

Abstract
MTMR2 is a 3-phosphatase specific for the phosphoinositides PI(3)P and PI(3,5)P2, which are mainly present on endosomes. Mutations in the MTMR2 gene in Schwann cells lead to a severe demyelinating peripheral neuropathy known as Charcot-Marie-Tooth disease type 4B1. MTMR2 expression is also detected in peripheral and central neurons, but neural functions of MTMR2 remain unclear. Here, we report that MTMR2 is localized to excitatory synapses of central neurons via direct interaction with PSD-95, a postsynaptic scaffolding protein abundant at excitatory synapses. Knockdown of MTMR2 in cultured neurons markedly reduces excitatory synapse density and function. This effect is rescued by wild-type MTMR2 but not by a mutant MTMR2 lacking PSD-95 binding or 3-phosphatase activity. MTMR2 knockdown leads to a decrease in the intensity of EEA1-positive early endosomes in dendrites but increases the intensity in the cell body region. Moreover, MTMR2 suppression promotes endocytosis, but not recycling, of the GluR2 subunit of AMPA receptors, which is an endosomal cargo. In addition, colocalization of internalized GluR2 with Lamp1-positive late endosomes/lysosomes is enhanced in the cell body area but not in dendrites. These results suggest that PSD-95-interacting MTMR2 contributes to the maintenance of excitatory synapses by inhibiting excessive endosome formation and destructive endosomal traffic to lysosomes.
Introduction
PSD-95/SAP90, an abundant excitatory postsynaptic scaffolding protein, has been implicated in the regulation of excitatory synapse formation, maturation, and plasticity (Montgomery et al., 2004; Funke et al., 2005; Fitzjohn et al., 2006; Okabe, 2007; Sheng and Hoogenraad, 2007; Keith and El-Husseini, 2008). Although various molecular mechanisms have been suggested to explain PSD-95-dependent synaptic regulation, we may be far from a comprehensive understanding of these mechanisms.
Phosphoinositides (PIs) are lipid signaling molecules that regulate cell signaling and membrane dynamics (Di Paolo and De Camilli, 2006). Myotubularin and related proteins constitute a large family of 3-phosphatases for PIs (Laporte et al., 2003; Clague and Lorenzo, 2005; Robinson and Dixon, 2006; Bolis et al., 2007). These proteins are conserved across a spectrum of eukaryotic species, including yeast, worms, flies, and mammals. In humans, there are 14 myotubularin family proteins—myotubularin 1 (MTM1) and 13 myotubularin-related proteins (MTMR1-13)—8 of which are catalytically active. MTM1, MTMR2, and MTMR13 have been associated with myopathy and neuropathies, highlighting their clinical importance (Laporte et al., 1996; Bolino et al., 2000; Azzedine et al., 2003; Senderek et al., 2003; Berger et al., 2006a; Previtali et al., 2007; Nicot and Laporte, 2008).
Mutations in the MTMR2 gene cause Charcot-Marie-Tooth disease type 4B1 (CMT4B1) (Bolino et al., 2000), an autosomal-recessive peripheral neuropathy characterized by childhood onset, progressive muscle weakness, reduced nerve conduction velocity, and nerve demyelination with myelin outfolding (Quattrone et al., 1996). MTMR2-deficient mice exhibit a CMT4B1-like neuropathy (Bolino et al., 2004; Bonneick et al., 2005), and ablation of MTMR2 in Schwann cells, but not in motor neurons, is sufficient to cause CMT4B1-like neuropathy (Bolino et al., 2004; Bolis et al., 2005). The interaction of MTMR2 with SAP97/Dlg1 (a relative of PSD-95) has been implicated in the regulation of membrane remodeling during myelination (Bolino et al., 2004; Bolis et al., 2009). In addition, most MTMR2 mutations in CMT4B1 patients disrupt the phosphatase activity of MTMR2 (Berger et al., 2002; Begley et al., 2003). MTMR2 is active toward two specific PIs, PI(3)P and PI(3,5)P2 (Berger et al., 2002; Kim et al., 2002; Laporte et al., 2002; Begley et al., 2003; Tronchère et al., 2004), which are thought to regulate the endosomal pathway (Robinson and Dixon, 2006). These results suggest that MTMR2-dependent regulation of membrane remodeling and endosomal trafficking may underlie the pathophysiology of CMT4B1.
In addition to Schwann cells, MTMR2 is expressed in peripheral nerves (Berger et al., 2002; Previtali et al., 2003). MTMR2 mRNAs are also expressed in the brain (Laporte et al., 1998), where it is detected in various principal neurons (Berger et al., 2002; Bolino et al., 2002). However, functions of MTMR2 in the CNS are not well understood. In addition, it is unclear whether and how MTMR2 is targeted to specific subcellular sites (i.e., sites of endosomal generation) for local regulation of endosomal trafficking.
Here, we report that MTMR2 is targeted to excitatory synapses via direct interaction with PSD-95 and show that MTMR2 is important for excitatory synapse maintenance by regulating endosomal trafficking.
Materials and Methods
Expression and shRNA constructs.
For expression constructs, regions of rat MTMR2 (full length, aa 1-643; Δ C, aa 1-640; C417S, phosphatase-inactive form) were subcloned into p3XFLAG-CMV7.1 (Sigma). For shRNA constructs, oligonucleotides targeting the following three regions of MTMR2 were subcloned into pSUPER.gfp/neo (Oligoengine); sh-M1, nt 970-988, 5′-AGTGTCAATGCTGTTGCCA-3′; sh-M1*, 5′-AGCGTCAACGCGGTTGCCA-3′; sh-M2, nt 191-208, 5′-TGAGGGAGGCAAACAAATT-3′; and sh-M3, nt 1615-1633, 5′-TCACTGTGGTCCTATATAA-3′. For the rescue expression constructs [wild type (WT), Δ C, and C417S], four point mutations (T972C, T975G, T978C, and T981G) were introduced to MTMR2 in p3XFLAG-CMV7.1 using a QuikChange kit (Stratagene). For GST-MTMR2, full-length MTMR2 was subcloned into pGEX4T-1. GW1-PSD-95-EGFP has been described previously (Arnold and Clapham, 1999).
Antibodies.
Rabbit polyclonal MTMR2 (#1490) and PSD-95 (#1399) antibodies were generated using GST–full-length MTMR2 and PSD-95 fusion proteins as immunogens, respectively. Specific antibodies were affinity-purified using PVDF membrane (GE Healthcare). The following antibodies have been described previously: PSD-95 (SM55 and 1399), PSD-93/Chapsyn-110 (B9594), SAP97 (B9591), SAP102 (1445), S-SCAM (1146), GRIP (C8399; pan-GRIP antibody), and EGFP (1167). Other antibodies were obtained from the following sources: PSD-95 (Affinity BioReagents); synaptophysin, β-tubulin, MAP2, and Flag (Sigma), EEA1 (BD Transduction Laboratory), and Lamp1 (StressGen).
Yeast two-hybrid screen and assays.
Two-hybrid screen and assays were performed using the L40 yeast strain. The C-terminal region of MTMR2 [aa 637-643 (wild type and point mutants) were generated by annealing oligonucleotides and subcloning them into pBHA (bait vector). PDZ domains in pGAD10 (prey vector) have been described previously (Kim et al., 1995; Wyszynski et al., 2002).
In situ hybridization.
To study the spatial expression of MTMR2 mRNAs, in situ hybridization was performed as described previously (Kim et al., 2004). The hybridization probe was prepared from pGEM7zf containing cDNA containing rat MTMR2 (436 bp; nucleotides 1691-2126; GenBank accession number XM-235822). 35S-labeled antisense riboprobes were prepared using Riboprobe System (Promega).
Immunoprecipitation.
In vivo coimmunoprecipitation were performed as described previously (Wyszynski et al., 1999). Deoxycholate extracts of the crude synaptosomal fraction of adult rat brain were incubated with MTMR2 (1490, 2 μg/ml) and PSD-95 (1399, 2 μg/ml) antibodies or IgG (2 μg/ml), followed by protein A-Sepharose precipitation. For in vitro immunoprecipitation, transfected HEK293T cells were extracted with PBS containing 1% Triton X-100 and incubated with FLAG-agarose (Sigma).
Neuron culture, transfection, and immunohistochemistry.
Cultured hippocampal neurons were prepared from embryonic day 18 rat brain. Neurons were placed on coverslips coated with poly-d-lysine (1 mg/ml) and maintained in Neurobasal medium supplemented with B27 (Invitrogen), 0.5 mm l-glutamine, and 12.5 μm glutamate, and grown in fresh medium without glutamate. Neurons were transfected using the calcium phosphate transfection kit (Clontech). For immunostaining, neurons were permeabilized in PBS containing 0.2% Triton X-100 and incubated with primary antibodies against MTMR2 (2 μg/ml) and PSD-95 (1:500), synaptophysin (1:200), MAP2 (1:500), EGFP (1:500), and Flag (1 μg/ml), followed by Cy3- or FITC-conjugated secondary antibodies.
Time-lapse imaging.
Cultured neurons expressing sh-M1 (or sh-vec) were mounted in a chamber (Zeiss) in which temperature (37°C) and CO2 concentration (10%) were maintained during the image acquisition. Images were acquired with an LSM510 confocal microscope (Zeiss) using a C-Apochromat ′ 63, 1.20 numerical aperture water-immersion objective. The laser power was attenuated to <3% to avoid fluorescence bleaching and neuronal toxicity. Images were captured every 10 min for 90 min.
Antibody feeding assay.
Live cultured neurons expressing HA-GluR2 were incubated with mouse HA antibodies (10 μg/ml) for 10 min at 37°C. After DMEM washing, neurons were returned to conditioned medium and incubated at 37°C for 10 min. Fixed neurons were incubated with Cy3-conjugated antibodies for surface GluR2, permeabilized, and labeled with Cy5- and FITC-conjugated antibodies for internalized GluR2 and EGFP (expressed from the shRNA vector), respectively.
Recycling assay.
Live labeling of HA-GluR2 was performed as in the antibody feeding assay. After brief washing in prewarmed DMEM, neurons were returned to normal conditioned medium and incubated for 10 min at 37°C to allow endocytosis. Remaining surface HA-GluR2-bound HA antibodies were acid stripped with 0.5 m NaCl/0.2 m acetic acid on ice for 4 min. Neurons were returned to normal conditioned medium and incubated for 30 min at 37°C to allow recycling. Neurons were fixed, and recycled receptors were labeled with Cy3-conjugated secondary antibodies. After permeabilization, internalized receptors were labeled with Cy5-conjugated secondary antibodies.
Endosomal sorting.
After surface labeling and endocytosis of HA-GluR2 as described in the antibody feeding assay, neurons were fixed in cold 4% formaldehyde/4% sucrose/1′ PBS, and incubated with anti-rabbit IgG donkey antibodies (10 μg/ml) to block HA-GluR2 remained at the surface. Then, neurons were permeabilized in cold 0.2% Triton X-100, followed by triple immunostaining for HA (internalized HA-GluR2), EEA1/Lamp1, and EGFP (for knockdown vector). Neurons were incubated with leupeptin (50 μg/ml) during the endosomal sorting experiments to block lysosomal degradations; neurons not treated with leupeptin were used for comparison.
Image acquisition and quantification.
Fluorescent images were acquired using a confocal microscope (LSM510). Neuronal images acquired from two or three independent experiments using the same parameter settings for all scans were analyzed using MetaMorph image analysis software (Universal imaging) and in part by a blind manner. Spines were defined as dendritic protrusions of 0.5–3 μm length, with or without a head. The number of excitatory synapses was measured by counting the number of PSD-95 or spines in contact with synapsin I on 12–24 neurons (∼200 μm total dendritic length per neuron). To quantify the number of EEA1- and Lamp1-positive endosomes, the number of the EEA1 and Lamp1 clusters in the cell body region and dendritic processes of cultured neurons were normalized to the cell body area and the dendritic length, respectively. As Lamp1 clusters were most abundant in proximal dendrites, dendrites within 50 μm from the cell body were used to analyze Lamp1 cluster density. Colocalization between internalized HA-GluR2 and endosomal markers was determined using the colocalization module of the MetaMorph program. Means from multiple individual neurons were averaged to obtain a population mean and SEM. The numbers (n) in figure legends represent cell numbers from two to four independent experiments. Statistical significance was determined by Student's t test or ANOVA.
Electrophysiology.
Cultured neurons were transfected with pSUPER. gfp/neo (sh-vec), or MTMR2 shRNAs, at day in vitro (DIV) 15. After 2 d, EGFP-expressing neurons were whole-cell voltage-clamped at −60 mV using an Axopatch 200B amplifier (Molecular Devices). mEPSCs were analyzed using the Mini Analysis Program (Igor).
Results
PSD-95 interacts with MTMR2
To identify proteins that contribute to PSD-95-dependent organization of excitatory synaptic structure and function, we searched for binding partners of PSD-95 using the second PDZ domain of PSD-95 as bait and a human brain yeast-two hybrid cDNA library. We identified MTMR2, a 3-phosphatase for PIs, as a novel binding partner of PSD-95 (Fig. 1A).
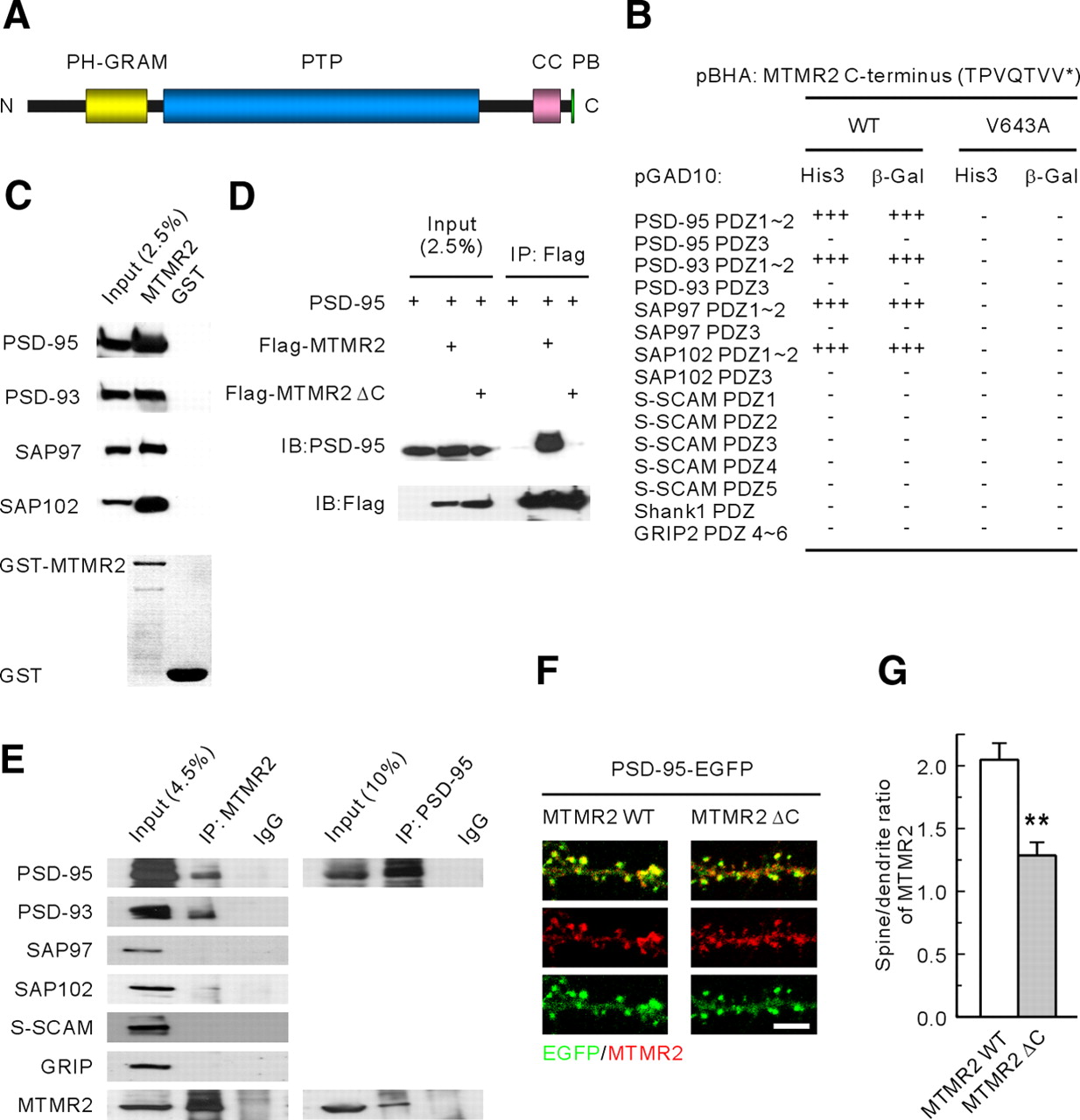
PSD-95 interacts with and promotes spine localization of MTMR2. A, Domain structure of MTMR2. PH-GRAM, glucosyltransferases, Rab-like GTPase activators and myotubularins; PTP, protein tyrosine phosphatase; CC, coiled coil; PB, PDZ binding motif. B, MTMR2 interacts with PSD-95 family proteins in yeast two-hybrid assays. The last seven amino acids of MTMR2 (WT or the C-terminal point mutant V643A) in the pBHA bait vector were tested for their binding to PDZ domains from PSD-95 family members and other PDZ proteins (S-SCAM, Shank1, and GRIP2) in the pGAD10 prey vector. HIS3 activity: +++, > 60%; ++, 30–60%; +, 10–30%; −, no significant growth. β-Galactosidase activity: +++, < 45 min; ++, 45–90 min; +, 90–240 min; −, no significant activity. C, Pulldown of PSD-95 family proteins by GST fusion proteins of MTMR2 (GST-MTMR2; full length). HEK293T cell lysates transfected with PSD-95 family members were pulled down by GST-MTMR2 and immunoblotted with antibodies against PSD-95, Myc (PSD-93), EGFP (SAP97), and Flag (SAP102). D, MTMR2 forms a complex with PSD-95 in HEK293T cells. HEK293T cells doubly transfected with PSD-95 and Flag-tagged MTMR2 (WT, or a Δ C mutant that lacks the PSD-95-interacting C terminus) were immunoprecipitated with Flag antibodies and immunoblotted with Flag and PSD-95 antibodies. E, MTMR2 forms a complex with PSD-95, PSD-93, and SAP102, but not with SAP97 or other PDZ proteins, in rat brain. F, G, MTMR2 WT shows a greater degree of spine localization than does MTMR2 Δ C. Cultured hippocampal neurons were transfected doubly with MTMR2 (WT or Δ C) and PSD-95-EGFP (DIV 15–16) and stained for EGFP and MTMR2. Spine localization of MTMR2 was quantified as the ratio of MTMR2 signals in a spine versus those in a neighboring dendrite. n = 8 cells for MTMR2 WT and 6 for MTMR2 Δ C. **p < 0.01. Scale bar, 5 μm.
In yeast-two hybrid assays, the C terminus of MTMR2, which ends with the class I PDZ domain-binding motif, QTVV, interacted with the first two PDZ domains of the PSD-95 family proteins, PSD-95/SAP90, PSD-93/Chapsyn-110, SAP97, and SAP102, but not with PDZ domains from S-SCAM, Shank1, or GRIP2 (Fig. 1B). A mutant MTMR2 with a C-terminal point mutation (V643A) eliminated the PDZ interactions. GST-MTMR2 fusion proteins pulled down all four PSD-95 family proteins (Fig. 1C). Wild-type MTMR2, but not a mutant MTMR2 lacking the three C-terminal PSD-95-binding residues (MTMR2 Δ C), formed a complex with PSD-95 in HEK293T cells (Fig. 1D). In rat brain, MTMR2 formed a complex with PSD-95, PSD-93, and SAP102, but not with SAP97, S-SCAM or GRIP (Fig. 1E).
A known function of PSD-95 is to promote synaptic localization of interacting proteins (Kim and Sheng, 2004; Funke et al., 2005; Sheng and Hoogenraad, 2007). In support of this hypothesis, WT MTMR2 showed greater spine localization than did MTMR2 Δ C, as measured by the ratio of MTMR2 signals in a spine to those in an adjacent dendrite (Fig. 1F,G). Expression levels of MTMR2 WT and MTMR2 Δ C, as measured by their fluorescence signals relative to that of PSD-95 in dendritic regions including the spines, were comparable (n = 8 for WT and 6 for Δ C, p = 0.97), suggesting that the difference in their spine localizations did not stem from altered expression. Together, these results suggest that PSD-95 directly interacts with MTMR2, and that this interaction promotes synaptic localization of MTMR2.
Expression patterns of MTMR2 mRNAs and proteins in the brain
We investigated the expression pattern of MTMR2 mRNA in the adult rat brain (Fig. 2A–C). Strong expression of MTMR2 mRNAs was found in the granular layer of olfactory bulb and dentate gyrus of hippocampal formation. Relatively strong expression was observed in the cerebral cortex (strong in the layer IV and piriform cortex), CA1-CA3 area of Ammon's horn, medial habenular nucleus of thalamus, ventromedial and arcuate nuclei of hypothalamus, medial amygdaloid nucleus, and granule cell layer of cerebellum.
Immunoblot analysis using MTMR2 antibodies generated in this study revealed that MTMR2 proteins are widely expressed in various rat brain regions (Fig. 2D). Expression levels of MTMR2 proteins gradually increased during postnatal rat brain development, similar to PSD-95 (Fig. 2E). In brain subcellular fractions, MTMR2 was mainly detected in synaptic fractions, including crude synaptosomal (P2) and synaptosomal membrane (LP1) fractions (Fig. 2F). Lower, but significant, levels of MTMR2 were also detected in synaptic cytosol (LS2) and synaptic vesicle (LP2) fractions. Much smaller amounts of MTMR2 were detected in the extra-crude synaptosomal (S2) fraction, which contains the cytosol (S3) and microsomes (P3). MTMR2 was detected in postsynaptic density (PSD) fractions, including PSD I and PSD II fractions, but unlike PSD-95, it was not detected in PSD III fractions (Fig. 2G), suggesting that MTMR2 is not tightly associated with the PSD at synapses.

Patterns of MTMR2 mRNA and protein expression. A–C, Distribution patterns of MTMR2 mRNAs in the brain. Coronal (A), horizontal (B), and sagittal (C) sections of adult rat brain (8 weeks) were hybridized with an MTMR2 riboprobe. Arc, Arcuate hypothalamic nucleus; CA1, CA1 field of Ammon's horn; CA3, CA3 field of Ammon's horn; Cb, cerebellum; CPu, caudate–putamen; Ctx, cerebral cortex; DG, dentate gyrus; Me, medial amygdaloid nucleus; MHb, medial habenular nucleus; Ob, olfactory bulb; VMH, ventromedial hypothalamic nucleus. Scale bar, 6 mm. D, Widespread expression of MTMR2 proteins in various brain regions, determined by immunoblotting. R, Other regions of the brain. β-Tubulin was used as a control. E, MTMR2 expression during rat brain development. E, Embryonic day; P, postnatal day; Wk, week. PSD-95 and β-tubulin were used as controls. F, Distribution patterns of MTMR2 in rat brain subcellular fractions. H, Homogenates; P2, crude synaptosomes; S2, supernatant after P2 precipitation; S3, cytosol; P3, light membranes; LP1, synaptosomal membranes; LS2, synaptosomal cytosol; LP2, synaptic vesicle-enriched fraction. SynPhy, synaptophysin. G, Enrichment of MTMR2 in PSD fractions. PSD fractions extracted with Triton X-100 once (PSD I) or twice (PSD II), or with Triton X-100 and a strong detergent Sarcosyl (PSD III), were immunoblotted with MTMR2 and PSD-95 antibodies.
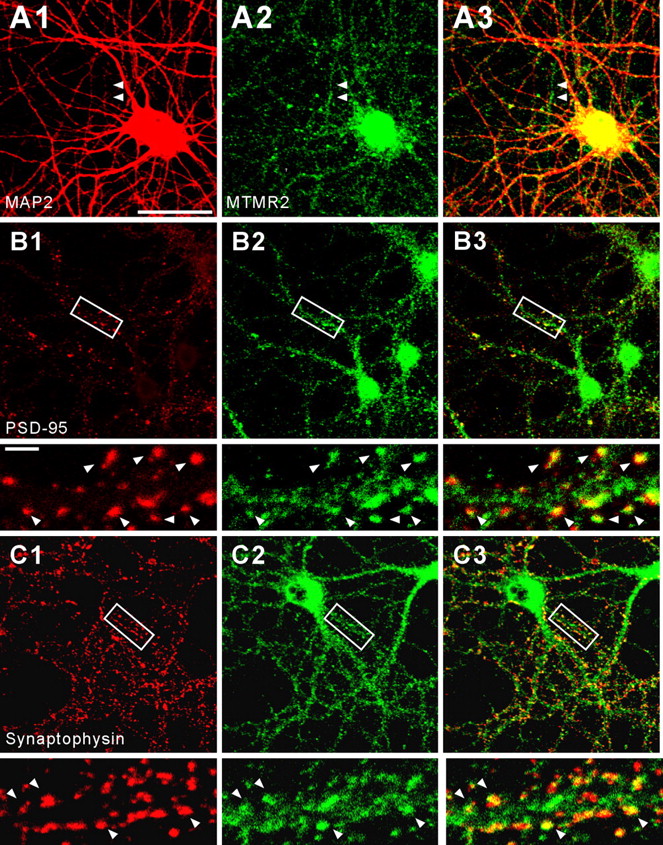
In cultured hippocampal neurons, MTMR2 was mainly detected in dendrites, although axons also showed weak MTMR2 expression (Fig. 3A). A majority of dendritic MTMR2 signals was detected in discrete clusters, some of which were colocalized with PSD-95 and synaptophysin (a presynaptic protein) (Fig. 3B,C), indicating their presence at excitatory synapses, while others were present in dendritic trunks.

Subcellular localization of MTMR2 in cultured hippocampal neurons. Cultured hippocampal neurons (DIV 18) were stained by double immunofluorescence staining for MTMR2 (A2, B2, and C2; green) and various subcellular markers (red); MAP2 (A1; a marker for dendrites), PSD-95 (B1; a marker for excitatory synapses), and synaptophysin (C1; a presynaptic marker). A3, B3, and C3 indicate merged images. MTMR2 distributes to MAP2-positive dendrites as well as MAP2-negative axons (A; MAP2-negative axons are indicated by arrowheads) and colocalizes with PSD-95 and synaptophysin (B, C; sites of colocalizations are indicated by arrowheads). Enlargements of the insets are shown below the original images. Scale bar, 50 μm; inset, 5 μm.
Knockdown of MTMR2 reduces excitatory synapse number and suppresses synaptic transmission
Myotubularins have been implicated in the regulation of endosomal trafficking, an important cell biological process known to regulate postsynaptic trafficking of membranes and receptors in neurons (Kennedy and Ehlers, 2006; Newpher and Ehlers, 2008). Therefore, we tested whether MTMR2 knockdown in cultured neurons had any effect on excitatory synaptic structure and function. To this end, we generated an MTMR2 shRNA (sh-M1) that reduces exogenous MTMR2 expression in HEK 293T cells by ∼80%, and endogenous MTMR2 expression in cultured hippocampal neurons by ∼55% (supplemental Fig. S1A–C, available at www.jneurosci.org as supplemental material).
MTMR2 knockdown in cultured hippocampal neurons (DIV 15–17) led to a marked reduction in the number of excitatory synapses, defined as PSD-95-positive dendritic spines, compared with neurons expressing empty shRNA vector (Fig. 4A,B). A similar reduction in the number of dendritic spines in contact with synapsin I (a marker for presynaptic nerve terminals) was observed upon MTMR2 suppression (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). Additionally, MTMR2 knockdown reduced the number of dendritic spines, but had no effect on their morphology (length and width) (supplemental Fig. S3A–D, available at www.jneurosci.org as supplemental material). The reduction in dendritic spine density may be attributable to a decrease in spine formation or maintenance. We observed that elimination of MTMR2 significantly enhanced the number of disappearing dendritic spines during the 90 min live imaging period, compared with control neurons transfected with the empty shRNA vector (supplemental Fig. S4A,B, available at www.jneurosci.org as supplemental material). In contrast, the number of newly appearing spines remained unaffected. These results suggest that MTMR2 knockdown reduces spine density by suppressing spine maintenance, and not formation.

Knockdown of MTMR2 reduces excitatory synapse number and suppresses excitatory synaptic function. A, MTMR2 knockdown reduces the number of excitatory synapses, defined as PSD-95-positive dendritic spines. Cultured hippocampal neurons were transfected with three independent shRNA constructs for MTMR2 (sh-M1, sh-M2, and sh-M3), a mutant sh-M1 with point mutations (sh-M1*) that is not active for MTMR2 mRNAs, or empty shRNA vector (sh-vec; control) (DIV 15–17), and stained for EGFP and PSD-95 for visualization of PSD-95-positive dendritic spines. Scale bar, 5 μm. B, Quantification of the effects of shRNA-mediated MTMR2 knockdown on the linear density of excitatory synapses. Histogram represents mean values ± SEM (sh-vec, n = 16 cells; sh-M1, n = 15; sh-M2, n = 16; sh-M3, n = 13; sh-M1*, n = 13; ***p < 0.001, ANOVA). C, MTMR2 knockdown reduces the frequency, but not amplitude, of mEPSCs. Cultured neurons transfected with sh-vec, sh-M1, or sh-M1* (DIV 15–17), were used to measure mEPSCs. D, E, Quantification of the effects of MTMR2 knockdown on the frequency and amplitude of mEPSCs.
To further characterize the effects of MTMR2 knockdown, we generated two additional MTMR2 shRNAs (sh-M2 and sh-M3), which target independent sites of MTMR2 (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). MTMR2 knockdown by sh-M2 and sh-M3 similarly reduced the density of excitatory synapses (PSD-95-positive spines) (Fig. 4A,B). In addition, a mutant MTMR2 shRNA that is not active against MTMR2 mRNAs (sh-M1*) (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material) did not reduce excitatory synapse number (Fig. 4A,B). These results suggest that the effects of MTMR2 knockdown are caused by specific degradation of MTMR2 mRNAs.
We next tested whether MTMR2 knockdown affects excitatory synaptic functions. MTMR2 knockdown in cultured neurons (DIV 15–17) caused a marked reduction in the frequency, but not amplitude, of miniature EPSCs (mEPSCs) (Fig. 4C–E). The lack of change in mEPSC amplitude may reflect the homeostatic maintenance of synaptic strength in the remaining spines. These results suggest that MTMR2 is required for both morphological and functional maintenance of excitatory synapses.
PSD-95 binding and phosphatase activities of MTMR2 are required for excitatory synapse maintenance
To identify molecular determinants of MTMR2 that are required for the maintenance of excitatory synapses, we rescued the effects of MTMR2 knockdown by coexpressing sh-M1 and sh-M1-resistant MTMR2 rescue constructs (supplemental Fig. S1E, available at www.jneurosci.org as supplemental material; WT and mutants), which are expressed at levels comparable to those of endogenous proteins. The WT MTMR2 rescue construct fully rescued the synapse-reducing effect of MTMR2 knockdown, restoring the number of excitatory synapses to a normal range (Fig. 5A,B). In contrast, the MTMR2 mutant that lacks phosphatase activity (C417S) failed to rescue the knockdown effects (Fig. 5A,B). MTMR2 Δ C, which lacks the ability to bind PSD-95, exhibited a reduced ability to rescue the knockdown phenotype compared with WT MTMR2 (Fig. 5A,B), but did partially rescue the knockdown effect, compared with MTMR2 C417S (Fig. 5A,B). This partial rescue activity may be attributable to the widespread distribution of the mutant protein at various subcellular sites, including dendritic spines. Expression levels of the three rescue constructs (MTMR2 WT, MTMR2 Δ C, and MTMR2 C417S) were comparable, as measured by MTMR2 signals in dendrites and cell bodies of the neurons transfected with these constructs, relative to those in neurons transfected with empty shRNA vector (WT, 85%, n = 13; Δ C, 98%, n = 23; C417S, 91%, n = 10 in the cell body; WT, 95%, n = 15; Δ C, 110%, n = 28; C417S, 89%, n = 10 in dendrites). These results suggest that MTMR2 contributes to the maintenance of excitatory synapses through mechanisms that depend on the PSD-95-binding and phosphatase activities of MTMR2.

Rescue of the effect of MTMR2 knockdown requires the PSD-95-binding and phosphatase activities of MTMR2. A, Rescue of MTMR2 knockdown by an sh-RNA-resistant MTMR2 expression construct. Both WT and mutant MTMR2 expression constructs were used to identify the molecular determinants important for MTMR2 function. Cultured hippocampal neurons were doubly transfected with sh-M1 plus MTMR2 [WT and mutants that lack PSD-95 binding (Δ C) or the phosphatase activity (C417S)], or singly with sh-M1 or sh-vec (DIV 15–17), and stained for EGFP, PSD-95, and MTMR2 to visualize PSD-95-positive dendritic spines and to monitor changes in MTMR2 expression. Note that MTMR2 images are shown in black and white instead of blue for image clarification. Scale bar, 5 μm. B, Quantification of the effects of the rescue constructs. Histogram represents mean ± SEM (sh-vec, n = 21 cells; sh-M1, n = 15; sh-M1 plus WT, n = 19; sh-M1 plus Δ C, n = 34; sh-M1 plus C417S, n = 15; **p < 0.01; ***p < 0.001, ANOVA).
MTMR2 knockdown leads to a decrease in the intensity of EEA1-positive early endosomes in dendrites but increases the intensity in the cell body region
How might the catalytic activity of MTMR2 contribute to the maintenance of excitatory synapses? PI(3)P and PI(3,5)P2, the main substrates of MTMR2, are localized primarily to the surfaces of early and late endosomes, respectively (Berger et al., 2002; Kim et al., 2002; Laporte et al., 2002; Begley et al., 2003; Tronchère et al., 2004; Robinson and Dixon, 2006; Nicot and Laporte, 2008). Accordingly, we examined whether early/late endosomes were altered in cultured neurons transfected with the MTMR2 knockdown construct by monitoring EEA1 (an early endosome-associated protein) and Lamp1 (a transmembrane protein enriched in late endosomes and lysosomes).
MTMR2 knockdown had no effect on the number of EEA1 or Lamp1 clusters in the cell body region (Fig. 6A,B). Interestingly, the intensity of total EEA1 clusters, but not that of Lamp1, normalized to the cell body area was increased by MTMR2 knockdown (Fig. 6A,B). In addition, the fluorescence intensities of EEA1 on individual clusters were significantly increased by MTMR2 knockdown, whereas Lamp1 intensities were unaffected (*p < 0.05 and p = 0.362, respectively). Because PI(3)P and Rab5 on early endosomes recruit EEA1 and stimulate endosome fusion (Simonsen et al., 1998), these results suggest that MTMR2 knockdown caused an increase in the concentration of PI(3)P, a substrate of MTMR2, on early endosomes.

MTMR2 knockdown leads to a decrease in the intensity of EEA1 clusters in dendrites but an increase in the cell body region. A, Cultured hippocampal neurons were transfected with sh-vec or sh-M1 (DIV 15–17), followed by immunostaining for EGFP (for sh vectors) and EEA1/Lamp1. Scale bars, 50 μm in soma and 10 μm in dendrites. B, Quantification of results in A. Total intensities and numbers of EEA1/Lamp1 clusters normalized to the cell body area or dendritic length in sh-M1-expressing cells were normalized to those in sh-vec-expressing control neurons. Soma/den, Somatic and dendritic. Histogram represents mean ± SEM (soma, sh-vec/EEA1, n = 32 cells; sh-M1/EEA1, n = 41; sh-vec/Lamp1, n = 26; sh-M1/Lamp1, n = 29; dendrites, sh-vec/EEA1, n = 29; sh-M1/EEA1, n = 25; sh-vec/Lamp1, n = 19; sh-M1/Lamp1, n = 20; **p < 0.01, Student's t test).
Interestingly, EEA1 clusters (normalized to dendritic length) were reduced in intensity, but not number, in the dendritic processes of sh-M1-expressing neurons, a result that contrasts with that obtained in the cell body area (Fig. 6A,B). Neither the number nor intensity of Lamp1 clusters in dendrites was changed by MTMR2 knockdown (Fig. 6A,B). These results indicate that MTMR2 knockdown decreases EEA1 intensity in dendrites but increases EEA1 intensity in the cell body without affecting the number of EEA1 clusters in dendrites or the cell body, suggesting the interesting possibility that MTMR2 knockdown causes dendritic early endosomes to move toward the cell body.
MTMR2 knockdown enhances endocytosis, but not recycling, of AMPA receptors
If early endosomes move down the dendrites toward the cell body under MTMR2 knockdown conditions, how might the number of EEA1 clusters in dendrites remain constant? One possibility is that MTMR2 knockdown increases the number of dendritic endosomes, for example by promoting endosomal production or suppressing endosomal recycling. To this end, we used GluR2, a subunit of AMPA receptors that translocates through endosomal pathways at excitatory synapses (Newpher and Ehlers, 2008). After surface labeling N-terminally HA-tagged GluR2 (HA-GluR2) by incubating live neurons with HA antibodies, endocytosis of the antibody-HA-GluR2 complex was monitored (antibody feeding assay in Materials and Methods). Interestingly, the amount of internalized GluR2 within a given time frame (20 min) was significantly increased in MTMR2-suppressed neurons, compared with control neurons expressing the empty knockdown vector (Fig. 7A,B). It should be noted that this difference was observed during a relatively short time window (20 min), which is normally used in endocytosis assays to minimize contamination of recycling events over longer time periods. Accumulation of these small changes over the time course of days may lead to significant changes, such as loss of synapses and dendritic spines.

MTMR2 knockdown enhances endocytosis, but not recycling, of the GluR2 subunit of AMPA receptors. A, B, MTMR2 knockdown enhances GluR2 endocytosis. Cultured neurons transfected with HA-GluR2 plus sh-vec (or sh-M1; DIV 16–20) were subjected to the antibody feeding assay (see Materials and Methods for details). The internalization index [ratio of internalized to total (internalized plus surface) receptors] in sh-M1-expressing cells was normalized to that in sh-vec-expressing control neurons. Histogram represents mean ± SEM (sh-vec, n = 25 cells; sh-M1, n = 17; **p < 0.01, Student's t test). C, D, MTMR2 knockdown does not affect GluR2 recycling. Cultured neurons transfected with HA-GluR2 plus sh-vec (or sh-M1; DIV 16–20) were subjected to the recycling assay (see Materials and Methods for details). The ratios of recycled/internalized (GluR2 remaining internalized after recycling) receptors in sh-M1- and sh-vec-expressing cells at 0 and 30 min time points were compared. Histogram represents mean ± SEM (sh-vec, 0 min, n = 22; 30 min, n = 16; sh-M1, 0 min, n = 19; 30 min, n = 15; ***p < 0.001, Student's t test).
The increase in the amount of internalized GluR2 following MTMR2 knockdown may be attributable to enhanced endocytosis or reduced recycling. In our experiments, MTMR2 knockdown had no effect on the recycling of internalized GluR2, relative to control (empty knockdown vector) (Fig. 7C,D), suggesting that suppression of MTMR2 selectively enhances endocytosis of AMPA receptors.
MTMR2 knockdown induces a large increase in the colocalization between internalized AMPA receptors and late endosomes/lysosomes mainly in the cell body
Enhanced EEA1 localization to early endosomes and increased production of GluR2-containing endosomes may lead to changes in the endosomal trafficking of internalized GluR2. Accordingly, we monitored whether internalized GluR2 shows enhanced colocalization with early endosomes or late endosomes/lysosomes in dendrites and the cell body. MTMR2 knockdown significantly increased the colocalization of internalized GluR2 with Lamp1 (late endosomes/lysosomes) in the cell body but not in dendrites in the presence of leupeptin, an inhibitor of lysosomal proteases (Fig. 8A,B). Conversely, Lamp1 showed increased colocalization with internalized GluR2 in the cell body but not in dendrites (Fig. 8A,B). The same experiments in the absence of leupeptin yielded essentially identical results with a small reduction in the colocalization (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). The absence of leupeptin did not significantly decrease the absolute levels of GluR2, although there was a tendency (data not shown). As for EEA1 (early endosomes), MTMR2 knockdown caused relatively small increases, compared with Lamp1, in the colocalization of internalized GluR2 with EEA1 in both dendrites and the cell body (Fig. 8A,B). These results suggest that MTMR2 knockdown promotes sorting of internalized AMPA receptors to the late endosomal/lysosomal pathway for protein degradation.

MTMR2 knockdown induces a large increase in the colocalization between internalized AMPA receptors and late endosomes/lysosomes mainly in the cell body region. A, Cultured neurons were doubly transfected with sh-M1 (or sh-vec) and HA-GluR2 (DIV 15–17), followed by antibody feeding, endocytosis, and triple staining for internalized HA-GluR2 (iHA-GluR2; green), EEA1/Lamp1 (red), and EGFP (shRNA; blue). The sorting experiment was performed in the presence of leupeptin to block lysosomal degradation. Scale bars, 50 μm in soma and 10 μm in dendrites. B, Quantification of colocalization between internalized GluR2 and EEA1/Lamp1 in the cell body (soma) and dendrites (den). The histogram represents mean ± SEM (soma, sh-vec/EEA1, n = 18 cells; sh-M1/EEA1, n = 26; sh-vec/Lamp1, n = 17; sh-M1/Lamp, n = 20; *p < 0.05; ***p < 0.001, Student's t test; dendrites, sh-vec/EEA1, n = 28; sh-M1/EEA1, n = 26; sh-vec/Lamp1, n = 19; sh-M1/Lamp1, n = 20; ***p < 0.001, Student's t test).
Discussion
Synaptic localization of MTMR2 by PSD-95
Our results indicate that PSD-95 directly interacts with and promotes spine localization of MTMR2, effectively enriching MTMR2 in dendritic spines. Consistent with this, a large fraction of MTMR2 is detected in synaptic membrane and PSD fractions. It should be noted that relatively small, but significant, amounts of MTMR2 are also detected in synaptic cytosol and synaptic vesicle-enriched fractions. This is consistent with the fact that MTMR2 is not enriched in PSD fractions to as great an extent as PSD-95. Therefore, it appears that the affinity of MTMR2 for PSD-95 is in a range that allows MTMR2 protein to be localized at the PSD, as well as in the cytosol and vesicles within the spine. A relatively small amount of MTMR2 was also detected in axons, indicating that MTMR2 acts at widespread subcellular sites in neurons.
There are 14 members of the myotubularin family of PI 3-phosphatases in humans. Although their biochemical functions have been relatively well characterized, whether and how they are targeted to distinct subcellular sites of major endosomal production has remained unclear. Our results indicate that MTMR2 is enriched at excitatory synapses. Importantly, we have identified a novel mechanism for this synaptic enrichment: interaction with the PDZ domain of a synaptic scaffolding protein. These results also suggest the novel involvement of PSD-95 in the MTMR2-dependent regulation of PI signaling at excitatory synapses.
A previous study has shown that MTMR2 in Schwann cells interacts with SAP97/Dlg1 (Bolino et al., 2004; Bolis et al., 2009), a PSD-95 family protein with a relatively widespread tissue distribution pattern compared with other PSD-95 family proteins (Lue et al., 1994; Müller et al., 1995). SAP97 is mainly detected at the node/paranode region in Schwann cells, and this localization is reduced in MTMR2-null mice (Bolino et al., 2004). This suggests that MTMR2 is important for the subcellular localization of SAP97/Dlg1, and that disruption of the MTMR2-SAP97 interaction may dysregulate cellular junctions or membrane dynamics at the node/paranode region, and as a consequence, lead to the myelin outfolding observed in CMT4B1. As our results indicate that MTMR2 interacts with PSD-95 and PSD-93/chapsyn-110, but not with SAP97, in the brain, MTMR2 appears to interact differentially with PSD-95 family proteins in different cell types. In addition, our finding that PSD-95 promotes synaptic localization of MTMR2 suggests the possibility that SAP97 might regulate nodal/paranodal localization of MTMR2.
MTMR2 is known to form heteromultimers with other myotubularins, including the catalytically inactive MTMR5/SBF1 and MTMR13/SBF2; these interactions enhance the phosphatase activity of MTMR2 (Kim et al., 2003; Robinson and Dixon, 2005; Berger et al., 2006b). A previous proteomic analysis identified MTMR5 as a component of the PSD, suggesting that MTMR5 is localized to excitatory synapses (Peng et al., 2004). In addition, MTMR13, which is also associated with the CMT4B2 demyelinating neuropathy (Azzedine et al., 2003; Senderek et al., 2003), is widely expressed in various tissues, including the brain (Azzedine et al., 2003). Therefore, these myotubularins might be indirectly recruited to excitatory synapses through MTMR2 and participate in the local regulation of endosomal trafficking.
Maintenance of excitatory synapses by MTMR2
Our results indicate that MTMR2 is required for the maintenance of excitatory synapses and dendritic spines. The MTMR2 knockdown-induced reduction in excitatory synapse number was rescued by WT MTMR2 but not by MTMR2 mutants that lack either PSD-95 binding or phosphatase activity. The 3-phosphatase activity of MTMR2 is active toward two specific PIs, PI(3)P and PI(3,5)P2, which are thought to be present mainly on early and late endosomes, respectively (Berger et al., 2002; Kim et al., 2002; Laporte et al., 2002; Begley et al., 2003; Tronchère et al., 2004; Robinson and Dixon, 2006; Nicot and Laporte, 2008). Therefore, it is expected that MTMR2 targeted to excitatory synapses by PSD-95 interaction might participate in the regulation of synaptic endosomal trafficking.
Consistent with this hypothesis, endosomal compartments are found in dendritic spines (Cooney et al., 2002), and active endosomal trafficking of synaptic membrane proteins occurs within the spine (Kennedy and Ehlers, 2006; Lau and Zukin, 2007; Shepherd and Huganir, 2007; Newpher and Ehlers, 2008). In addition, specialized endocytic zones are present in dendritic spines at sites adjacent to the PSD (Blanpied et al., 2002). As the endocytic zone and the PSD are physically coupled by dynamin-3 (Lu et al., 2007), PSD-95-bound MTMR2 may be ideally localized to regulate the endosomal production and traffic occurring at an adjacent endocytic zone.
How might endosomal dysregulation contribute to the loss of excitatory synapses? One possibility is that excessive production of endosomes, which contain synaptic surface proteins as well as membranes, leads to synaptic loss. In support of this possibility, our data indicate that MTMR2 knockdown leads to an increase in endocytosis (not recycling) of GluR2-containing AMPA receptors, a well known endosomal cargo, at excitatory synapses (Fig. 7). Little is known about the potential involvement of MTMR2 in regulating the very early steps of endocytosis or endosome production, although studies in non-neural cells suggest this possibility. In L6 cells, the MTMR2 substrate PI(3)P is generated in the plasma membrane by insulin stimulation, which acts via activation of the small GTPase TC10 (Maffucci et al., 2003). PI(3)P increases the affinity of the AP2 endocytic complex for the tyrosine-based endocytic motif in target receptors (Rapoport et al., 1997). In addition, the class II PI 3-kinase C2α, which produces PI(3)P from PI, is localized to clathrin-coated pits and is activated by clathrin binding (Gaidarov et al., 2001). Deletion of let-512, a Caenorhabditis elegans homolog of the class III PI 3-kinase Vps34, leads to defects in endocytosis (Xue et al., 2003). These results suggest that PI(3)P can be generated in the plasma membrane and may have function at early stages of endocytosis. It is thus possible that MTMR2, which dephosphorylates PI(3)P to PI, may act as a negative regulator of early endocytic processes.
Alternatively, MTMR2 knockdown may lead to synaptic loss by interfering with normal endosomal trafficking. Specific PI species in endosomal surfaces, often in combination with small GTPases such as Rab5, function as an identity code that directs their interaction with effector proteins and mediates trafficking to various subcellular compartments (Zerial and McBride, 2001; Behnia and Munro, 2005). For instance, PI(3)P and Rab5 on early endosomes recruit EEA1 and promote endosome fusion (Simonsen et al., 1998). In support of a role for PIs in endosomal trafficking, overexpression of MTMR2 in yeast, which reduces levels of PI(3)P and PI(3,5)P2 in endosomes, induces abnormal accumulation of enlarged vacuoles (Laporte et al., 2002). Knockdown or pharmacological inhibition of the class III PI 3-kinase Vps34, which reduces endosomal PI(3)P levels, inhibits the endosomal pathway (Petiot et al., 2003; Johnson et al., 2006). Interestingly, MTMR2 knockdown in epithelial cells increases the amount of PI(3)P in late endosomes and blocks the egress of the epidermal growth factor receptor from late endosomes (Cao et al., 2008). These results suggest the interesting possibility that MTMR2 knockdown might cause EEA1-containing early endosomes to move from dendrites to the cell body. Our results indicate that MTMR2 knockdown induces a decrease in the intensity of EEA1 clusters in dendrites and increases the intensity in the cell body area (Fig. 6). In addition, MTMR2 knockdown markedly enhances colocalization of internalized GluR2 with Lamp1-containing late endosomes/lysosomes mainly in the cell body region (Fig. 8). These results suggest that EEA1-associated early endosomes in the absence of MTMR2 expression are likely to move from dendrites to the cell body area. If this is the case, there should be a decrease in the number of EEA1 clusters in dendrites. However, there was no change in the number of dendritic EEA1 clusters (Fig. 6). A possible explanation is that early endosome generation in dendrites is enhanced, as demonstrated by increased generation of GluR2-containing endosomes with MTMR2 knockdown (Fig. 7). It is conceivable that the continuous dendrite-to-soma flow of EEA1 molecules could eventually lead to a decrease in the intensity of EEA1 clusters in dendrites. Last, our results suggest that abnormal endosomal traffic induced by MTMR2 knockdown may trap AMPA receptors in defective endosomal compartments, promoting their lysosomal degradation.
In conclusion, our results suggest that PSD-95 promotes the synaptic localization of MTMR2, and that synaptically targeted MTMR2 maintains excitatory synapses by inhibiting excessive endosomal production and destructive trafficking to lysosomes.
Footnotes
-
This work was supported by the National Creative Research Initiative Program of the Korean Ministry of Science and Technology (to E.K.). A part of this work was technically supported by the core facility service of the 21C Frontier Brain Research Center.
- Correspondence should be addressed to Eunjoon Kim at the above address. kime{at}kaist.ac.kr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}