

Abstract
Individuals with the neurofibromatosis type 1 (NF1) inherited cancer syndrome exhibit neuronal dysfunction that predominantly affects the CNS. In this report, we demonstrate a unique vulnerability of CNS neurons, but not peripheral nervous system (PNS) neurons, to reduced Nf1 gene expression. Unlike dorsal root ganglion neurons, Nf1 heterozygous (Nf1+/−) hippocampal and retinal ganglion cell (RGC) neurons have decreased growth cone areas and neurite lengths, and increased apoptosis compared to their wild-type counterparts. These abnormal Nf1+/− CNS neuronal phenotypes do not reflect Ras pathway hyperactivation, but rather result from impaired neurofibromin-mediated cAMP generation. In this regard, elevating cAMP levels with forskolin or rolipram treatment, but not MEK (MAP kinase kinase) or PI3-K (phosphatidylinositol 3-kinase) inhibition, reverses these abnormalities to wild-type levels in vitro. In addition, Nf1+/− CNS, but not PNS, neurons exhibit increased apoptosis in response to excitotoxic or oxidative stress in vitro. Since children with NF1-associated optic gliomas often develop visual loss and Nf1 genetically engineered mice with optic glioma exhibit RGC neuronal apoptosis in vivo, we further demonstrate that RGC apoptosis resulting from optic glioma in Nf1 genetically engineered mice is attenuated by rolipram treatment in vivo. Similar to optic glioma-induced RGC apoptosis, the increased RGC neuronal death in Nf1+/− mice after optic nerve crush injury is also attenuated by rolipram treatment in vivo. Together, these findings establish a distinctive role for neurofibromin in CNS neurons with respect to vulnerability to injury, define a CNS-specific neurofibromin intracellular signaling pathway responsible for neuronal survival, and lay the foundation for future neuroprotective glioma treatment approaches.
Introduction
Neurofibromatosis type 1 (NF1) is one of the most common inherited cancer syndromes affecting the nervous system. Children with NF1 are born with one mutated (nonfunctional) and one functional copy of the NF1 gene in every cell in their body (NF1 heterozygosity), but develop low-grade central and peripheral nervous system (PNS) tumors after somatic inactivation of the remaining functional NF1 allele in specific cells (e.g., glial cells). Within the CNS, glial neoplasms affecting the optic nerve (ON)/chiasm (optic glioma) arise in 15% of children with NF1 (Listernick et al., 1997). Nearly half of children with NF1-associated optic gliomas have visual impairment (Listernick et al., 1997), and few regain vision after treatment (Dalla Via et al., 2007). Additionally, 50–70% of children with NF1 exhibit learning disabilities that limit scholastic performance (Eliason, 1986; North et al., 1995, 1997; Ozonoff, 1999; Hyman et al., 2005). To model these learning disabilities, previous studies used Nf1 genetically engineered mice (GEMs) to demonstrate that the learning and memory abnormalities seen in Nf1+/− mice result from Nf1 heterozygosity in neurons, not glial cells (Costa et al., 2002; Cui Y. et al., 2008).
The role of the NF1 protein, neurofibromin, in neuronal function has been primarily studied in PNS neurons. Pioneering studies by Parada and associates first demonstrated that complete Nf1 inactivation in peripheral ganglion cells resulted in relative neurotrophin independence, leading to inappropriate (increased) neuronal survival (Vogel et al., 1995). In contrast, previous studies by our laboratory focusing on neuronal and glial differentiation from neural stem cells (NSCs) in vitro and in vivo demonstrated that Nf1−/− neurons exhibited decreased neurite lengths, resulting in reduced secondary somatosensory cortical thickness (Hegedus et al., 2007). However, unlike PNS neurons whose relative neurotrophin independence results from Ras hyperactivation, NSCs expressing an activated Ras allele form neurons with normal neurite lengths in vitro and in vivo, suggesting that Ras hyperactivation was not responsible for this abnormal neuronal phenotype in Nf1-deficient cells.
The purpose of the current study was to resolve the apparent differences between central and peripheral system neuronal vulnerability to changes in Nf1 gene dose, and to define the molecular mechanism underlying these differences. Herein, we show that PNS neurons are not significantly affected by Nf1 heterozygosity in vitro, whereas Nf1+/− neurons from either the hippocampus or retina have dramatically shorter neurite lengths and growth cone areas, as well as increased apoptosis in vitro. We further demonstrate that these phenotypes in CNS neurons result from impaired neurofibromin regulation of cAMP. Finally, we use the Nf1+/−GFAPCKO (conditional knock-out) optic glioma GEM strain to show that correcting the cAMP signaling defect resulting from reduced neurofibromin function in retinal neurons ameliorates the retinal ganglion cell (RGC) apoptosis associated with tumor formation in vivo. Together, these findings demonstrate that CNS and PNS neurons are differentially sensitive to the effects of Nf1 heterozygosity, and establish cAMP as an important target for future therapeutic drug design aimed at reducing CNS neuronal dysfunction in individuals with NF1.
Materials and Methods
Chemicals, reagents, and antibodies.
All chemicals were purchased from Sigma unless otherwise indicated: class III β-tubulin (Tuj-1; 1:1000 dilution; Covance), CD90.2 (1:250 dilution; eBioscience), forskolin (0.01 mm), rolipram (200 μm), 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene (U0126; 10 μm), 2′,5′-dideoxyadenosine (DDA; 100 μm), and 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002; 30 μm). All drug treatments in vitro were performed for the entire culture period, with the exception of LY294002, which was added only during the final 3 d of the experiment.
Cell culture.
Hippocampal cultures were prepared as described previously (Clarris et al. 1994) with Hibernate-E used for dissection medium. Hippocampi were dissociated in HBSS containing 1% papain (Worthington Biochemicals) and 5 U/ml DNase (Invitrogen), transferred to a solution containing 1% ovomucoid (Worthington Biochemicals), and plated in DMEM plus 10% fetal calf serum for 4 h before medium was switched to neural basal medium plus B27 and 2 mm l-glutamine for 3 d.
Dorsal root ganglion (DRG) cultures were obtained and cultured as reported previously (Brown et al. 2009). Dissociated cultures dissociated in 0.02% trypsin/EDTA were grown in C10-2 medium for 48 h. For the oxidative stress experiments, DRG neurons were grown in neural basal medium containing B27, 50 ng/ml nerve growth factor (NGF), and 2 mm l-glutamine for 3 d.
Wild-type (WT) or Nf1 heterozygous (Nf1+/−) RGCs were generated from postnatal day 4 (P4)–P5 mice according to established protocols, and were cultured for 6 d before fixation (Barres et al. 1988; Shoge et al. 1999).
Primary astrocyte cultures were prepared as described previously for mixed glial-RGC culture experiments, and cultured for 3 d before fixation (Dugan et al., 1999; Sandsmark et al., 2007).
Retinal explant cultures were prepared with the RGC layer facing down and contacting the substrate (Hansen et al. 2004). Magnetic cell sorting was used to isolate RGCs (∼94% purity) using established protocols (Kerrison and Zack, 2007): retinal tissues were dissected in Mg2+/Ca2+-free HBSS and dissociated in HBSS containing 1% papain (Worthington Biochemicals) and 5 U/ml DNase (Invitrogen), transferred to a solution containing 1% ovomucoid (Worthington Biochemicals), and triturated before magnetic capture affinity separation. Dissociated cells were then magnetically depleted of non-RGCs using a CD11b/c antibody, followed by immunoenrichment of CD90.2-positive RGCs. After centrifugation, purified RGCs were resuspended in neural basal culture medium and plated on poly-d-lysine- (10 μg/ml) and laminin-coated (9.6 μg/ml) glass coverslips (22 mm; Fisher Scientific) in 35 mm dishes at a density of 15,000 cells per coverslip. RGCs were cultured in neurobasal medium and supplemented with B27, selenium, putrescine, triiodothyronine, transferrin, progesterone, pyruvate (1 mm), glutamine (2 mm), ciliary neurotrophic factor (10 ng/ml), brain-derived neurotrophic factor (BDNF; 50 ng/ml), and insulin (5 μg/ml) at 37°C and 10% CO2.
Excitotoxic death and oxidative stress experiments.
CNS (hippocampal and forebrain) and PNS (DRG) neuronal cultures were prepared as described above. After day in vitro (DIV) 10 for CNS neurons and DIV 3 for PNS neurons, the culture medium was replaced with medium lacking phenol red. Excitotoxic cell death was assessed after exposure to medium containing 50 μm glutamate for 24 h. Death from oxidative stress was assessed after exposure to medium containing 100 μm H2O2 (CNS neurons) or 60 μm H2O2 (PNS neurons) for 24 h. At the end of the incubation period, the medium was collected for lactate dehydrogenase (LDH) measurements, and the cells were fixed in 4% paraformaldehyde for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining (apoptosis).
LDH measurements.
Media removed from the CNS and PNS cultures were analyzed in triplicate using a commercial LDH cytotoxicity assay kit (Promega). Fresh medium was used as a negative control, whereas LDH (1:5000 dilution) served as a positive control. LDH levels were quantitated using the μQuant plate reader at an absorbance of 490 nm (Bio-Tek Instruments).
Immunofluorescence.
Cells were prepared as described previously (Brown and Bridgman, 2009) using PBS instead of cacodylate buffer. Images were taken using an Olympus confocal or an inverted microscope equipped with a Cooke Sensicam. TUNEL was accomplished according to the manufacturer's instructions (Roche Diagnostics). Fixed cultures were imaged using phase confocal microscopy and then scored for growth cone area, neurite length, or TUNEL staining using ImageJ.
cAMP measurements.
Dissected whole retinas snap frozen in liquid nitrogen were triturated in ice-cold 5% trichloroacetic acid (10 μl per mg tissue) and centrifuged at 1000 × g for 10 min at 4°C. Supernatants supplemented with equal volume of 0.1 m HCl were extracted with water-saturated ether thrice before desiccation in a vacuum centrifuge. cAMP levels were determined using a cAMP enzyme immunoassay kit (Assay Designs).
Optic nerve crush injury.
Surgeries were conducted on 3-month-old mice. All surgeries were performed on the left eye, leaving the right eye as a control. Animals were anesthetized with ketamine (80 mg/kg) and xylazine (8 mg/kg) intraperitoneally. The conjunctiva was then incised at the limbus, and the subtenon space was bluntly dissected posteriorly. Next, the muscle cone was entered and the optic nerve was exposed. The axons of the optic nerve were crushed with fine forceps for 10 s, 1 mm posterior to the globe under direct visualization. Retinal ganglion cell axon crush resulted in a separation of the proximal and distal optic nerve end within an intact meningeal sheath. This procedure spares the meningeal vessels that carry arterial circulation to the retina. Any animal in which the meningeal vessels were compromised was killed immediately. Ophthalmic ointment with neomycin-polymyxin-hydrocortisone was applied to the wound for postsurgical pain management.
Eye injections.
Eye injections were preformed as described previously (Jaubert-Miazza et al., 2005). Briefly, 3-month-old mice received either drug or control solution injected into their left eye 3 d after ON crush injury on the same day. A Hamilton syringe, filled with 5 μl of solution containing either Rolipram (272 μm) or DMSO (Sigma), was inserted into the hole made by the first pipette. Three days later, mice were deeply anesthetized (isofluorane vapor or Nembutal, i.p.), transcardially perfused with heparinized Ringer's solution (30 ml) followed by 4% paraformaldehyde in 0.1 m PBS, pH 7.4 (40 ml), and tissues processed for analysis.
Rolipram treatment.
Nf1+/−GFAPCKO mice were treated at 2 months of age with rolipram (5 mg/kg/d in the drinking water) for 30 d. After perfusion, the eyeballs were placed in 1% agar in PBS and oriented with the retro-orbital optic nerve horizontal to the cutting surface. Five micrometer paraffin sections were generated for TUNEL labeling using a Roche Diagnostics kit. The number of TUNEL+ cells in the retinal ganglion cell layer was determined as a percentage of DAPI+ cells for at least four mice per group. This experiment was performed twice on independent cohorts of mice with identical results.
Structural analysis of growth cone areas and neurite lengths.
Growth cone areas were measured using ImageJ, starting at the neck and tracing around the growth cone. The mean, standard error, and p value were calculated from at least 40 randomly selected growth cones per condition. The investigators were always blinded to the genotype of the growth cones. Neurite lengths were also measured using ImageJ, and only neurons with intact growth cones were included. Neurites were traced starting at the outer edge of the cell body so that the cell body size would not skew measurements. As before, at least 40 randomly selected neurites per condition were used to calculate the mean, standard error, and p value.
Statistical analyses.
Student's t tests were used to determine significant changes in growth cone area and neurite length. A 2 × 2 contingency table analysis with Fisher's exact test was used to compare cell death frequencies. Mean and SEM were used for all graphs. All in vitro experiments were performed in a blinded fashion at least three times with identical results.
Results
Nf1 heterozygosity results in impaired hippocampal neuron morphology and survival
To determine the effect of reduced Nf1 expression on CNS neurons, we used dissociate cultures of hippocampal neurons from approximately embryonic day 13.5 (E13.5) mouse embryos. Each culture plate was derived from a single embryo, and the investigator was blinded to the genotype until after the scoring was complete. After 3 d in culture, cells were fixed and labeled with the Tuj-1 neuronal marker and TUNEL to measure apoptotic cell death. Cultures were then scored for growth cone area, neurite length, and cell death. Surprisingly, the growth cone areas from Nf1+/− hippocampal neurons were ∼40% smaller than their WT counterparts (p = 0.0001; N = 40) (Fig. 1A). Given the importance of growth cones in neuronal target finding during development and regeneration (Lankford et al., 1990), neurons with reduced growth cone spreading may also have attenuated neuronal processes. As predicted, Nf1+/− hippocampal neurite lengths were 25% shorter than their WT counterparts (p = 0.02; N = 47) (Fig. 1B). Moreover, Nf1+/− hippocampal neurons also exhibited increased cell death in vitro compared to WT neurons (p = 0.05) (Fig. 1C). Collectively, these findings demonstrate that reduced neurofibromin expression results in impaired hippocampal neuron function in vitro.

Nf1+/− hippocampal neurons have reduced growth cone areas, neurite lengths, and cell survival in vitro. A, Nf1+/− hippocampal neuron growth cones (labeled with Tuj-1) are smaller than their WT counterparts (p = 0.0001; N = 40). Scale bar, 50 μm. B, Neurite lengths, identified by Tuj-1 immunostaining, are significantly shorter in Nf1+/− hippocampal neurons (p = 0.02; N = 47). Scale bar, 50 μm. C, Nf1+/− hippocampal neurons exhibit more TUNEL+ labeling compared to their WT counterparts (p = 0.05). Error bars indicate SEM. Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01).
Nf1 heterozygosity has minimal effects on PNS neuronal function
Previous research has shown that complete Nf1 inactivation confers relative trophic factor-independent survival on DRG neurons and that heterozygosity has no effect on the survival of DRG neurons, with or without NGF (Vogel et al., 1995). In light of the effects of Nf1 heterozygosity on CNS neurons, we sought to determine whether the observed growth and survival deficits reflected differences in sensitivities to Nf1 gene dose in CNS versus PNS neurons. DRG neurons were prepared from the same ∼E13.5 mouse embryos used to generate the above hippocampal neuron cultures. As before, the investigator was blinded to the genotypes until all data were compiled. In contrast to hippocampal neurons, there was a small increase in growth cone areas in Nf1+/− DRG neurons compared to their WT counterparts, which was the opposite of what we observed with hippocampal neurons (p = 0.05; N = 40) (Fig. 2A). Similar to previous reports, there were no differences in neurite lengths or apoptosis between Nf1+/− and WT DRG neurons (Fig. 2B,C). These findings confirm earlier studies, and demonstrate striking differences in sensitivity to Nf1 heterozygosity in CNS and PNS neuronal populations.

Nf1+/− DRG neurons have normal growth cone areas, neurite lengths, and cell survival in vitro. A, Nf1+/− DRG neuronal growth cones (labeled with Tuj-1) are marginally larger than their WT counterparts (p = 0.0001; N = 40). B, Neurite lengths, identified by Tuj-1, are equivalent in WT and Nf1+/− DRG neurons (N = 42). Scale bar, 20 μm. C, Nf1+/− and WT DRG neurons have equivalent numbers of TUNEL+ neuronal cells. Error bars indicate SEM. The asterisks denotes a statistically significant difference (p < 0.05).
Nf1 heterozygosity results in impaired RGC neuron morphology and survival
To strengthen our observation that Nf1 heterozygosity in CNS neurons results in abnormal neuronal phenotypes, we next chose to examine another population of CNS neurons relevant to children with NF1. We elected to study RGC neurons based on our finding of retinal layer neuronal death in a genetically engineered mouse model of NF1-associated optic glioma (Hegedus et al., 2009). For these studies, we used both dissociate RGC cultures and retinal explants with identical results. Retinal explant cultures from P5 mice contained a near-pure (≥98%) population of RGC neurons, as demonstrated using the CD90.2 RGC marker (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Cells were fixed and stained after 6 d in culture, and growth cone spreading was assessed in a blinded fashion as before.
As seen with the hippocampal neurons, Nf1+/− RGC neurons had 57% smaller growth cones compared to their WT counterparts (p = 0.0001; N = 40) (Fig. 3A). Neurite length was assessed in Thy1.2-magnetic bead affinity-purified dissociate cultures: as before, Nf1+/− RGC neurite lengths were 35% shorter than their WT counterparts (p = 0.0001; N = 40) (Fig. 3B). To assess apoptotic cell death, retinal explants were grown on a bed of astrocytes, to facilitate RGC migration from the explant. The percent of TUNEL+, CD90.2+ cells was 9% higher in Nf1+/− compared to WT RGC neurons, independent of the Nf1 status of the astrocytes (p = 0.05) (Fig. 3C). In combination with the results obtained with hippocampal and DRG neurons, these data confirm that CNS neurons are uniquely sensitive to reduced Nf1 gene expression.

Nf1+/− RGCs exhibit reduced growth cone areas, neurite lengths, and cell survival in vitro. A, Nf1+/− RGC neuronal growth cones (labeled with Tuj-1) are smaller than their WT counterparts (p = 0.0001; N = 40). Scale bar, 20 μm. B, Nf1+/− RGC neurite lengths, after magnetic cell sorting and Tuj-1 immunostaining, are significantly shorter than their WT counterparts (p = 0.0001; N = 40). Scale bar, 20 μm. C, Nf1+/− retinal explants exhibit more TUNEL+ neuronal cells regardless of astrocyte genotype (p = 0.0001 for WT astrocytes; p = 0.0004 for Nf1−/− astrocytes). Error bars indicate SEM. Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01).
To extend our findings to other conditions that result in neuronal death, we also examined the impact of Nf1 heterozygosity on apoptosis resulting from excitotoxicity (CNS neurons) and oxidative stress (CNS and PNS neurons). Using glutamate-induced excitotoxicity, we found that Nf1+/− forebrain neurons exhibit increased LDH release and apoptosis (28% increase relative to untreated cultures) compared with their WT counterparts (12% increase relative to untreated cultures) (Fig. 4A). Similarly, Nf1+/− forebrain neurons exhibit increased LDH release and apoptosis in response to H2O2-induced oxidative stress (21% increase relative to untreated cultures) compared to WT neurons (8% increase relative to untreated cultures) (Fig. 4B). However, in contrast, Nf1+/− DRG neurons exhibited levels of LDH release and apoptosis indistinguishable from WT neurons in response to H2O2-induced oxidative stress (both exhibited a 30% increase relative to untreated cultures) (Fig. 4C). These results, coupled with our finding that Nf1+/− CNS, but not PNS, neurons have reduced neurite lengths, growth cone areas, and survival, demonstrate that Nf1 heterozygosity confers a vulnerability to cell death in the CNS.

Nf1+/− forebrain neurons have increased cell death in response to excitotoxic and oxidative stress in vitro. A, Nf1+/− forebrain neurons exhibit increased LDH release (left; p = 0.001; N = 5) and apoptosis (percentage of TUNEL+ cells; right; p = 0.0001; N = 5) after exposure to 50 μm glutamate compared to their WT counterparts. B, Nf1+/− forebrain neurons exhibit increased LDH release (left; p = 0.05; N = 9) and apoptosis (percentage of TUNEL+ cells; right; p = 0.001; N = 9) after exposure to 100 μm H2O2 compared to their WT counterparts. C, Nf1+/− and WT DRG neurons exhibit similar levels of LDH release and percentage of TUNEL+ neuronal cells after exposure to 60 μm H2O2. Error bars indicate SEM. Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01, ***p < 0.001).
cAMP levels, not Ras hyperactivation, are responsible for the neuronal dysfunction seen in Nf1+/− RGCs in vitro
Whereas previous studies have demonstrated that Nf1−/− PNS neurons exhibit increased cell survival resulting from impaired neurofibromin Ras regulation, we have shown that reduced Nf1-deficient CNS neuronal neurite lengths are the consequence of neurofibromin control of cAMP levels and not Ras regulation. To determine whether cAMP or Ras deregulation was primarily responsible for the neuronal abnormalities seen in Nf1+/− CNS neurons, we initially examined activated mitogen-activated protein kinase (MAPK; phospho-MAPK) (Fig. 5A) and Akt (phospho-Akt) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) expression by double-labeling immunofluorescence, and found no differences in Ras pathway activation between WT and Nf1+/− retinal ganglion layer neurons in situ. Furthermore, reducing MAPK activation with the U0126 MAP kinase kinase (MEK) inhibitor did not restore growth cone spreading in Nf1+/− RGC neurons (Fig. 5B). The concentration of U0126 used in these experiments was sufficient to reduce, but not eliminate, MAPK activation in the retina (Fig. 5C). Similarly, pharmacologic inhibition of phosphatidylinositol 3-kinase (PI3-K; LY294002) activation in Nf1+/− RGC neurons did not restore growth cone spreading in Nf1+/− RGC neurons (Fig. 6A). Finally, to directly evaluate the impact of activated Ras in RGC neurons, we used a mouse strain [brain lipid-binding protein (BLBP)-Cre] to drive constitutively active KRasG12D in neurons (Hegedus et al., 2007, 2009). Although neurons from BLBP-Cre; KRasG12D mice exhibit high levels of Ras and Ras pathway activity (Hegedus et al., 2007, 2008), RGC neurons from these mice had growth cone areas indistinguishable from WT mice (Fig. 6B). Together, these data indicate that the Ras pathway, although important for Nf1−/− PNS neuronal function, is not primarily responsible for the abnormalities observed in Nf1+/− CNS neurons.

MAPK pathway activation is not responsible for the abnormal Nf1+/− RGC neuronal phenotypes. A, No differences in the number or intensity of pMAPK+, NeuN+ Nf1+/− RGC neurons were observed compared to their WT counterparts (arrows). B, Treatment with the MEK inhibitor U0126 (10 μm) does not restore Nf1+/− RGC neuronal growth cone areas to WT levels (N = 24). C, Treatment of whole retina explants with U0126 (10 μm) is sufficient to reduce pMAPK levels. Error bars indicate SEM. Asterisks denote statistically significant differences (**p < 0.01).

Ras pathway hyperactivation is not responsible for the abnormal CNS Nf1+/− neuronal phenotypes. A, Treatment with the PI3-K inhibitor LY294002 (30 μm) did not restore Nf1+/− RGC neuronal growth cone areas to WT levels (p = 0.0001; N = 40). B, Expression of constitutively active KRasG12D in neurons had no effect on RGC growth cone area (N = 40). Error bars indicate SEM. Asterisks denote a statistically significant difference (**p < 0.01).
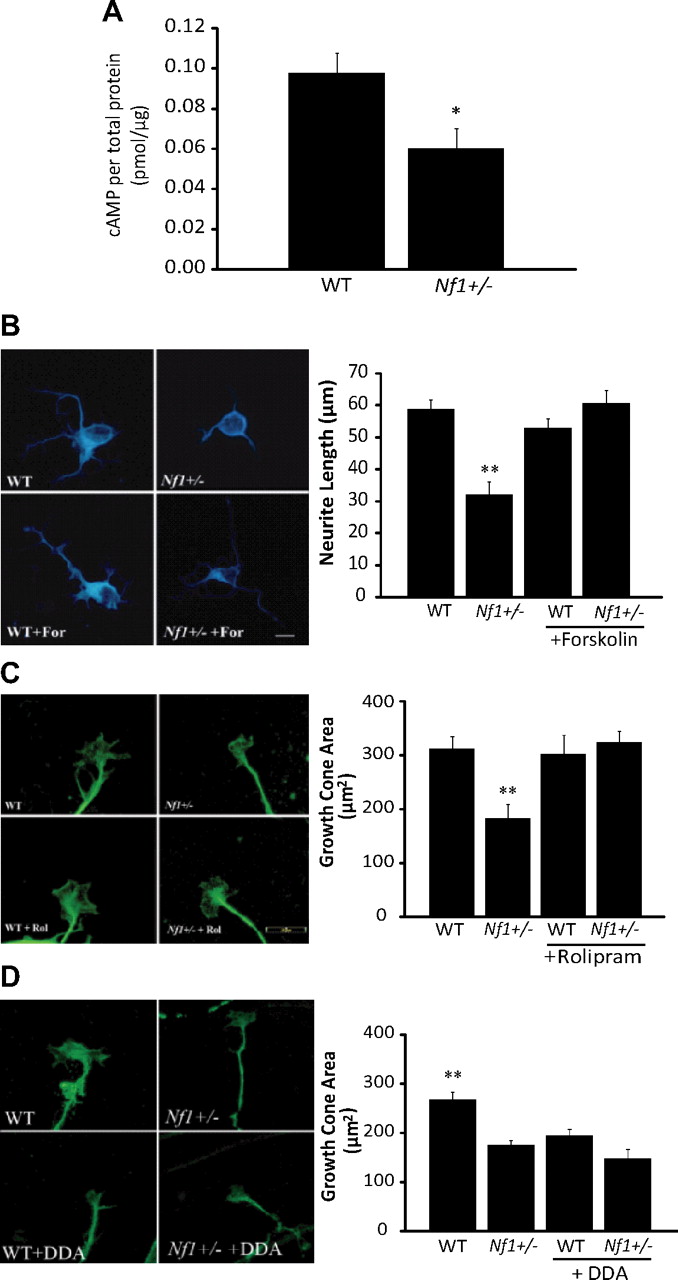
To determine whether cAMP might be responsible for the neuronal abnormalities observed in Nf1+/− RGC neurons, we first measured cAMP levels in whole retina preparations. We found that cAMP levels were significantly lower in Nf1+/− RGCs than in their WT counterparts (p = 0.02; N = 8) (Fig. 7A). Second, we restored cAMP levels using two methods: adenylyl cyclase activation (forskolin) (Fig. 7B, supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) and phosphodiesterase-4 inhibition (rolipram) (Fig. 7C). After either forskolin or rolipram treatment, Nf1+/− RGC neuronal growth cone areas and neurite lengths were restored to WT levels. Identical effects were observed using embryonic RGC neurons (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material) and hippocampal neurons (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). Third, lowering cAMP levels in WT RGC neurons using DDA (adenylyl cyclase inhibitor) resulted in reduced growth cone areas (Fig. 7D), similar to what we observed in Nf1+/− RGC neurons.

Nf1+/− neurite outgrowth and growth cone spreading defects are cAMP dependent. A, cAMP levels are significantly reduced in Nf1+/− whole retinal preparations compared to those from WT retinas (p = 0.02; N = 8). B, Forskolin (10 μm) treatment rescues Nf1+/− RGC neurite outgrowth lengths to WT levels (N = 40). Scale bar, 20 μm. C, Rolipram (200 μm) treatment of Nf1+/− retinal explant RGCs restores growth cone areas to WT levels (N = 40). D, Inhibition of adenylyl cyclase with 100 μm DDA reduces WT RGC explant growth cone areas to levels observed in Nf1+/− RGC neurons (N = 40). Error bars indicate SEM. Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01).
Next, to relate neurite length to apoptosis, we assayed cell survival after rolipram or forskolin treatment. Since neurite number has previously been correlated with cell survival (Oshitari et al., 2002), we examined the impact of cAMP-elevating treatments on RGC explant neurite number. As before, rolipram restored neurite numbers to WT levels (Fig. 8A). We then quantified the number of caspase-3-immunoreactive RGC neurons with one or more neurites to determine whether restoration of cAMP levels in Nf1+/− RGC neurons would increase the number of neurons early during the process of programmed cell death. Similarly, rolipram or forskolin treatment reduced the number of caspase-3-immunoreactive RGC neurons with one or more neurites to levels observed in WT RGC neurons (supplemental Fig. 1D,E, available at www.jneurosci.org as supplemental material). Finally, we directly assayed apoptosis using the TUNEL method. After treatment with either rolipram or forskolin, RGC neuronal death was reduced to WT levels (p = 0.0001) (Fig. 8B). Together, these data show that reduced cAMP levels are responsible for the abnormal phenotypes observed in Nf1+/− RGC neurons.

Rolipram treatment rescues Nf1+/− RGC death in vitro. A, Nf1+/− retinal explant neurons show reduced neurite numbers (normalized to the distance around the explant perimeter; p = 0.0035). Rolipram (200 μm) treatment restores Nf1+/− explant neurite numbers to WT levels. B, Forskolin (10 μm) or rolipram (200 μm) treatment reduces Nf1+/− retinal explant neuronal apoptosis (p = 0.0001). Error bars indicate SEM. Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01).
Nf1 heterozygosity predisposes to increased RGC apoptosis after optic nerve crush and optic glioma formation in vivo
We performed two experimental paradigms to examine the impact of Nf1 heterozygosity on RGC neuronal survival in the intact animal. First, we used a standard optic nerve crush injury model to determine whether Nf1 heterozygosity leads to increased RGC apoptosis. Three days after optic nerve crush, we determined the number of apoptotic cells in the retinal ganglion layer. Compared to WT mice, apoptotic RGC neuronal death in Nf1+/− mice was increased by 15% (p = 0.01) (Fig. 9A).

Nf1+/− RGC neuronal apoptosis is induced by optic nerve crush and optic glioma formation in vivo. A, Three days after optic nerve crush, Nf1+/− mice have greater numbers of TUNEL+ (green) and NeuN+ (red) neurons in the RGC layer (white arrows) than their WT counterparts (p = 0.01). B, The increased retinal ganglion layer neuronal apoptosis associated with optic glioma formation in Nf1+/−GFAPCKO mice (white arrows) is reduced by treatment with rolipram in vivo (p = 0.0001). C, The increased retinal ganglion layer neuronal apoptosis associated with optic nerve crush injury 6 d after injury in Nf1+/− mice (white arrows) is reduced by treatment with rolipram in vivo (p = 0.006). Error bars indicate SEM. Asterisks denote statistically significant differences (**p < 0.01).
Second, we used a novel GEM model of NF1-associated optic glioma (Bajenaru et al. 2003; Zhu et al. 2005): nearly 100% of these Nf1+/− mice lacking neurofibromin expression in GFAP+ cells (Nf1+/−GFAPCKO mice) develop gliomas affecting the prechiasmatic optic nerves and chiasm by 3 months of age. Moreover, Nf1+/−GFAPCKO mice exhibit axonal damage in the retro-orbital optic nerve proximal to the site of glioma formation and demonstrate RGC death (Hegedus et al. 2009) (Fig. 9B). No RGC neuronal apoptosis was observed in Nf1+/− mice or mice lacking Nf1 expression in GFAP+ cells (Hegedus et al. 2009).
Restoration of cAMP reverses Nf1+/− RGC neuronal apoptosis after optic glioma formation and optic nerve crush in vivo
Given the increased susceptibility of Nf1+/− RGCs to cell death in vitro and in vivo, we next sought to determine whether elevating cAMP levels in Nf1+/−GFAPCKO mice might reduce the RGC neuronal death resulting from optic glioma. After 30 d of rolipram treatment beginning at 2 months of age, RGC apoptosis was significantly attenuated (p = 0.0001) (Fig. 9B). No significant change in the total number of cells was seen (data not shown). These results demonstrate that RGC death in the setting of an optic glioma can be reduced by raising cAMP levels in vivo.
To extend these findings to another paradigm of neuronal injury, we treated Nf1+/− and WT mice with ether 200 μm rolipram or DMSO (vehicle) after optic nerve crush injury. Similar to Nf1+/−GFAP CKO mice with optic glioma, rolipram treatment reduced RGC neuronal apoptosis in Nf1+/− mice by 32% compared to vehicle-treated controls (p = 0.006) (Fig. 9C). Collectively, these results support a model in which Nf1+/− CNS neurons are susceptible to injury in a cAMP-dependent manner.
Discussion
Children with NF1 have neuronal abnormalities that primarily affect the CNS, including learning, memory, and behavioral problems as well as optic nerve dysfunction resulting from optic glioma formation. The predominance of symptoms within the CNS of children with NF1 suggests that neurofibromin is a critical regulator of CNS neuron function, and that CNS neurons might be uniquely sensitive to changes in NF1 gene expression. In this study, we use a combination of in vitro and in vivo experimental paradigms to demonstrate that CNS neuronal populations (hippocampal and retinal ganglion neurons), but not PNS dorsal root ganglion neurons, are dramatically affected by reduced Nf1 gene expression (Nf1 heterozygosity). Moreover, we show that the selective vulnerability of CNS neurons to Nf1 heterozygosity reflects impaired neurofibromin regulation of cAMP levels. These findings highlight the dramatic differences in cellular responses to Nf1 gene dosage and neurofibromin-regulated intracellular signaling, and provide new mechanistic insights into the selective vulnerability of CNS neurons to injury.
Whereas previous studies have shown that Nf1 heterozygosity can increase proliferation, motility, angiogenic properties, and differentiation in numerous nervous (Daginakatte and Gutmann, 2007; Kim et al., 1997; Bennett et al., 2003) and nonnervous (McDaniel et al., 2008; Ingram et al., 2000; Atit et al., 1999) system cell types, there is comparatively little information about the impact of reduced neurofibromin expression on neuronal function. This is particularly germane to children and adults with NF1 whose brains are composed of NF1+/− neurons. Previous investigations focused on PNS neurons as a model system and found no effect of Nf1 heterozygosity on DRG, nodose, trigeminal, or superior cervical ganglion neurons (Vogel et al., 1995). In contrast, unlike their PNS neuronal counterparts, we show that Nf1+/− hippocampal and retinal ganglion neurons have reduced neurite lengths, growth cone areas, and survival. Consistent with our finding that Nf1 heterozygosity in CNS neurons results in altered neuronal function, mice heterozygous for an inactivating Nf1 mutation in synapsin-I-expressing neurons in the brain exhibit learning and memory deficits (Cui et al., 2008).
In addition to the differences we found in sensitivity to Nf1 gene dosage, CNS and PNS neurons demonstrate diametrically opposed responses to complete Nf1 inactivation: Nf1-deficient DRG neurons survived in short-term culture without exogenous NGF, whereas WT and Nf1+/− DRG neurons died. Similarly, sensory neurons isolated from Nf1−/− embryos survived and extended neurites in the absence of neurotrophins, whereas wild-type or Nf1+/− neurons died rapidly unless NGF or BDNF was supplied in vitro (Vogel et al., 1995, 2000). In addition, Nf1−/− neurons in Nf1 conditional knock-out mice have increased neurite outgrowth and collateral branching after dorsal root injury in vivo (Romero et al., 2007). Moreover, in the present study, we showed that DRG neuronal apoptosis induced by oxidative stress was increased equivalently in Nf1+/− and WT neurons. In striking contrast to these findings in PNS neurons, we demonstrated in this report that Nf1+/− hippocampal and retinal ganglion neurons have reduced neurite extension, growth cone areas, and survival in response to several distinct injury conditions in vitro (oxidative and excitotoxic stress) and in vivo (optic glioma formation and optic nerve crush). These new observations are consistent with our earlier findings that Nf1-deficient neurons differentiated from neural stem cells have shorter neurites than their wild-type counterparts in vitro and in vivo (Hegedus et al., 2007).
Previous studies have shown that PNS neurite extension and survival is regulated by the Ras signaling pathway (Vogel et al., 2000; Aletsee et al., 2001). Neurofibromin functions in many cell types as a negative regulator of Ras activity (Basu et al., 1992; DeClue et al., 1992; Bollag et al., 1996; Hiatt et al., 2001; Dasgupta et al., 2005; Xu et al., 2007): Nf1 inactivation leads to increased Ras activity and deregulated signaling through the Ras-mediated Akt and MAPK effector proteins. In this regard, the increased neurite extension and survival observed in Nf1−/− PNS neurons could be reversed with neutralizing Ras antibodies (Vogel et al. 2000), consistent with earlier reports demonstrating that BDNF-induced nodose ganglion and NGF-induced DRG neuronal survival are blocked by Ras neutralizing antibodies (Borasio et al., 1993). Further examination of the responsible Ras effector pathways revealed that PI3-K, rather than ERK, activation was required for NGF-induced embryonic sensory neuron survival (Klesse and Parada, 1998). Similar dependence on PI3-K activation was also observed in mouse DRG neurons, such that inhibition with LY294002 blocked normal axonal growth cone morphology, neurite outgrowth, and survival (Edström and Ekström, 2003).
In CNS neurons, we found no increased Akt activation in Nf1+/− RGC neurons and no effect of MEK or PI3-K inhibition on RGC neurite outgrowth or survival. Similarly, RGC neurons expressing an activated KRas allele had normal growth cone areas. These findings agree with previous reports demonstrating that while Nf1 loss in brain neurons is associated with increased MAPK activation in vivo (Zhu et al., 2001; Hegedus et al., 2007), Ras pathway hyperactivation is not responsible for the reduced neurite lengths seen in Nf1−/− neurons. In a previous study, we demonstrated that expression of either an activated KRas (KRasG12D) or Akt (myr-Akt1) allele had no effect on neurite length in vitro or in vivo (Hegedus et al., 2007). Rather, the reduced neurite lengths observed in vitro and in vivo were reversed by agents that elevate intracellular cAMP levels (forskolin, rolipram), consistent with the role of neurofibromin as a positive regulator of cAMP in the brain (Tong et al., 2002; Dasgupta et al., 2003). In addition, replacement of the neurofibromin Ras-GAP domain in Nf1 mutant mice resulted in restoration of MAPK activity levels, but had no effect on the hypothalamic neuron dysfunction seen in these mice (Hegedus et al., 2008). Collectively, these findings further support our current observations in Nf1+/− RGC and hippocampal neurons that neurofibromin regulates CNS neurite lengths in a cAMP-dependent manner.
In contrast to our results demonstrating that cAMP is responsible for abnormal CNS neuronal function, a previous study showed that 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor treatment corrected the learning deficits and long-term potentiation abnormalities in Nf1+/− mice (Li et al., 2005). However, HMG-CoA reductase inhibitors affect secondary lipid modifications on numerous other proteins in addition to Ras. Although one target of isoprenylation is monomeric G-proteins (e.g., Ras), heterotrimeric G-proteins can also undergo isoprenylation. In this regard, atorvastatin can modulate β-adrenergic cAMP generation and signaling by isoprenylation of G-protein γ subunits (Mühlhäuser et al., 2006). In keeping with these findings, lovastatin has been shown to inhibit cAMP-stimulated chloride secretion (Ecay and Valentich 1993) and prolactin/growth hormone gene expression through a cAMP-dependent mechanism (Lasa et al., 1997). Similarly, simvastatin increases cAMP levels in a dose-dependent fashion, which can be abolished using the mevalonate metabolite geranylgeranyl pyrophosphate or the H-89 protein kinase A inhibitor (Maeda and Horiuchi 2009). Finally, atorvastatin and simvastatin can induce cerebellar granule cell death without affecting Akt or MAPK signaling (März et al., 2007). In light of these previous results and our current findings, it is possible that the observed effect of lovastatin on Nf1+/− mouse learning and memory reflects modulation of cAMP levels, rather than Ras activity, in CNS neurons.
One of the clinically important implications of the present report is the ability to significantly attenuate the Nf1+/− RGC neuronal death associated with optic glioma formation and optic nerve crush injury with rolipram. The sensitivity of RGC neurons to cAMP is underscored by numerous previous studies: RGC neuronal death caused by tetradotoxin can be reversed by elevating cAMP levels in vitro (Kaiser and Lipton 1990), whereas RGC neuronal survival and axonal regeneration after axotomy or optic nerve crush can be increased by the introduction of nonhydrolyzable cAMP analogs (Cui et al., 2003; Monsul et al., 2004). Our finding that Nf1+/− RGC neuron survival reflects deregulated cAMP signaling is intriguing in light of previous studies from our laboratory showing that Nf1 loss in astrocytes leads to inappropriate cell survival as a consequence of impaired cAMP generation (Warrington et al., 2007). In contrast to neurons, increased cAMP levels are associated with reduced astrocyte survival (Dugan et al., 1999; Warrington et al., 2007). After treatment with rolipram or forskolin, Nf1−/− astrocyte survival is reduced to wild-type levels in vitro. Moreover, rolipram treatment in vivo similarly results in reduced optic glioma proliferation (N. Warrington, S. Gianino, D. H. Gutmann, and J. B. Rubin, unpublished observation). The observation that cAMP-elevating drugs not only reduce optic glioma growth but also attenuate RGC neuronal apoptosis raises the exciting possibility that this therapeutic strategy may have both anticancer and neuroprotective effects in the management of children with NF1-associated optic glioma.
Footnotes
This work was supported in part by National Cancer Institute Grant U01-CA141549-01 to D.H.G. This work was also supported by the Bakewell Neuroimaging Core and National Institutes of Health Neuroscience Blueprint Interdisciplinary Center Core Grant P30 (NS057105 to Washington University). We appreciate the expert technical assistance provided by Ryan Emnett and Crystal White.
- Correspondence should be addressed to Dr. David H. Gutmann, Department of Neurology, Washington University School of Medicine, Box 8111, 660 South Euclid Avenue, St. Louis, MO 63110. gutmannd{at}neuro.wustl.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}