) and M2 (CD206 and Arg1) phenotypic markers as a function of time throughout the rostrocaudal extent of spinal contusion lesions (Figs. 2, 3) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). At 1–3 dpi, the ratio of M1/M2 cells was approximately equivalent throughout the lesion (Figs. 2, 3). Some cells coexpressed M1 and M2 markers (data not shown), suggesting coinduction of parallel but independent transcriptional mechanisms (Wang et al., 1995). However, after the first week, the M1/M2 ratio increased markedly, mostly because labeling for M2 phenotypic markers was reduced or eliminated (Figs. 2, 3).

Macrophages with an M1 phenotype dominate sites of spinal cord injury. A , CD16/32+ M1 macrophages and arginase 1+ M2 macrophages coexist at the lesion epicenter during the first week after injury; however, only M1 macrophages persist until 28 dpi. B , Quantitation of macrophages expressing M1 and M2 phenotypic markers as a function of time after SCI. C , When expressed as a ratio of M1/M2 cells, there is an obvious shift toward an M1 macrophage phenotype after the first week after injury. [M1 and M2 markers; red, AF546 and nuclear stain with 4′,6′-diamidino-2-phenylindole (DAPI); blue]. Scale bar, 20 μm.

M1 macrophages dominate zones of Wallerian degeneration after SCI. A , Similar to the macrophage response that evolves at the lesion epicenter (see Fig. 2), M1 (CD86+) and M2 (Arg 1) macrophages coexist within the dorsal funiculus at 3 dpi, but only M1 macrophages persist until 28 dpi (dotted line indicates the border of the gray matter and dorsal funiculus). B , Quantitation of macrophages expressing M1 and M2 markers in the dorsal funiculus at different times after SCI. C , When expressed as a ratio of M1/M2 cells, there is an obvious shift toward an M1 macrophage by 3 dpi. (M1 and M2 markers; red, AF546 and nuclear stain with DAPI; blue). Scale bar, 20 μm.
The injured spinal cord microenvironment downregulates the M2 macrophage phenotype
Microglia/macrophages possess an M2 phenotype in the intact CNS (Ponomarev et al., 2007). Consequently, the loss of an M2 phenotype as a function of time after SCI suggests that lesion-associated factors (e.g., cytokines, oxygen tension, chemokines, etc.) may cause existing microglia/macrophages and/or newly emigrating monocytes to differentiate into M1 macrophages. Indeed, macrophage phenotype and function are dynamically regulated by the tissue microenvironment with recent data showing that macrophages can change their phenotype in response to sequential stimuli (Stout et al., 2005). To test whether the microenvironment of the injured spinal cord polarizes macrophage phenotype, M2 macrophages generated from EGFP+ bone marrow were microinjected into naive or injured spinal cord at 7 dpi (n = 3 per group). An M2 phenotype was confirmed before intraspinal injection using molecular and phenotypic criteria (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (Fig. 4). Three days after injection, i.e., 10 d after SCI, confocal microscopy reveals that the M2 phenotype of injected cells is maintained only when macrophages are injected into intact spinal cord. Indeed, when EGFP+ M2 macrophages were exposed to the contusion lesion (or tissue immediately adjacent to the lesion), the percentage of cells that maintained their M2 phenotype was reduced 20–40% (Fig. 4) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Collectively, these data suggest that lesion-derived factors influence the terminal differentiation of macrophages, suppressing or downregulating an M2 phenotype while inducing or maintaining M1 macrophages.

The M2 macrophage phenotype is downregulated in the injured spinal cord. The phenotype of EGFP+ M2 macrophages was confirmed ex vivo ( A–D ), and then cells were microinjected into areas of intact ( F–I ) or injured (7 dpi; J–M ) spinal cord. The percentage of EGFP+ cells expressing arginase 1 (or CD206) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) was reduced when injected into injured spinal cord (n = 3 per group; *p < 0.05 vs intact for arginase 1) ( E ). Confocal images of EGFP+ cells stained with antibodies labeling the M2 marker arginase 1 (red) reveal SCI-dependent reduction in the M2 phenotype (compare G–I with K–M ). I , M , High-powered images of boxed areas in H and L , respectively. Notice loss of arginase 1 labeling inside EGFP+ macrophages in injured spinal cord. Instead, a small population of EGFP-negative parenchymal cells express low levels of arginase 1. Scale bar: F–H , J–L , 20 μm; I , 7.5 μm; M , 5.5 μm.
M1 and M2 macrophages exert distinct effects on neuron survival and axon growth
From the above data, it is clear that M1 macrophages predominate at sites of SCI. This “M1 bias” could exacerbate secondary neurodegeneration and may explain why wound healing is inefficient after SCI. Indeed, M1 cells release oxidative metabolites (e.g., nitric oxide) and proteases that can kill neurons and glia (supplemental Fig. 5, available at www.jneurosci.org as supplemental material), whereas M2 cells facilitate tissue repair (Goerdt and Orfanos, 1999). To test whether these unique functions of M1 and M2 macrophages confer differential effects on neuron survival and axon growth, primary cortical or DRG neurons were grown in the presence of media conditioned by M1 or M2 macrophages. Previously, we have documented that media isolated from unstimulated macrophages has little effect on axon growth or neurotoxicity (Gensel et al., 2009). For this reason and because macrophages are always present in some activated form in the lesioned spinal cord, the effects of M1 or M2 MCM were compared with each other or media control.
As shown in Figure 5 A, M1 but not M2 MCM is toxic to cortical neurons. Because some toxic mediators released by macrophages are labile with short half-lives (e.g., oxygen and nitrogen free radicals), it is possible that media transfer assays underestimate the neurotoxic potential of M2 macrophages. Thus, we repeated this assay by overlaying cortical neuron cultures with activated M1 or M2 macrophages grown on trans-well inserts. Images in Figure 5, B and C, confirm that neurotoxicity is a feature specific to M1 macrophages. In contrast, M2 macrophages caused no overt pathology (Fig. 5 C).

M1 and M2 macrophages exert disparate effects on neuron survival and axon growth. M1 macrophages are neurotoxic in vitro ( A–C ). Regardless of whether cortical neuron survival (7 d in vitro) was assessed in a media transfer assay ( A ) or in response to M1 or M2 macrophages in a trans-well system ( B , C ), M1 macrophages were neurotoxic. Cortical neuron survival was quantified using a MAP2 ELISA ( A ; *p < 0.05 vs control and M2 MCM). M2 macrophages had no adverse effect on neurons ( A , C ). MCM from M1 and M2 cells also trigger distinct neurite outgrowth patterns in DRG neurons ( D–G ). Sholl analysis was used to measure neurite outgrowth length and complexity ( D ). M1 MCM stimulated a short arborizing growth pattern with axons usually terminating within 500 μm of the cell soma ( D–G ). Conversely, M2 MCM induced long unipolar or bipolar axonal extensions with less branching evident near the soma and overall length often exceeding 1 mm ( D–G ) (*p < 0.05 for E , G ; data are representative of 2 independent experiments; n = 15–20 DRGs analyzed).
To evaluate the potential of M1 and M2 macrophages to influence axon growth/sprouting, M1 or M2 MCM was overlaid onto adult DRG neurons as described previously (Gensel et al., 2009). Although both M1 and M2 MCM promoted axon growth relative to control media (p < 0.0001; data not shown), their growth-promoting effects were distinct. Neurons stimulated with M1 MCM extended short, highly branched neurites, whereas those stimulated with M2 MCM exhibited a unipolar or bipolar phenotype with their axons projecting over long distances, some >1200 μm or more than two times the distance covered by DRGs stimulated with M1 MCM (Fig. 5 D–G). The growth-promoting effects of M1 or M2 macrophages were not a result of residual cytokines left over from the differentiation protocol. When applied to DRG neurons in vitro, neither recombinant IL-4 (20 ng/ml; concentration used to create M2 macrophages) or IFNγ/LPS (20 ng/ml and 100 ng/ml, respectively) had any effect on neurite outgrowth (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). These data corroborate previous work by Golz et al. (2006) using higher concentrations of IL-4 in DRG explant cultures.
M2 macrophages overcome axon growth inhibition by CSPG and myelin
CSPG and MAG are potent inhibitors of axon growth (Canning et al., 1996; Domeniconi et al., 2002). In Figure 5, we show that M1 and M2 macrophages can enhance axon growth on a permissive substrate, i.e., laminin. However, functionally significant regeneration will only be realized in vivo if macrophage-derived factors can promote axon growth on inhibitory substrates that dominate sites of CNS injury.
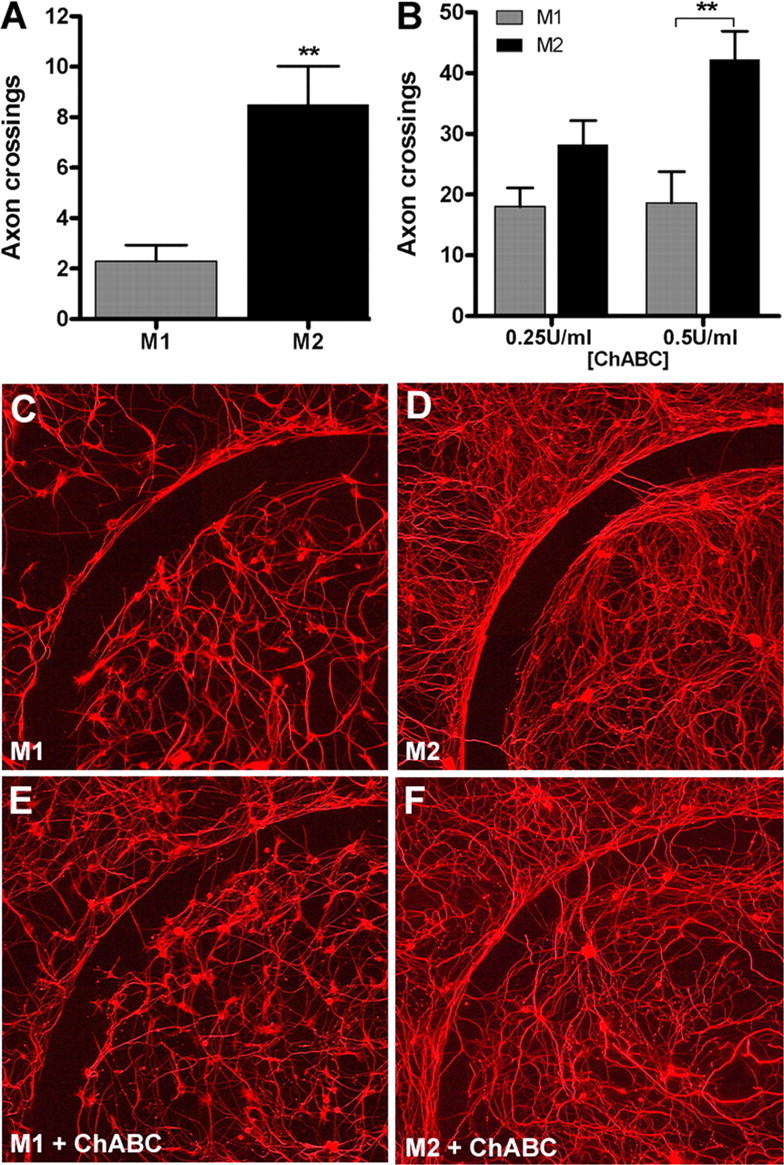
Using a novel in vitro model of the glial scar (Tom et al., 2004), we found that M2 macrophages promote a modest increase in axon growth across an inhibitory CSPG gradient (p < 0.01 vs M1 MCM) (Fig. 6 A). Others have shown that chABC can be used in vivo and in vitro to degrade inhibitory proteoglycan substrates and promote axon sprouting and regeneration (Bradbury et al., 2002; Steinmetz et al., 2005). When M1 or M2 MCM was combined with chABC, axon growth across the CSPG gradient was increased threefold to fivefold relative to that achieved by M1 or M2 MCM alone (Fig. 6 B–F). Importantly, M2 MCM was more effective at synergizing with chABC to promote axon growth (Fig. 6 B). A qualitative inspection of the laminin-enriched core of the spot assays shows that the overall density of axonal projections was consistently greater when cultures were exposed to M2 MCM (Fig. 6 C–F). This is consistent with the effects of M2 MCM on cortical neurons (Fig. 5) and further supports the notion that M2 macrophages promote axon growth without causing concurrent neurotoxicity.

M2 macrophages promote axon growth across an inhibitory CSPG gradient. Adult DRG neurons were treated with M1 or M2 MCM for 5 d in vitro, and then axons crossing over an aggrecan barrier were quantified. M2 MCM consistently enhanced DRG axon crossings compared with M1 MCM (p < 0.01; A , C , D ). M2 MCM-mediated axon growth was augmented in the presence of increasing concentrations of chABC (**p < 0.01 at 0.5 U/ml chABC vs M1 MCM; B , E , F ). Data are representative of three independent experiments; n = 12 spots per condition.
We next evaluated the relative ability of M1 and M2 MCM to influence axon growth on an inhibitory myelin substrate. Specifically, we grew DRG neurons on a live cell monolayer engineered to express MAG (Domeniconi et al., 2002) in the presence of M1 or M2 MCM. In general, very few neurons in either culture extended neurites on the MAG substrate. However, the number of neurites extending from a given cell as well as the overall length of the neurite was consistently enhanced in response to M2 MCM relative to M1 MCM (Fig. 7).

M2 macrophages enhance axon growth on an inhibitory myelin (MAG) substrate. DRG neurons plated on a monolayer of CHO cells engineered to express MAG (Domeniconi et al., 2002) were treated with M1 or M2 MCM for 48 h, and then neurite outgrowth was assessed. M2 MCM increased the number ( A ) and length ( B ) of neurites extending from DRG neurons ( A ; *p < 0.05). Representative β-tubulin III+ DRG neurons are provided showing the median number and length of neurites elicited by M1 or M2 MCM ( C ). These data represent mean of n = 20–25 neurons analyzed from an individual experiment.
Discussion
A robust and protracted macrophage response accompanies all forms of CNS trauma, yet the functional significance of this response is unknown. After SCI, depletion or inhibition of CNS macrophages consistently confers neuroprotection and promotes functional recovery (Giulian and Robertson, 1990; Blight, 1994; Popovich et al., 1999; Saville et al., 2004). Paradoxically, the controlled activation or even augmentation of this response can enhance various indices of CNS repair (e.g., axon growth/sprouting, remyelination, etc.) (Rapalino et al., 1998; Yin et al., 2003, 2006; Vallières et al., 2006; Barrette et al., 2008). These divergent effects may be explained by the induction of a macrophage response that is both phenotypically and functionally heterogeneous.
Based on transcriptional profiling, proteomics, and functional assays, macrophages are typically classified on a linear scale with classically activated macrophages (M1) representing one extreme of activation and alternatively activated macrophages (M2) the other (for review, see Gordon, 2003; Mosser and Edwards, 2008). M1 macrophages, activated via toll-like receptors (TLRs) and IFNγ, produce proinflammatory cytokines and oxidative metabolites that facilitate their role as indiscriminate killers of microbes and tumor cells (Gordon, 2003; Mantovani et al., 2004). Conversely, M2 macrophages form in the presence of IL-4. M2 cells downregulate inflammation and facilitate wound healing, in part through the induction of scavenger receptors and matrix degrading enzymes that enhance phagocytosis and promote tissue remodeling (Gordon, 2003; Mantovani et al., 2004).
As with most models, the M1/M2 classification scheme undoubtedly oversimplifies the complexity of the CNS macrophage response. Indeed, multiple subsets of monocyte-derived macrophages are known to contribute to distinct stages of wound healing (Geissmann et al., 2003; Auffray et al., 2007), and recent data suggest that a fully differentiated macrophage subpopulation can reversibly change its phenotype and function in response to signals in the microenvironment (Stout and Suttles, 2004; Mosser and Edwards, 2008). Still, despite the limitations of linear classification, this model provides a useful conceptual framework in which to define the continuum of posttraumatic CNS macrophage activation.
As shown in the present report, M1 macrophages dominate the lesion site and nearby spared tissue, even at chronic postinjury survival times. An M2 macrophage response also was observed; however, this was short-lived, dissipating within 3–7 d after injury. Normally, during the course of cutaneous wound healing, there is a well defined shift in macrophage effector functions. Early on, proinflammatory, proteolytic macrophages predominate. These cells ensure that the wound is sterile, degrade lesion borders, and remove effete neutrophils. The latter is important for signaling subsequent phases of wound healing, including the recruitment of macrophages that promote new blood vessel growth and extracellular matrix deposition. Distinct patterns of gene expression indicate that these functional changes involve a shift from M1 to M2 macrophages in the wound site (Deonarine et al., 2007). A similar response has been described in the healing myocardium and is dependent on the mobilization of distinct monocyte subsets from the circulation (Nahrendorf et al., 2007). Our present data indicate that an analogous M1-to-M2 macrophage shift does not occur after SCI. In fact, despite the induction of wound healing genes in the injured spinal cord (Velardo et al., 2004), chronic inflammation is a hallmark of spinal contusion/compression pathology, and, in all examples of clinical and experimental SCI, macrophages dominate the lesion site indefinitely (Fleming et al., 2006; Chang, 2007; Donnelly and Popovich, 2008). Based on our present data, these appear to be of the M1 phenotype. However, it is not clear how or why this phenotype predominates.
IFNγ, a key mediator in promoting the development of M1 macrophages, is upregulated after SCI. Importantly, increased expression of IFNγ and its receptor is maintained at chronic postinjury intervals. Currently, the source and mechanisms responsible for regulating IFNγ in chronically injured spinal cord are unknown. Other proinflammatory mediators, including high mobility group box protein (HMGB1) and tumor necrosis factor-α (TNFα), also are increased at sites of CNS injury (Yakovlev and Faden, 1994; Wang et al., 1996; Streit et al., 1998; Kobori et al., 2002; Kim et al., 2006) and can promote formation of M1 macrophages (Mosser and Edwards, 2008). HMGB1 activates macrophages via TLRs (Park et al., 2004, 2006), resulting in nuclear factor κB (NF-κB)-mediated production of proinflammatory cytokines and iNOS activation, i.e., hallmarks of M1 macrophage activation (Medzhitov, 2001). TNFα elicits a similar proinflammatory phenotype via NF-κB activation (Hohmann et al., 1990). The M1 phenotype may also persist by default because signals that stimulate or maintain M2 macrophages are expressed at low or undetectable levels in the spinal cord. Specifically, we were unable to detect significant basal levels or injury-associated increases in intraspinal IL-4, IL-13, or IL-10.
We predict that the polarized M1 macrophage response impairs recovery from SCI, in part through propagating secondary neurodegenerative cascades and promoting retraction of dystrophic axons at the site of injury (Horn et al., 2008). For example, M1 macrophages synthesize and release cytokines, reactive oxygen species, proteolytic enzymes [e.g., matrix metalloproteinases (MMPs)], and nitric oxide, all of which are able to kill neurons and glia (Block et al., 2007). The release of one or more of these factors is likely responsible for causing toxicity in cortical neuron cultures (Fig. 5). Interestingly, it was shown recently that macrophages may cause axonal retraction after SCI, in part through the release of MMP9 (Busch et al., 2009). Despite their enhanced capacity for cell killing, M1 macrophages also trigger a growth program in adult DRG neurons that manifests as short-distance, arborizing neuritic growth. This type of growth is reminiscent of the afferent sprouting that occurs at sites of spinal cord injury and is believed to cause neuropathic pain, spasticity, and autonomic dysreflexia (Weaver et al., 2001; Hofstetter et al., 2005; Hou et al., 2008, 2009).
In contrast, sustaining the M2 response that is induced early after SCI (or eliciting M2 macrophages at later postinjury intervals) could enhance clearance of necrotic debris without causing toxicity while also promoting axonal regeneration. A phenotypic hallmark of M2 macrophage is increased expression of the mannose receptor (CD206) (Gordon, 2003). CD206 binds late-stage apoptotic and necrotic cells and facilitates removal of dying cells without causing bystander damage (Nauta et al., 2003). This may explain why media conditioned by M2 macrophages was not neurotoxic (Fig. 5). M2 macrophages also enhance growth of adult DRG axons, even in the presence of potent growth-inhibitory molecules. Although MMPS are released by both M1 and M2 macrophages, the composition of MMPs (and other proteases) is likely to be distinct with M2-derived proteases favoring the degradation of axon growth inhibitory molecules without causing overt toxicity. For example, MMP1 and MMP12 are increased in M2 macrophages, whereas MMP9 is downregulated (Chizzolini et al., 2000; Gordon, 2003; Shimizu et al., 2004; Gratchev et al., 2005; Kahnert et al., 2006). Also, as a result of increased arginase 1 activity, M2 macrophages synthesize high levels of polyamines that work downstream of cAMP to overcome myelin inhibition of axon growth in vitro (Cai et al., 2002). In this way, M2 macrophages could modify the extracellular matrix and prime neuronal metabolism to overcome growth inhibition.
Regardless of the mechanism, the axon growth stimulated by M2 macrophages is phenotypically distinct and is quantitatively more impressive than that caused by M1 macrophages, especially in terms of the overall length of axon growth. Currently, we do not know why M1 and M2 macrophages trigger distinct regenerative phenotypes in sensory axons. Based on work from Smith and Skene (1997) and Liu and Snider (2001), along with the knowledge that M1 and M2 macrophages have divergent secretory repertoires, we predict that M1 and M2 macrophages engage distinct intracellular signaling cascades in neurons. Ongoing studies will attempt to discriminate between the macrophage-derived factors that induce an “elongating” axonal phenotype, similar to that triggered by a “conditioning lesion,” and those that trigger “arborizing” or “sprouting” axon growth.
To our knowledge, these data are the first to document the functional heterogeneity of the innate immune response after SCI. The clinical implications are noteworthy because it may be possible to use drugs to manipulate the natural course of CNS macrophage activation to minimize secondary injury and promote axon regeneration. For example, IL-4 and IL-10, both signals that promote M2 macrophage formation, promote modest neuroprotection after SCI (Bethea et al., 1999; Lee et al., 1999). Whether these cytokines influenced CNS macrophage phenotype or function was not determined. Peroxisome proliferator-activated receptor γ agonists, used clinically to treat diabetes, also can promote M2 macrophage differentiation (Odegaard et al., 2007) and have been shown to be neuroprotective in preclinical models of SCI (McTigue et al., 2007). However, before the repair potential of M2 or “wound healing” macrophages can be engaged effectively, it will be useful to first understand the molecular and cellular cues that naturally drive macrophages toward an M1 phenotype after SCI. By doing so, we may reveal additional or complementary therapeutic targets, some of which may prove useful in ameliorating the destructive effects of macrophages in other forms of traumatic or ischemic CNS injury.
Footnotes
-
This work was supported by the National Institutes of Health–National Institute of Neurological Disorders and Stroke, the Craig H. Neilsen Foundation, and The Paralysis Project of America. We thank Drs. Ron Hart for assisting with the microarray data analysis, Sarah Busch and Jerry Silver for helping us to establish CSPG spot assays, and Marie Filbin and Marco Domeniconi for providing MAG CHO cells and helping us to establish the neurite growth assay on myelin.
- Correspondence should be addressed to Phillip G. Popovich, Ohio State University Medical Center, 786 Biomedical Research Tower, 460 West 12th Avenue, Columbus, OH 43210. phillip.popovich{at}osumc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}