

Abstract
α-Synuclein is central in Parkinson's disease pathogenesis. Although initially α-synuclein was considered a purely intracellular protein, recent data suggest that it can be detected in the plasma and CSF of humans and in the culture media of neuronal cells. To address a role of secreted α-synuclein in neuronal homeostasis, we have generated wild-type α-synuclein and β-galactosidase inducible SH-SY5Y cells. Soluble oligomeric and monomeric species of α-synuclein are readily detected in the conditioned media (CM) of these cells at concentrations similar to those observed in human CSF. We have found that, in this model, α-synuclein is secreted by externalized vesicles in a calcium-dependent manner. Electron microscopy and liquid chromatography–mass spectrometry proteomic analysis demonstrate that these vesicles have the characteristic hallmarks of exosomes, secreted intraluminar vesicles of multivesicular bodies. Application of CM containing secreted α-synuclein causes cell death of recipient neuronal cells, which can be reversed after α-synuclein immunodepletion from the CM. High- and low-molecular-weight α-synuclein species, isolated from this CM, significantly decrease cell viability. Importantly, treatment of the CM with oligomer-interfering compounds before application rescues the recipient neuronal cells from the observed toxicity. Our results show for the first time that cell-produced α-synuclein is secreted via an exosomal, calcium-dependent mechanism and suggest that α-synuclein secretion serves to amplify and propagate Parkinson's disease-related pathology.
Introduction
Genetic and biochemical data indicate that an increase in the levels of expression of the wild-type (WT) α-synuclein protein is sufficient to cause neurodegeneration in Parkinson's disease (PD) (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004). The aberrant function of α-synuclein is not understood, although there is evidence that abnormal folding and aggregation may play a role and that the toxic α-synuclein species may be oligomeric intermediates (Conway et al., 2000, 2001; Goldberg and Lansbury, 2000; Olanow et al., 2004; Vekrellis et al., 2004). Until recently, α-synuclein was considered to exert its pathogenic effects intracellularly. However, El-Agnaf et al. (2003, 2006) showed that α-synuclein species can be detected in human plasma and CSF and that it could be secreted into the medium of cultured neuronal cells. In another report, monomeric and oligomeric α-synuclein were shown to be secreted from differentiated human neuroblastoma cells and primary cortical neurons (Lee et al., 2005). Using a similar model, Sung et al. (2005) demonstrated that secreted α-synuclein from SK-N-BE cells reduces the viability of these cells and can be cleaved by matrix metalloproteases. These results suggest that the pathogenic actions of α-synuclein extend to the extracellular space and neighboring cells. In support for this hypothesis, exogenously added recombinant α-synuclein to cell culture medium can be internalized by the recipient cells (Sung et al., 2001; Ahn et al., 2006; Lee et al., 2008; Luk et al., 2009) and cause cell death (Du et al., 2003; Albani et al., 2004; Zhang et al., 2005). Such studies have used very high concentrations of recombinant α-synuclein and cationic liposomes to assist its uptake.
The pathophysiological role of the secreted α-synuclein forms remains essentially unknown. In one study (Zhang et al., 2005), it was demonstrated that the interaction between recombinant α-synuclein and microglia could activate these cells, which in turn induces neurotoxicity. It has also been suggested that α-synuclein aggregates can transmit pathology via neuron-to-neuron interactions (Desplats et al., 2009). The secretion of α-synuclein has been reported to be insensitive to brefeldin A (BFA) (Lee et al., 2005), suggesting that it is secreted via an endoplasmic reticulum (ER)/Golgi-independent, pathway. Electron microscopy and density gradient ultracentrifugation suggested that the vesicles containing α-synuclein have morphologies and sedimentation properties similar to the dense core vesicles (Lee et al., 2005), but their exact identities remain unknown. To investigate the underlying mechanisms of neurotoxicity caused by cell-secreted α-synuclein species, we have generated WT α-synuclein–Tet-off inducible SH-SY5Y cells. We show that α-synuclein is associated with externalized membrane vesicles, suggestive of a vesicle-based exporting mechanism. Our results show that different species of naturally secreted α-synuclein cause cell death to neuronal cells. Immunodepletion of α-synuclein from the medium rescues this death. Finally, we demonstrate that treatment of the conditioned medium (CM) with oligomer-disrupting compounds reduces the secreted α-synuclein-related cytotoxicity.
Materials and Methods
Reagents.
All reagents were obtained from Sigma unless otherwise specified. Doxycycline (Dox) was purchased from Clontech. Recombinant α-synuclein was from Millipore Bioscience Research Reagents. Cytochalasin D (CytoD) and protein G agarose beads were purchased from Calbiochem.
Cell culture.
The generation of the stable SH-SY5Y cell lines inducibly expressing WT α-synuclein was described previously (Vekrellis et al., 2009). SH-SY5Y cells were cultured in RPMI 1640 medium containing 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mm l-glutamine. Cells overexpressing either WT α-synuclein or β-galactosidase (bGAL) were cultured in the presence of 250 μg/ml G418 and 50 μg/ml hygromycin B. α-Synuclein expression was switched off by the addition of doxycycline (0.5 μg/ml). Stock cultures were kept in the presence of doxycycline. Neuronal differentiation was performed with the addition of 10 μm all-trans retinoic acid for 5 d. Cultures of rat (embryonic day 18) cortical neurons were prepared as described previously (Vogiatzi et al., 2008). Dissociated cells were plated onto poly-d-lysine-coated 12-well dishes at a density of ∼250.000/cm2. Cells were maintained in Neurobasal medium (Invitrogen), with B27 serum-free supplements (Invitrogen), 0.5 mm l-glutamine, and penicillin/streptomycin (as above). More than 98% of the cells cultured under these conditions represent postmitotic neurons (Rideout et al., 2001). Mouse BV2 microglia cells were cultured in RPMI 1640 medium containing 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mm l-glutamine.
Preparation of conditioned medium and cell extract.
SH-SY5Y cells inducibly expressing WT α-synuclein were cultured in 140-mm-diameter dishes in RPMI 1640 medium containing 10% FBS in the presence or absence of doxycycline (0.5 μg/ml) until 70–80% confluent. The medium was then replaced with RPMI 1640 medium containing 2% FBS. After 48 h, the culture supernatant (CM) was collected and centrifuged at 4000 × g for 10 min at 4°C to remove cell debris. For Western blotting and size exclusion chromatography (SEC), the CM was concentrated using 3 kDa cutoff Amicon Ultra filters (Millipore). For toxicity assays, the CM was used without concentration. In some cases, CM was treated with 0.4 μm of either Congo Red (CR) or scyllo-inositol (SI) for 4 h at 4°C before its addition to recipient cells.
For extraction of cellular proteins, cells were harvested, washed twice with ice-cold PBS, and lysed with STEN lysis buffer (50 mm Tris, pH 7.6, 150 mm NaCl, 0.1% SDS, 1% NP-40, and 2 mm EDTA) on ice for 15 min. Protein content was estimated using the Bradford method (Bio-Rad).
Isolation of externalized membrane vesicles.
The isolation of the externalized membrane vesicles was performed as described previously (Tanudji et al., 2002). Briefly, the CM was first centrifuged at 4000 × g for 10 min at 4°C to remove dead cells and debris, and the supernatant was further centrifuged at 100,000 × g for 2 h at 4°C. The supernatant (S100) was collected, and the pellet (P100) containing the externalized vesicles was reconstituted in 25 μl of radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% NP-40, 0.5% Na-deoxycholate, and 0.1% SDS).
Preparation of exosome-depleted medium.
The depletion of the medium from bovine serum-derived exosomes was performed as described previously (Théry et al., 2006). Briefly, RPMI 1640 medium containing 20% FBS, penicillin/streptomycin, and l-glutamine was centrifuged at 100,000 × g for 16 h at 4°C. The supernatant was carefully removed and sterilized by filtering through a 0.2 μm filter (Whatman) and stored at 4°C until additional use in exosome preparation.
Purification of exosomes.
Exosomes were prepared as described previously (Théry et al., 2006). Briefly, SH-SY5Y cells were cultured in two 140 mm dishes in 10% FBS. At 48 h before the exosome preparation, culture medium was replaced with exosome-depleted medium diluted 10-fold with RPMI 1640 medium containing only penicillin/streptomycin and l-glutamine. Culture supernatants of cells grown for 48 h were collected and spun at 300 × g for 10 min to remove cells. The supernatants were then sequentially centrifuged at 2000 × g for 10 min, 10,000 × g for 30 min, and 100,000 × g for 90 min. The pellet (P100) containing exosomes was washed once with cold PBS and centrifuged again at 100,000 × g for 90 min. P100 was resuspended in 50 μl of RIPA buffer. All centrifugations were performed at 4°C.
Electron microscopy.
Externalized membrane vesicles were isolated from 15 ml of CM, and electron microscopy was performed as described previously (Théry et al., 2006) with some modifications. Briefly, vesicles were fixed with 2% paraformaldehyde in 100 mm Na2HPO4, applied to a Formvar-coated copper grid, and negatively stained with 2% uranyl acetate. Microphotographs were obtained using a Phillips EM 300 electron microscope.
Mass spectrometric protein analyses.
The proteins were extracted from the exosomes using an acetone–TCA precipitation procedure followed by washes with acetonitrile and tetrahydrofuran. The resulting protein pellets were subjected to reduction, alkylation, and in-solution phase proteolysis with trypsin as described previously (Kouri et al., 2009). The resulting tryptic digest was analyzed with an Agilent 1200 nano-liquid chromatography system interfaced with an Agilent 6330 MSD ion trap mass spectrometer system retrofitted to a nanoelectrospray ionization source [liquid chromatography–mass spectrometry (LC-MS)]. The capillary column used for the peptide separation was the Agilent C18 Zorbax at 1.8 μm particle size, 75 μm inner diameter, and 150 mm length. Before the analyses, the LC-MS system was tuned and calibrated to meet the specifications of the manufacturer for mass accuracy and detection sensitivity. The obtained product ion MS2 peptide spectra were searched against the Swiss Prot nonredundant sequence database. The search engine used was Protein Pilot (version 2.0.1) with the ESI-Trap instrument option, Search Effort at Thorough ID, and a detection protein threshold >1.3 (>95% confidence).
Treatment of vesicles with Na2CO3.
To remove non-integral membrane proteins, exosomes were treated with Na2CO3 100 mm, pH 11, for 30 min at 4°C (Fujiki et al., 1982). After centrifugation at 50,000 × g, integral exosomal membrane proteins were recovered in the pellet fraction, whereas the exosomal lumen proteins remained in the soluble fraction.
35S radiolabeling.
SH-SY5Y cells inducibly expressing WT α-synuclein were cultured in 140-mm-diameter dishes in RPMI 1640 medium containing 10% FBS in the presence or absence of Dox until 70–80% confluent. Cells were starved from methionine and cysteine for 15 min at 37°C. After starvation, 2.5 mCi of 35S-Protein Labeling mix (PerkinElmer Life and Analytical Sciences) were added for 3 h. Cells were rinsed twice with plain RPMI 1640 medium and grown overnight in fresh unlabeled culture medium with excess of l-methionine and l-cysteine (10 mm final concentration). The CM, containing radiolabeled secreted proteins, was collected, cleared from cell debris, concentrated, and analyzed by immunoblotting. To detect intracellular radiolabeled proteins, cells were first washed for 2 min with trypsin–EDTA to remove non-integral membrane-bound proteins. After centrifugation at 400 × g for 5 min, cell pellets were lysed in STEN lysis buffer and used for immunoprecipitation.
Immunoprecipitation of labeled α-synuclein.
Cell lysates were precleared with protein-G agarose beads for 1 h at 4°C. Agarose beads were removed by centrifugation at 1000 × g for 5 min at 4°C, and anti-α-synuclein antibody (Syn-1, mouse monoclonal, 5 μg/mg protein; BD Biosciences) was added for 6 h at 4°C by rotation. At the end of this period, protein-G agarose beads were added and the mixture was incubated overnight at 4°C by rotation. Labeled α-synuclein was detected by gel autoradiography.
Immunodepletion of CM.
Conditioned medium (7 ml) was immunodepleted of α-synuclein by immunoprecipitation with anti-α-synuclein antibody (0.5 μg/ml CM) as described above. Immunodepleted CM was collected and sterilized by filtering through a 0.2 μm filter (Whatman) before being applied on recipient cells. Control immunodepletion was performed with either anti-c-myc antibody (mouse monoclonal; Santa Cruz Biotechnology) or protein-G agarose alone.
Uptake of radiolabeled proteins.
Cells were treated overnight with CM containing radiolabeled secreted proteins (see above). After 2 min treatment with trypsin–EDTA to remove nonspecific surface protein binding, cells were examined for the internalization of labeled α-synuclein by immunoprecipitation and gel autoradiography.
Subcellular fractionation.
After overnight application of CM containing 35S-labeled secreted proteins, recipient cells were treated with trypsin–EDTA, washed once with PBS, and centrifuged at 400 × g for 5 min at 4°C. Cell pellet was resuspended in a detergent-free buffer containing 0.25 m sucrose, 10 mm HEPES, pH 7.4, 1 mm EDTA, 5 mm MgCl2, and 1× cocktail protease inhibitors (Roche). Homogenization was achieved first by four freeze–thaw cycles, followed by three sets of 15 bounces of a Teflon homogenizer. Finally, the homogenate was passed a few times through an insulin syringe. For nuclei sedimentation, the homogenate was centrifuged at 2000 × g for 15 min at 4°C. To isolate microsomes, the supernatant was further centrifuged at 100,000 × g for 2 h at 4°C. Pellets were washed twice with PBS containing protease inhibitors and reconstituted in RIPA buffer. Nuclear, microsomal, and cytosolic fractions were finally analyzed for the presence of internalized α-synuclein by immunoprecipitation and autoradiography.
Toxicity assays.
Toxicity was assessed by using the LIVE/DEAD viability/cytotoxicity kit (Invitrogen). Briefly, cells were stained by 2 μm calcein-AM, which labels living cells, and 1 μm ethidium homodimer (EthD-1), which labels dead cells, for 30 min. Hoechst 33258 dye was used at the same time for total nuclei staining. After staining, cells were washed once with normal RPMI 1640 medium and immediately visualized on a Leica DMIRB inverted fluorescence microscope. Images were captured under 20× magnification with a Leica DFC-350FX digital cooled CCD camera using the LAS AF software. Toxicity was defined in each image as the percentage of dead cells versus the total number of cells, counting at least 150 cells per image.
Size exclusion chromatography.
For SEC, 8–9 mg of protein of concentrated CM diluted in PBS were injected into a Superdex 200 10/300 GL column (GE Healthcare) previously equilibrated with PBS. Elution was performed at a constant flow rate of 0.25 ml/min in PBS. The 250 μl fractions were collected and analyzed by Western immunoblotting for the presence of α-synuclein. High-molecular-weight (HMW) and low-molecular-weight (LMW) fractions containing α-synuclein were separately pooled and concentrated using 3 kDa cutoff filters (Millipore). PBS was exchanged to 50 mm ammonium acetate buffer, pH 7.5, by means of ultrafiltration, and the samples were further lyophilized to dryness. Finally, samples were reconstituted in 250 μl of RPMI 1640 medium containing 10% FBS, sterilized by filtering, and applied to recipient cells in 2× concentration.
Western immunoblotting.
Denaturing gel electrophoresis was performed in 12% SDS-PAGE gels in Tris–glycine buffer. Immunoblotting was performed using the following antibodies: anti-α-synuclein (rabbit polyclonal from Santa Cruz Biotechnology; C-20 or Syn-1 monoclonal from BD Biosciences), anti-β-actin (mouse monoclonal; Sigma), anti-γ-tubulin (mouse monoclonal; Sigma), anti-BSA (mouse monoclonal; Antibody Shop), anti-Alix (mouse monoclonal; Santa Cruz Biotechnology), anti-heat shock protein 70 (Hsp70) (mouse monoclonal; Stressgen), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (mouse monoclonal; Sigma), anti-14-3-3β (rabbit polyclonal; Santa Cruz Biotechnology), anti-extracellular signal-regulated kinase (ERK) (rabbit polyclonal; Santa Cruz Biotechnology), anti-ubiquitin (mouse monoclonal; Millipore Bioscience Research Reagents), anti-cofilin (goat polyclonal; Santa Cruz Biotechnology), anti-flotillin-1 (mouse monoclonal; Santa Cruz Biotechnology), anti-annexin II (rabbit polyclonal; Santa Cruz Biotechnology), anti-c-myc (mouse monoclonal; Sigma), anti-cleaved caspase-3 (rabbit monoclonal; Cell Signaling Technology), and anti-19S Rpt6 proteasome subunit (mouse monoclonal; Biomol). All immunoblots represent one of at least three experiments. Quantification of bands on Western immunoblots was performed using Gel Analyser software (Biosure). Differences in protein expression levels were quantified after standardization of all values using the appropriate loading controls (γ-tubulin, β-actin, ERK, and BSA). All statistical analyses were performed using the Student's t test, and p values of <0.05 were considered significant.
Immunocytochemistry and confocal microscopy.
For confocal microscopy, cells grown on glass coverslips were fixed with 4% paraformaldehyde for 15 min. After PBS washes, cells were blocked with 10% normal goat serum (NGS) in PBS. Primary anti-GM130 antibody (mouse monoclonal, 1:100; BD Biosciences Transduction Laboratories) was applied for 16 h in PBS containing 2% NGS and 0.1% Triton X-100. Mouse cyanine 3 (Cy3) secondary antibody (Jackson ImmunoResearch) was added for 1 h in PBS/Triton X-100/NGS. Alternatively, cells were stained for 16 h with Alexa 633–phalloidin (1:500; Invitrogen). Cell nuclei were stained with Hoechst 33258 dye. Coverslips were mounted in Mowiol mounting media and visualized under 63× magnification using a Leica TCS SP5 confocal microscope combined with dual (tandem) scanner.
Results
α-Synuclein is constitutively and physiologically secreted
In our SH-SY5Y cell system, the expression of WT α-synuclein is tightly regulated through the Tet-off regulatory system (Vekrellis et al., 2009). To investigate whether α-synuclein is released from our cells, we induced α-synuclein expression by culturing cells in the absence of Dox for 8 d and examined the culture medium (CM) for the presence of secreted α-synuclein (Fig. 1A). CM was concentrated and Western immunoblotted with Syn-1 antibody. Secreted monomeric (Fig. 1A) and oligomeric (Fig. 1B) α-synuclein was clearly detected in the CM and accumulated over time (Fig. 1A, right). This was suggestive of α-synuclein being secreted by SH-SY5Y cells in a constitutive manner (Fig. 1C). We confirmed that the presence of α-synuclein in the CM was not attributable to cell death by measuring the percentage cell death before CM collection. Using the LIVE/DEAD Cytotoxicity kit, cell death was <5% in cells expressing or not expressing α-synuclein (percentage cell death, 4.1 ± 0.9 or 3.8 ± 0.3% for WT-positive and WT-negative cells, respectively, mean ± SD; n = 3). We further confirmed the physiologic secretion of α-synuclein by analyzing the CM for the presence of cytosolic proteins, such as ubiquitin (Fig. 1A), protein kinase Cα (PKCα), and 19S Rpt6 proteasome subunit (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Both poly-ubiquitinated proteins and monomeric ubiquitin were absent in the CM of SH-SY5Y cells (Fig. 1A). We also examined the CM from SH-SY5Y cells overexpressing bGAL in a similar manner. No bGAL was detected in the CM of these cells (data not shown), arguing that overexpression alone was not sufficient to lead to α-synuclein release. In agreement with a physiological mode of release, secreted α-synuclein was detected in low levels in control (+Dox) SH-SY5Y cells (see Figs. 1B, 3A,B) (supplemental Figs. S1A, available at www.jneurosci.org as supplemental material). We also quantified the levels of α-synuclein in the CM after 48 h using recombinant α-synuclein as standard. The concentration of secreted α-synuclein was estimated to be 2–12 nm, which resembles the levels so far detected in biological fluids (data not shown) (Borghi et al., 2000; El-Agnaf et al., 2003, 2006).

α-Synuclein is constitutively released by a nonclassical secretory pathway. A, SH-SY5Y cells were cultured in the presence or absence of Dox for 8 d in 10% FBS. After that period, the culture medium was replaced with 2% FBS medium for the indicated times. Cell lysates (CL; left) and concentrated CM (right) were analyzed by Western immunoblotting with antibodies to α-synuclein or ubiquitin (Ubi). ERK and BSA levels were used as loading controls. B, SH-SY5Y cells were cultured as in A, and the CM was collected after 48 h and analyzed by Western immunoblotting with C-20 antibody. C, Representative plot showing the levels of secreted α-synuclein, estimated as the percentage ratio of the extracellular α-synuclein versus the intracellular α-synuclein, over time. Each point represents the mean ± SD of three measurements. D, α-Synuclein-expressing cells were treated with 2 μg/ml BFA (+) or without (−) for 6 h. The levels of intracellular α-synuclein (CL) and extracellular α-synuclein (CM) were assessed by Western immunoblotting. ERK and BSA were used as loading controls.
Having shown that our cells physiologically secrete α-synuclein, we went on to investigate the manner of this secretion. It has been suggested previously that α-synuclein exocytosis is mediated by a nonclassical, BFA-independent, secretory mechanism (Lee et al., 2005). Treatment of SH-SY5Y cells with 2 μg/ml BFA, an effective inhibitor of the classical ER/Golgi-dependent pathway, for 6 h resulted in a robust disassembly of the Golgi complex (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). BFA-induced disruption of the classical secretory pathway did not alter the levels of secreted α-synuclein (percentage secreted α-synuclein, 3.5 ± 0.3 and 2.1 ± 1.1% for CTL untreated and BFA-treated cells, respectively, mean ± SD; n = 3), suggesting that α-synuclein indeed follows an unconventional secretory pathway (Figs. 1D, 2B).

α-Synuclein is released by a calcium-dependent endocytic mechanism. A, Representative immunoblots showing the levels of intracellular and extracellular α-synuclein after no treatment (−) or a 6 h treatment (+) with compounds that either affect Ca2+ concentration or impair the endosomal pathway. Actin and BSA levels were used as loading controls. B, Graph summarizing the effect of various chemical compounds on α-synuclein secretion after the 6 h treatments. Data are shown as mean ± SD (n = 3). *p < 0.05, statistically significant differences (independent t test). C, α-Synuclein-expressing cells were treated with various chemical compounds for 6 h. Cell viability was assessed with EthD-1/Hoechst staining. Data are presented as mean ± SD (n = 3) and statistically analyzed by an independent t test. D, α-Synuclein-expressing cells were treated for 6 h with vehicle, PSI (10 nm), H2O2 (0.5 μm), or MPP+ (0.25 mm). After treatment, cell homogenates and concentrated CM were analyzed by Western immunoblotting using C-20 antibody. Actin and BSA were used as loading controls. One representative immunoblot from each treatment is shown. The graph is summarizing the results of three independent experiments (n = 3; mean ± SD, one-way ANOVA test followed by Tukey's test). Stauro, Staurosporin; Thapsi, thapsigargin; Meth, methionine; Iono, ionomycin.
Externalization of α-synuclein is sensitive to changes in temperature and serum content
Facilitated protein secretion, such as classical ER/Golgi transport, is generally a temperature-dependent process (Saraste et al., 1986). However, passive diffusion through a pore in the cell membrane is not affected by changes in temperature (Melchior and Gerace, 1995). In addition, heat shock conditions can induce the release of some proteins exported via leaderless secretory pathways (Rubartelli et al., 1990; Jackson et al., 1992; Lindstedt et al., 1993). The effect of heat shock on α-synuclein export was tested by culturing α-synuclein-expressing cells at 40°C for 6 h. Under these conditions, an upregulation of Hsp70 was observed (supplemental Fig. S1C, top, available at www.jneurosci.org as supplemental material), whereas cell viability remained unchanged (data not shown). CM from CTL and heat-shocked cells was concentrated and subjected to SDS-PAGE and immunoblotting analysis for the presence of secreted α-synuclein. The release of α-synuclein was greatly increased under heat shock conditions (percentage secreted α-synuclein, 3.4 ± 0.4 and 11.6 ± 3.0% for CTL and heat-shocked cells, respectively, mean ± SD; n = 3) (supplemental Fig. S1C, top, available at www.jneurosci.org as supplemental material) but not in lower temperatures such as 25°C (supplemental Fig. S1C, bottom, available at www.jneurosci.org as supplemental material). These results suggest that the externalization of α-synuclein does not occur through passive diffusion across the plasma membrane.
It has been reported that unconventional protein secretion is affected by factors present in serum (Chang et al., 1997; Tanudji et al., 2002). To examine this possibility, α-synuclein-expressing cells were cultured in medium containing 0, 2, and 5% FBS for 6 h. Quantification of cell death verified that, under these conditions, serum deprivation did not affect cell viability (percentage cell death, 2.2 ± 0.6, 2.8 ± 0.2, 2.3 ± 0.6, and 3.2 ± 0.3% for 0, 2, 5, and 10%, respectively, mean ± SD; n = 3). However, the release of α-synuclein observed was inversely related to the serum content (percentage secreted α-synuclein, 2.8 ± 0.4 and 21.7 ± 1.5% for 2 and 0% FBS, respectively, mean ± SD; n = 3) (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material).
α-Synuclein is released by a calcium-dependent endosomal mechanism
To dissect the secretory mechanism of α-synuclein in more detail, we tested whether the levels of extracellular α-synuclein were altered after treatment with chemical compounds that have been reported previously to influence the nonclassical secretion of other proteins. Because the importance of the actin network on endocytosis/exocytosis and vesicle trafficking has been well established (Lanzetti, 2007), we first investigated the role of actin cytoskeleton on α-synuclein secretion. For this purpose, cells expressing α-synuclein were treated for 6 h with 2 μm of the actin polymerization inhibitor CytoD (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). The CM from CTL and CytoD-treated cells were concentrated and analyzed by SDS-PAGE and immunoblotting for secreted α-synuclein. Actin depolymerization did not affect α-synuclein externalization to a significant extent (percentage secreted α-synuclein, 2.5 ± 1.7 and 2.9 ± 1.1% for CTL and CytoD-treated cells, mean ± SD; n = 3) (Fig. 2B). Under these conditions, cell viability was not compromised (Fig. 2C).
Protein kinases play a prominent role in signal transduction pathways that could facilitate protein trafficking, including unconventional secretion (Seino and Shibasaki, 2005). We have used staurosporin, a nonselective, broad kinase inhibitor (Klinkspoor et al., 1996; Smolian et al., 2001), to test whether protein kinases are involved in the unconventional secretion of α-synuclein. α-Synuclein-expressing cells were treated for 6 h with 100 nm staurosporin, and the CM were collected and concentrated. This treatment inhibited the phosphorylation of p-MARCKS (myristoylated alanine-rich protein kinase C substrate), a specific substrate of PKC, without affecting the levels of the enzyme (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Again, cell viability was not compromised (Fig. 2C). Protein kinase inhibition did not change the levels of extracellular α-synuclein (percentage secreted α-synuclein, 2.5 ± 0.8 and 3.0 ± 1.4% for CTL and staurosporin-treated cells, mean ± SD; n = 3), indicating that protein kinases do not mediate α-synuclein export (Fig. 2B).
Calcium is fundamental in regulating exocytosis, early endosome fusion, and exosome-mediated release (Mills et al., 2001; Savina et al., 2003; Barclay et al., 2005). To determine whether changes in Ca2+ levels modulate α-synuclein secretion, we first treated cells with 0.5 mm EDTA for 6 h, and the CM were collected and concentrated. Secreted α-synuclein levels were assessed by SDS-PAGE and immunoblotting. Removal of extracellular Ca2+ did not change the levels of secreted α-synuclein (percentage secreted α-synuclein, 3.8 ± 1.7 and 3.1 ± 0.5% for CTL and EDTA-treated cells, mean ± SD; n = 3) (Fig. 2A,B). However, when cells were treated with 100 nm thapsigargin (Thastrup et al., 1990), which raises cytosolic Ca2+ concentration, we observed a significant increase in the levels of secreted α-synuclein (percentage secreted α-synuclein, 2.6 ± 0.3 and 7.7 ± 2.2% for CTL and thapsigargin-treated cells, mean ± SD; n = 3) (Fig. 2A,B). This result was confirmed using the calcium ionophore ionomycin. Treatment of α-synuclein-expressing cells with 100 nm ionomycin for 6 h led to a significant increase in the levels of extracellular α-synuclein (percentage secreted α-synuclein, 3.1 ± 0.8 and 6.6 ± 0.9% for CTL and ionomycin-treated cells, mean ± SD; n = 3) (Fig. 2A,B). As expected, ionomycin treatment also increased the levels of secreted oligomeric α-synuclein (supplemental Fig S2C, available at www.jneurosci.org as supplemental material). To further verify that intracellular Ca2+ is important for α-synuclein secretion, we used the membrane-permeable Ca2+-chelator BAPTA-AM. Treatment with 2 μm BAPTA-AM for 6 h decreased the levels of secreted α-synuclein, further supporting the idea that intracellular Ca2+ acts as a key regulator of α-synuclein export (percentage secreted α-synuclein, 3.4 ± 0.4 and 1.8 ± 0.5% for CTL and BAPTA-AM-treated cells, mean ± SD; n = 3) (Fig. 2A,B). It should be noted that all compounds used for manipulation of Ca2+ concentration did not affect cell viability (Fig. 2C).
We next investigated whether the endocytic pathway is implicated in the process of α-synuclein externalization. Weak bases have been shown to prevent endosomal/lysosomal acidification, ultimately resulting in osmotic swelling and vacuolation of lysosomes (Poole and Ohkuma, 1981). We have initially used the lysosomotropic amine methylamine, to impair the lysosomal pathway (Lindstedt et al., 1993; Geetha and Wooten, 2008). We found that blockage of the lysosomal pathway by 2 mm methylamine caused a profound increase in the levels of secreted α-synuclein (percentage secreted α-synuclein, 3.4 ± 0.4 and 10.8 ± 2.4% for CTL and methylamine-treated cells, mean ± SD; n = 3) (Fig. 2A,B) without affecting cell viability (Fig. 2C). This indicated that the endocytic pathway could be involved in α-synuclein secretion. We verified this finding by using chloroquine (CQ), which potentially perturbs membrane trafficking from endosomes to lysosomes (Andrei et al., 1999; Savina et al., 2003; Yuyama et al., 2008). Treatment with 25 μm CQ for 6 h, which resulted in obvious vacuolation (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material) but not cell death (Fig. 2C), induced a significant increase in the levels of secreted α-synuclein, although the increase was not as profound as with methylamine (percentage secreted α-synuclein, 3.6 ± 0.5 and 5.0 ± 0.5% for CTL and CQ-treated cells, mean ± SD; n = 3) (Fig. 2A,B). We concluded that the endocytic pathway is involved in α-synuclein secretion.
Cellular stressors do not lead to alterations of α-synuclein secretion
Oxidative stress has been shown to increase α-synuclein levels and apoptosis in a variety of cell models (Sherer et al., 2002). To examine the effect of oxidative stress on α-synuclein secretion, we treated α-synuclein-expressing cells with 0.5 μm H2O2 and 0.25 mm 1-methyl-4-phenylpyridinium (MPP+) for 6 h and assessed extracellular α-synuclein levels by immunoblotting using the C-20 antibody. Our results suggest that oxidative stress does not affect the levels of secreted α-synuclein in our cell system (percentage secreted α-synuclein, 2.44 ± 0.63, 3.48 ± 0.52, and 2.28 ± 0.44% for CTL, H2O2-treated, and MPP+-treated cells, mean ± SD; n = 3) (Fig. 2D). In another study (Lee et al., 2005), it was demonstrated that proteasome inhibition increases intracellular and extracellular α-synuclein aggregates. To address a role of proteasomal function in α-synuclein secretion, we treated α-synuclein-expressing cells with the potent proteasome inhibitor I (PSI) (10 nm) for 6 h. Immunoblotting of the concentrated CM with the C-20 antibody revealed that proteasome inhibition did not alter extracellular α-synuclein levels (percentage secreted α-synuclein, 2.44 ± 0.63 and 2.86 ± 0.46% for CTL and PSI-treated cells, mean ± SD; n = 3) (Fig. 2D).
In these studies, we ensured that the levels of the pharmacological stressors used did not lead to cytotoxicity (Fig. 2C), so as to avoid the confounding factor of leakage of α-synuclein from dying cells. We cannot therefore exclude the possibility that higher levels of these stressors could impact, in addition to viability, α-synuclein secretion.
α-Synuclein is exported in association with membrane vesicles that have the hallmarks of exosomes
We addressed the question of whether the nonclassical α-synuclein secretion occurred via externalized membrane vesicles (Cooper and Barondes, 1990; Denzer et al., 2000; van Niel et al., 2006). Extracellular membrane vesicles were precipitated from the CM of α-synuclein-expressing and nonexpressing CTL cells by centrifugation at 100,000 × g. The supernatant (S100) and the pellet (P100) were analyzed by SDS-PAGE and immunoblotting for the presence of α-synuclein. α-Synuclein was readily detected in both the soluble and the membrane fraction of the CM, suggesting that α-synuclein is secreted, at least in part, in association with externalized membrane vesicles (Fig. 3A). Interestingly, oligomeric forms of α-synuclein were also detected in the vesicle P100 fraction (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). To ensure that the presence of doxycycline itself or expression of α-synuclein does not interfere with the generation of these externalized vesicles, we analyzed whole P100 pellets from the CM of α-synuclein expressing (−dox) and nonexpressing CTL (+dox) cells by highly sensitive Coomasie blue staining and found that protein patterns were identical (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material).

α-Synuclein exocytosis is mediated by membrane vesicles that resemble exosomes. A, α-Synuclein-expressing cells and nonexpressing CTL cells were cultured in 2% FBS for 48 h. After ultracentrifugation of the CM, the membrane (P100) and the soluble (S100) fractions were analyzed for α-synuclein with the polyclonal C-20 antibody. B, P100 pellets were reconstituted in RIPA buffer and analyzed by Western immunoblotting with antibodies against the proteins indicated. TfR, Transferrin receptor. C, P100 pellet representing pure exosomes was analyzed using antibodies against α-synuclein (C-20) and Alix. D, Externalized membrane vesicles were prepared as in A. After fixation, vesicles were negatively stained with 2% uranyl acetate and observed by electron microscopy. Scale bar, 100 nm. E, Analysis of externalized membrane vesicles by mass spectrometry. The annotated product ion MS2 spectrum of the tryptic peptide EGVVQGVASVAEK traceable to α-synuclein is depicted. F, The P100 fraction containing exosomes was treated with Na2CO3. After a 50,000 × g centrifugation, the integral membrane proteins were recovered in the pellet (P), whereas non-integral and lumen proteins remained in the supernatant (S). P and S were analyzed for α-synuclein by immunoblotting with the C-20 antibody.
Recent studies have suggested that exosome-mediated export could play a critical role in the processing of proteins associated with neurodegenerative diseases, because β-amyloid (Aβ) peptides and prions were found to be externalized from cells via this pathway (Fevrier et al., 2004; Rajendran et al., 2006; Vella et al., 2008). To biochemically analyze the extracellular α-synuclein-containing vesicles, the vesicular fraction (P100) from the CM of α-synuclein-expressing and nonexpressing CTL cells was prepared and subjected to SDS-PAGE and immunoblotting. We found that P100 was positive for the exosome-specific proteins Alix and flotillin, as well as for Hsp70, GAPDH, and annexin II, proteins thought to be related with exosomes (Fevrier et al., 2004; Fauré et al., 2006; Rajendran et al., 2006; Théry et al., 2006; Trajkovic et al., 2008) (Fig. 3B). In addition, proteins reported to be excluded from exosomes, such as 14-3-3β and cofilin (Simpson et al., 2008), were both absent from P100 (Fig. 3B) (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). Unlike cofilin, which was shown to be absent from the S100 fraction, 14-3-3β was detected in the same fraction consistent with reports (Berg et al., 2003) showing the presence of 14-3-3 family members extracellularly (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). P100 also did not contain Rab5, a marker for early endosomes, or the transferrin receptor, a general marker for endosomal recycling (Fig. 3B).
To confirm that the CM of α-synuclein-expressing cells contained exosomes, we isolated pure exosomes from the CM using a validated differential ultracentrifugation procedure (Théry et al., 2006). Western immunoblotting revealed that purified, Alix-positive, exosomes (P100) contained α-synuclein (Fig. 3C). The morphological characteristics of α-synuclein-positive vesicles were analyzed by electron microscopy. Electron micrographs of P100 revealed uniformly rounded, cap-shaped vesicles with diameters in the range of 50–140 nm (mean size, 89.1 ± 2.3 nm, mean ± SEM; n = 77), further arguing for the exosomal nature of these vesicles (Fig. 3D) (supplemental Fig. S3D, available at www.jneurosci.org as supplemental material). These results are in agreement with previous reports (Théry et al., 2002, 2006; Fauré et al., 2006; Vingtdeux et al., 2007; Yuyama et al., 2008). The protein composition of externalized exosomes was also analyzed using mass spectrometry. Proteomic analysis confirmed the presence of α-synuclein in the exosome pellet of α-synuclein-expressing cells (Fig. 3E). In agreement with previous studies (Wubbolts et al., 2003; Valadi et al., 2007; Simpson et al., 2008; Gonzalez-Begne et al., 2009), LC-MS proteomic analysis of our exosome preparation identified common, as well as cell-type-specific, proteins (supplemental table, available at www.jneurosci.org as supplemental material).
Together, these data demonstrate that α-synuclein is exported via membrane vesicles that possess the characteristics of exosomes. To examine the topology of α-synuclein found in the exosomes, we treated the P100 fraction with Na2CO3. Interestingly, α-synuclein was present both in the membrane (P) and the lumen (S) of Na2CO3-treated exosomes (Fig. 3F).
Medium from α-synuclein-expressing cells is toxic to differentiated SH-SY5Y cells and primary cortical neurons
Recombinant α-synuclein can cause cell death when added to recipient cells (Sung et al., 2001; Zhang et al., 2005; Ahn et al., 2006). We wanted to examine whether the physiologically secreted α-synuclein from our cells could affect cell viability. α-Synuclein-expressing and nonexpressing CTL cells, as well as bGAL-expressing cells, were cultured for 48 h in 2% FBS, and the CM was applied to differentiated SH-SY5Y cells for 24 h. Application of the CM from α-synuclein-expressing cells induced drastic morphological changes to recipient cells, characteristic of cellular degeneration such as process retraction and membrane blebbing (Fig. 4A). Cell death was determined to be ∼17% (percentage cell death, 16.8 ± 0.6%, mean ± SD; n = 4) (Fig. 4B,C). Cells treated with CM from uninduced SH-SY5Y cells or bGAL-expressing cells did not exhibit any abnormalities or cell death, suggesting that the observed cytotoxicity could be associated with the presence of α-synuclein in the CM. Using recombinant α-synuclein, we estimated the concentration of α-synuclein in the CM. Interestingly, cell death correlated with the amount of secreted α-synuclein present in the CM (Fig. 4D). Neuronal death was verified by assessment of caspase-3 activation in the CM-treated differentiated SH-SY5Y cells. After treatment with CM, the recipient cells were lysed and analyzed by Western immunoblotting using an antibody against the cleaved caspase-3. Caspase-3 activation was only evident when cells were treated with CM containing secreted α-synuclein (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material).

Secreted α-synuclein induces cell death to differentiated SH-SY5Y cells. A, α-Synuclein-expressing and uninduced CTL cells, as well as bGAL-expressing cells, were cultured in 2% FBS for 48 h. CM was collected and applied to differentiated SH-SY5Y cells for 24 h. Representative phase micrographs of the recipient cells are shown. Scale bar, 50 μm. B, CM was prepared and used to treat recipient SH-SY5Y cells as in A. After 24 h, the recipient cells were stained with EthD-1 (red) and Hoechst (blue). Scale bar, 50 μm. C, Percentage cell death was determined by counting the percentage ratio of EthD-1-positive cells versus the Hoechst-positive cells. Quantitative analysis demonstrated a statistically significant increase in cell death when recipient cells were treated with CM from α-synuclein-expressing cells (n = 4; mean ± SD, one-way ANOVA test followed by Tukey's test, *p < 0.001). D, CM was collected from α-synuclein-expressing cells as in A and applied to differentiated SH-SY5Y cells for 24 h. Cell death was quantified by EthD-1/Hoechst staining. The amount of secreted α-synuclein present in the CM was determined by Western immunoblotting and recombinant α-synuclein as standard. The average of three experiments is shown. E, CM from α-synuclein-expressing or uninduced CTL cells were applied to cycling SH-SY5Y cells for 24 h, and the recipient cells were stained with EthD-1 and Hoechst. Quantitative analysis showed a reduction in cell viability from α-synuclein-rich CM (n = 4; mean ± SD, independent t test, *p < 0.01).
CM from α-synuclein-expressing and nonexpressing CTL cells was also applied to cycling SH-SY5Y cells. Treatment of cycling cells with secreted α-synuclein-containing CM for 24 h affected cell viability (percentage cell death, 9.3 ± 1.2 and 4.8 ± 1.3% for CM from α-synuclein-expressing and CTL cells, respectively, mean ± SD; n = 4) but to a lower extent compared with differentiated recipient sister cultures (Fig. 4E). Treatment of rat primary cortical neurons for 24 h with secreted α-synuclein-rich CM resulted in a profound increase in neuronal degeneration, similar to that of differentiated SH-SY5Y cells (percentage cell death, 20.5 ± 3.8%, mean ± SD; n = 3) (Fig. 5A,B). Importantly, effective immunodepletion of α-synuclein from the CM using the α-synuclein-specific antibody Syn-1 (Fig. 5C), before application on recipient neurons, significantly reduced neuronal loss (Fig. 5A,B), indicating that secreted α-synuclein is, to a great extent, responsible for the observed neurotoxicity. Control immunodepletion using an irrelevant antibody did not rescue primary neurons from the secreted α-synuclein-induced toxicity (Fig. 5B), further arguing that the death phenotype was attributable to the presence of α-synuclein in the CM.

Immunodepletion of α-synuclein from the CM reduces CM-induced toxicity. A, α-Synuclein-expressing and nonexpressing CTL cells were cultured in 2% FBS for 48 h. The CM was cleared from cell debris and immunoprecipitated with antibodies to α-synuclein or c-myc. Immunodepleted and control CM were applied to rat embryonic cortical neurons for 24 h. Recipient neurons were stained with EthD-1 (red) and Hoechst (blue). Scale bar, 10 μm. B, Quantification of the cell death revealed significant increases in cytotoxicity when α-synuclein was present in the CM. Immunodepletion of α-synuclein from the CM was protective against the death phenotype. Data are presented as mean ± SD (n = 3) and analyzed with one-way ANOVA test followed by Tukey's test; *p < 0.001 comparing CM from WT+ with CM from WT−; #p < 0.001 comparing CM from WT− with α-synuclein-immunodepleted CM (WT-/Syn-1 IP). C, Cell lysates (CL) of α-synuclein-expressing (−dox) or nonexpressing CTL (+dox) cells were immunoprecipitated and Western immunoblotted with the Syn-1 antibody. α-Synuclein was successfully immunoprecipitated. α-Synuclein present in the CM of these cells was similarly immunoprecipitated. Under such conditions, the CM was almost completely depleted of α-synuclein (CM/IP/Syn-1). As expected, control immunoprecipitation using the A-14 c-myc antibody (CM/IP/c-myc) had no effect on α-synuclein levels.
Extracellular α-synuclein is internalized by proliferating SH-SY5Y cells but not differentiated SH-SY5Y cells or cortical neurons
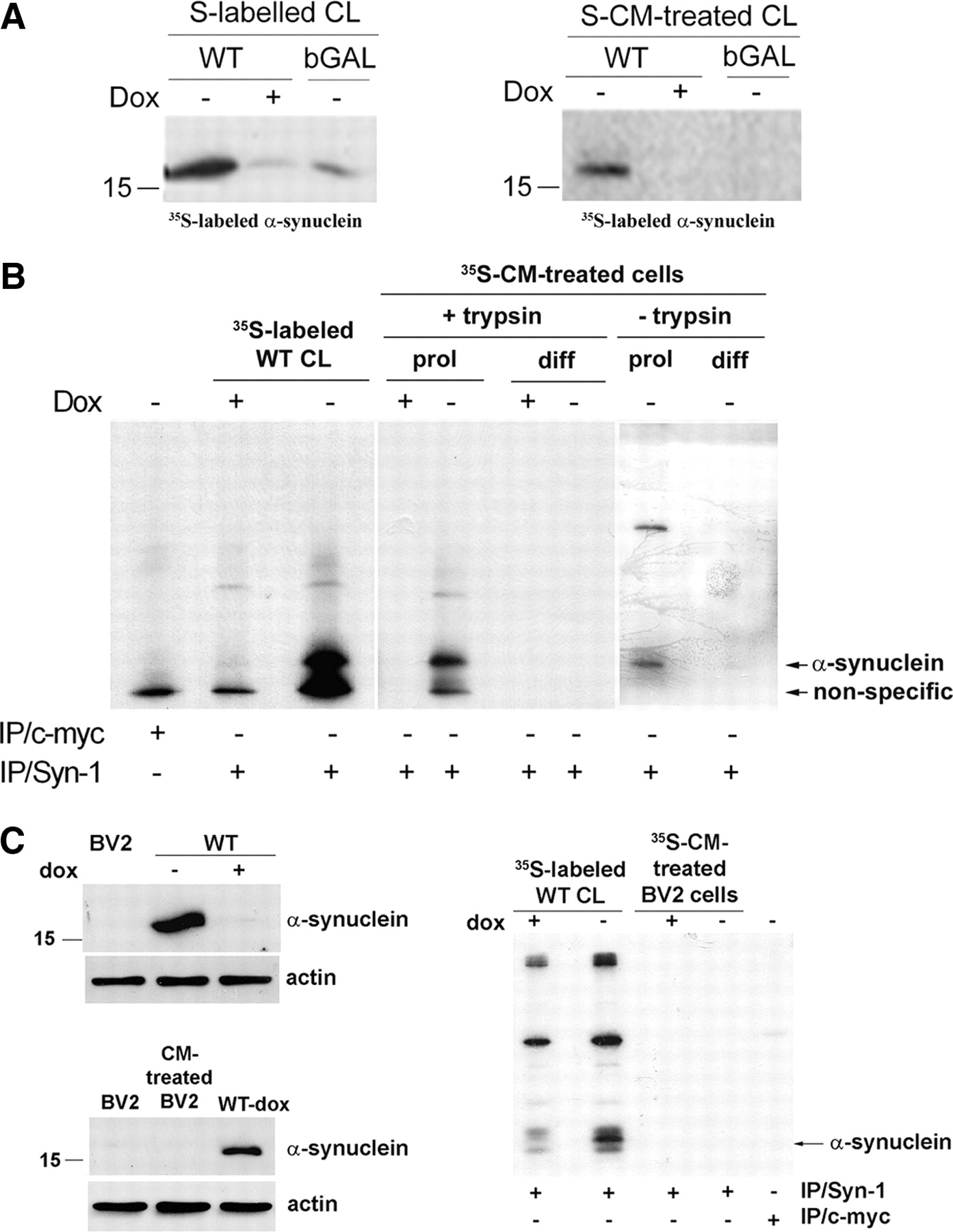
It has been reported that recombinant α-synuclein can be internalized by cells via endocytosis (Sung et al., 2001; Zhang et al., 2005; Ahn et al., 2006). We used 35S-labeled CM as a source of secreted α-synuclein to investigate whether cell-produced externalized α-synuclein was uptaken by recipient proliferating cells. Labeled CM from bGAL-expressing cells was used as control. After 35S labeling of intracellular α-synuclein (Fig. 6A, left), cells were grown in fresh medium for 16 h, and the CM containing secreted labeled α-synuclein was applied onto proliferating SH-SY5Y cells for 24 h. After a 2 min treatment with trypsin–EDTA to remove nonspecific surface protein binding, cells were lysed and immunoprecipitated with Syn-1 antibody. Autoradiography demonstrated that labeled α-synuclein could indeed be uptaken by recipient cycling cells (Fig. 6A, right). The uptake of extracellular α-synuclein did not result in the formation of intracellular inclusions as was confirmed by negative Thioflavin S staining (data not shown). Interestingly, inhibition of the lysosomal pathway of recipient cells by 1 mm NH4Cl for 24 h exacerbated the toxicity of CM from α-synuclein-expressing cells, suggesting that uptaken α-synuclein may be degraded by the lysosome (supplemental Fig S4B, available at www.jneurosci.org as supplemental material). Subcellular fractionation demonstrated the presence of labeled α-synuclein in the microsomal fraction of the recipient cells (data not shown). To investigate whether neurons also uptake secreted α-synuclein, we applied the CM containing labeled α-synuclein to differentiated SH-SY5Y cells and rat cortical neurons. Unlike proliferating cells, cell-produced labeled α-synuclein could not be detected inside neuronal cells even after prolonged film exposure (Fig. 6B). To rule out the possibility that the levels of internalized α-synuclein were too low to be detected, we applied “cold” CM from α-synuclein-expressing cells on rat cortical neuronal cultures for 16 h and checked for the presence of internalized human α-synuclein. Lysates (600 μg) were immunoprecipitated with the Syn-1 antibody and Western blotted with the human-specific LB 509 antibody. Internalization of human α-synuclein was not evident even under these conditions (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material). However, a very small amount of uptaken α-synuclein could be detected in differentiated SH-SY5Y when trypsin–EDTA treatment was replaced by extensive (three times) washing with cold PBS (Fig. 6B).

Externalized α-synuclein is uptaken by proliferating SH-SY5Y cells but not neuronal cells. A, α-Synuclein-expressing (−dox) or nonexpressing CTL (+dox) cells and bGAL-expressing (bGAL-dox) cells were 35S labeled. CM from the labeled cells, containing labeled secreted proteins (see Materials and Methods), was applied to recipient cycling SH-SY5Y cells. Endogenously expressed (left) or uptaken (right) labeled α-synuclein was detected in recipient cell extracts by gel autoradiography after immunoprecipitation of the cell lysates with Syn-1 antibody. B, As in A, but c-myc immunoprecipitation was used as a negative control and CM from cells, containing abundant labeled secreted proteins, was applied to both recipient proliferating (prol) and differentiated (diff) SH-SY5Y cells. The recipient cells were either treated with trypsin–EDTA (+trypsin) or washed thoroughly (−trypsin). Gels were exposed for 30 d. C, Left, BV2 cell lysate was analyzed for the presence of α-synuclein by Syn-1 immunoblotting compared with cells expressing (−dox) or nonexpressing (+dox) α-synuclein. CM from α-synuclein-expressing cells (WT-dox) was collected and applied to BV2 cells. Uptake of α-synuclein by recipient BV2 cells was assessed in the cell lysates by Western immunoblotting using the Syn-1 antibody. Actin was used as loading control. Right, α-Synuclein-expressing (−dox) or nonexpressing CTL (+dox) cells were 35S labeled (35S-labeled WT CL). CM from the labeled cells was applied to recipient cycling BV2 cells, which were subsequently treated with trypsin–EDTA (35S-CM-treated BV2 cells). Cell-produced (first and second lanes) or uptaken labeled α-synuclein (third and fourth lanes) was detected in cell extracts by gel autoradiography after immunoprecipitation of the cell lysates with Syn-1 antibody. c-myc immunoprecipitation was used as a negative control (fifth lane).
It has been reported that recombinant α-synuclein can activate microglia cells (Zhang et al., 2005; Su et al., 2008). To examine whether naturally secreted α-synuclein could be uptaken by microglia cells, murine BV2 microglia, which normally do not express α-synuclein (Fig. 6C, left), were treated with CM rich in α-synuclein or labeled α-synuclein for 24 h. Uptaken α-synuclein was assessed by immunoprecipitation and Western blotting. As depicted in Figure 6C, BV2 cells did not uptake extracellular α-synuclein.
Both oligomeric and monomeric α-synuclein species are found extracellularly
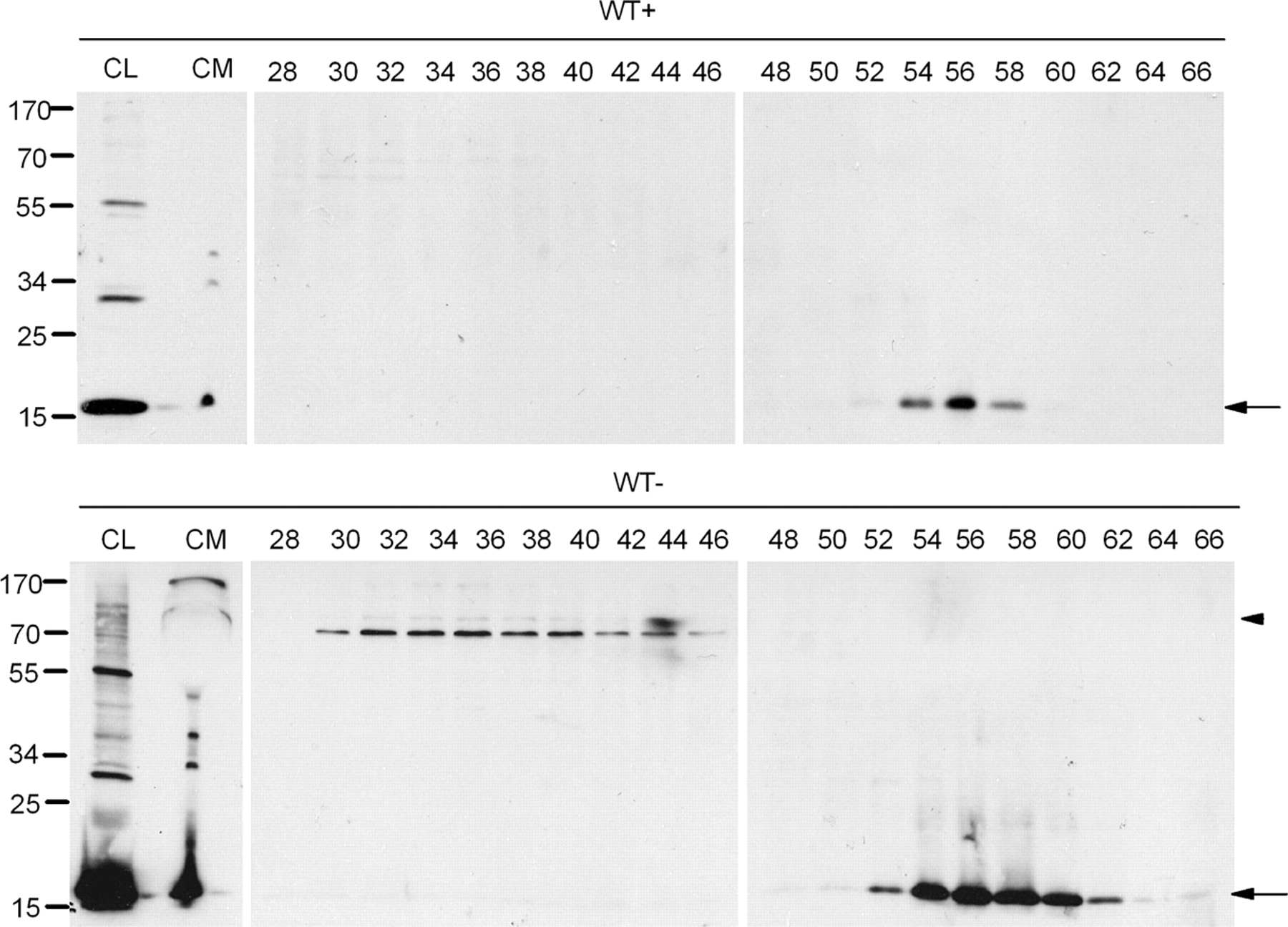
It has been reported that the presence of soluble oligomers of α-synuclein may account for the pathogenic actions of α-synuclein, although the mechanism through which this occurs remains unresolved (Vekrellis et al., 2004). Interestingly, α-synuclein oligomeric species were readily detected in the CM of α-synuclein-expressing cells after concentration and immunoblotting with the polyclonal C-20 antibody (Figs. 1B, 7). These results were confirmed using the monoclonal Syn-1 antibody (data not shown). To provide additional evidence for the identity of these species, the concentrated CM from α-synuclein-expressing and nonexpressing CTL cells was analyzed by SEC (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). SEC fractions were then subjected to SDS-PAGE and immunoblotting for the presence of α-synuclein using the C-20 antibody. Using this technique, HMW (>580 kDa) and LMW (<38 kDa) α-synuclein species, migrating under reducing conditions as (potentially) tetrameric and monomeric α-synuclein, respectively, were effectively identified and separated (Fig. 7). Similar results were obtained with the Syn-1 antibody (data not shown).

Extracellular α-synuclein oligomeric and monomeric species are present in CM. α-Synuclein-expressing (WT−) and nonexpressing CTL (WT+) cells were cultured in 2% FBS for 48 h. The CM was cleared from cell debris, concentrated, and fractionated by SEC. The cell lysates (CL), the concentrated CM, and the SEC fractions corresponding to WT+ and WT− cells were further analyzed for α-synuclein with the Syn-1 antibody. Arrows show α-synuclein present in LMW fractions, and arrowhead shows α-synuclein present in HMW fractions.
Treatment of CM with oligomer-interfering compounds partially rescues recipient cells from α-synuclein-induced toxicity
To investigate whether extracellular α-synuclein oligomers are critical for the observed secreted α-synuclein-induced cytotoxicity, we treated the CM with two oligomer-interfering compounds, CR and SI. CR has been shown to effectively disrupt preformed oligomeric/aggregated forms of various proteins, including mutant α-synuclein or Huntingtin (Carter and Chou, 1998; Sánchez et al., 2003; Emmanouilidou et al., 2010). SI, a cell-permeable sugar, can successfully neutralize the adverse effects of Aβ oligomeric species, possibly by stabilizing a nontoxic conformation of Aβ oligomers (McLaurin et al., 2000; Townsend et al., 2006). CM from α-synuclein-expressing cells and nonexpressing CTL cells was treated with 0.4 μm CR or SI for 4 h at 4°C. SDS-PAGE and immunoblotting analysis of such CM after concentration revealed that, consistent with previous findings, treatment of the CM with CR caused an obvious reduction of extracellular α-synuclein oligomeric species and a simultaneous increase of monomeric α-synuclein, possibly attributable to the collapse of oligomers to monomers (Fig. 8A). No change in the levels of the α-synuclein oligomers was observed after SI treatment of the CM (Fig. 8A). In addition, CR or SI treatments did not affect the membrane integrity of exosomes present in the CM (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material). When CR- or SI-treated CM was applied to differentiated SH-SY5Y cells for 4 h, this resulted in a significant, ∼50%, reduction of toxicity from secreted α-synuclein (Fig. 8B). When CM was treated with chiroinositol (CI), an inactive isomer of SI, there was no promotion of survival of the recipient cells (Fig. 8B), indicating that the rescue effect of SI was indeed related with its anti-oligomeric action. These data suggest that naturally secreted α-synuclein oligomers are, at least in part, responsible for the α-synuclein-induced cytotoxity in recipient cells.

Treatment of secreted α-synuclein-rich CM with oligomer-interfering compounds reduces CM-induced toxicity. A, α-Synuclein-expressing cells were cultured in 2% FBS for 48 h. The CM was cleared from cell debris and equal parts were treated with vehicle, CR (0.4 μm), or SI (0.4 μm) for 4 h at 4°C. CM was then concentrated and analyzed for α-synuclein with the C-20 antibody. After treatment, the levels of secreted α-synuclein oligomers were quantified (n = 3; mean ± SD, one-way ANOVA test, *p < 0.01). B, CM was collected and treated as in A. After treatment with the compounds, CM was applied to differentiated SH-SY5Y cells for 24 h. Survival was assayed by EthD-1/Hoechst staining. Data are shown as mean ± SD (n = 3). Statistical analysis was performed using the one-way ANOVA test followed by Tukey's test (*p < 0.001 comparing CM, WT+ with CM, WT−; #p < 0.001 comparing CM, WT− with CM, WT−/CR or CM, WT−/SI).
SEC fractions containing either HMW or LMW secreted α-synuclein species reduce the viability of differentiated SH-SY5Y cells
Our data using CR and SI supported the notion that α-synuclein oligomers could be involved in secreted α-synuclein-induced toxicity. Because we detected several distinct α-synuclein species in the CM (Fig. 7), we wanted to examine whether the toxicity observed could be attributed to particular α-synuclein species. For this purpose, α-synuclein-expressing and nonexpressing CTL cells were cultured in 2% FBS for 48 h, and the CM was concentrated and analyzed by SEC. HMW and LMW fractions containing α-synuclein were pooled, buffer exchanged, and lyophilized. After their reconstitution in growth medium, HMW and LMW fractions were applied separately on differentiated SH-SY5Y cells for 48 h. Interestingly, both HMW and LMW fractions induced cell degeneration to recipient cells (Fig. 9A) almost to the same extent (percentage cell death, 8.9 ± 0.5 and 11.8 ± 1.6% for HMW and LMW fractions, respectively, mean ± SD; n = 3) (Fig. 9B).

Isolated secreted α-synuclein oligomeric and monomeric species decrease the viability of differentiated SH-SY5Y cells. A, α-Synuclein-expressing and uninduced CTL cells were cultured in 2% FBS for 48 h. After removal of cell debris, the CM was fractionated by SEC. Extracellular HMW and LMW α-synuclein-containing species were collected, lyophilized, and applied to differentiated SH-SY5Y cells for 48 h. Representative phase micrographs are shown. Scale bar, 50 μm. B, Quantification graph presenting the increase in cell death after application of extracellular HMW or LMW α-synuclein species for 48 h (n = 3; mean ± SD, one-way ANOVA test followed by Tukey's test, *p < 0.001, **p = 0.001).
Discussion
Because of the lack of an ER signaling peptide from its sequence, α-synuclein was considered to be exclusively localized and exert its pathogenic effects intracellularly. However, there is emerging evidence suggesting that α-synuclein can be secreted in the extracellular space, thereby affecting the homeostasis of neighboring neurons possibly by a neuron-to-neuron transmission mechanism (Lee, 2008; Desplats et al., 2009). In the current study, using an inducible WT α-synuclein SH-SY5Y cell line, we investigated the mechanism of α-synuclein secretion and the effects of this secretion in the homeostasis of recipient neurons. Importantly, the levels of secreted α-synuclein (2–12 nm) in our cell system were similar to those reported in biological fluids (Borghi et al., 2000; El-Agnaf et al., 2003). Our data strongly support that α-synuclein is physiologically secreted in the extracellular space in association with membrane vesicles. The protein composition, morphology, and size of these vesicles were found to be similar to exosomes, small (40–100 nm), endosome-derived vesicles secreted by a multitude of cell types during fusion of multivesicular bodies with the plasma membrane. In support of this idea, impairment of the endocytic pathway by weak bases resulted in a profound increase in α-synuclein secretion. Exosome-mediated exocytosis is a calcium-dependent process (Raposo et al., 1997; Savina et al., 2003, 2005). By using Ca2+ ionophores/chelators, we have found that α-synuclein secretion is greatly affected by changes in intracellular, but not extracellular, calcium concentration. However, we cannot exclude the possibility that alternative mechanisms for α-synuclein secretion, such as secretory vesicle-mediated exocytosis, may also operate in our cell system. Indeed, part of intracellular α-synuclein was found previously to be localized in secretory vesicles (Lee et al., 2005).
Exosomes are considered to be biologically active entities that can interact with recipient cells in a variety of ways, such as endocytosis, receptor-ligand binding, attachment, or fusion with the plasma membrane (Théry et al., 2002; Vincent and Magee, 2002; Keller et al., 2006; van Niel et al., 2006). Alternatively, extracellular degradation of the exosomal membrane by proteases or lipases could allow the release of proteins from the exosomal lumen to the extracellular matrix (Mehul and Hughes, 1997; Hughes, 1999). Initially, the release of exosomes was considered to be a cellular mechanism of clearing unnecessary proteins (Pan et al., 1985; Johnstone et al., 1987; van Niel et al., 2006; Vella et al., 2008). The novel finding that α-synuclein can be exported from the cell via the exosomal pathway provides a common pathway for the delivery of a potentially toxic protein in the extracellular space, thereby spreading its pathogenic actions in neighboring healthy cells. Interestingly, exosomes bearing prions were efficient initiators of prion propagation in uninfected recipient cells and produced prion disease when inoculated into mice (Fevrier et al., 2004; Vella et al., 2007, 2008). β-Amyloid peptides of intracellular origin were found to be released in association with exosomes (Rajendran et al., 2006). Moreover, cultured cortical neurons can secrete exosomes (Fauré et al., 2006). In this context, our data support the Trojan Horse hypothesis in which neurodegeneration results from the progressive propagation of pathogenic events, cell by cell, throughout brain areas (Caughey, 2000; Ghidoni et al., 2008; Lee, 2008). The finding that grafted healthy neurons can gradually develop α-synuclein pathology as host neurons many years after transplantation provides additional support for a prion-like mechanism for the spread of PD (Brundin et al., 2008). However, it remains unclear whether internalization of released α-synuclein or some other factor is responsible for the development of α-synuclein deposits in the transplanted cells.
We show that CM containing soluble secreted α-synuclein can cause cell death to healthy recipient cells that is associated with caspase-3 activation. Degeneration was greatly amplified when the recipient cells have a neuronal phenotype, such as differentiated SH-SY5Y cells or primary cortical neurons. The cell death was associated with secreted α-synuclein because immunodepletion of α-synuclein from the CM significantly ameliorated the cytotoxic effects. Importantly, death correlated with extracellular α-synuclein concentration.
CM from WT-expressing SH-SY5Y cells was found to contain soluble monomeric and oligomeric α-synuclein. We isolated HMW and LMW secreted α-synuclein species and evaluated their toxicity. Both species decreased the viability of recipient differentiated neuroblastoma cells. Our data indicate that presence of either cell-derived HMW or LMW α-synuclein species extracellularly can decrease cell viability, suggesting that high n-oligomeric or low n-monomeric α-synuclein could act in a synergistic way to trigger neurodegeneration. Importantly, the molar amount of HMW α-synuclein was much lower than that of LMW α-synuclein, suggesting that, on a molar basis, HMW oligomeric α-synuclein exerted greater cytotoxicity.
Further supporting a critical role for the extracellular α-synuclein oligomers, treatment of the CM with oligomer-interfering compounds, such as CR and SI, partially rescued the recipient cells from α-synuclein-induced toxicity. In a number of studies, CR has been used to disrupt β-sheet-rich species, including α-synuclein, huntingtin, and Aβ (Heiser et al., 2000; Sánchez et al., 2003; Inouye and Kirschner, 2005). Indeed, CR dissociated the α-synuclein oligomeric species contained in the CM. Although CR did not affect the integrity of the exosomes in the CM, we cannot exclude the possibility that CR may also act by preventing the interaction of exosomes with the plasma membrane. The mechanism by which SI interacts with β-sheet conformations is not known. In our hands, SI did not disrupt the extracellular α-synuclein oligomeric species, nor did it affect exosomal integrity. This is consistent with our previous finding that SI does not disrupt intracellular α-synuclein oligomeric species despite its survival-promoting effects (Vekrellis et al., 2009). It has been hypothesized that SI could stabilize a nontoxic oligomeric conformation preventing the production of the more toxic protofibrils (McLaurin et al., 2000; Townsend et al., 2006).
Critical to the understanding of the toxic effects of secreted α-synuclein is the issue of whether it is internalized by recipient cells. In our hands, secreted α-synuclein appears to be readily uptaken by cycling SH-SY5Y cells but not to any significant degree by neuronal cells or microglia. This difference may point to cell-type-specific mechanisms of uptake. This may be especially applicable to oligomeric species, which, because of their low abundance, cannot be detected by autoradiography in our hands. Recent evidence suggests that α-synuclein uptake by neuronal cells depends highly on its fibrilarization state. Furthermore, cationic liposomes have been shown to be necessary for the delivery of fibrillar recombinant α-synuclein inside neuronal cells (Luk et al., 2009). Because we can only detect soluble but not fibrilar α-synuclein species in the medium of our cells, it is possible that these do not efficiently enter recipient neurons. Lee et al. (2008) have also suggested that aggregated α-synuclein is internalized more efficiently, and via a different pathway, compared with the monomeric protein. However, caution is needed in the interpretation of these results, because they are based on very high amounts of the recombinant protein.
Although we cannot exclude the possibility that secreted α-synuclein exerts its toxic effects by uptake into recipient cells, in which it could impact cellular homeostasis in various ways, for example via disruption of protein degradation pathways (Sánchez et al., 2003; Inouye and Kirschner, 2005), the fact that these effects are more pronounced in neuronal cells in which limited uptake is detected raises the possibility that they are mediated extracellularly at the level of cell membrane. It is tempting to speculate that extracellular secreted α-synuclein could trigger degeneration via a specific signaling process. This process could be initiated by an interaction of secreted α-synuclein with the plasma membrane, possibly through a specialized, so far unidentified, receptor. Alternatively, and according to the amyloid pore model, the interaction of secreted α-synuclein with cellular membranes could result in pore formation, which greatly compromises membrane integrity, and lead to neuronal degeneration via the creation of significant alterations in the equilibrium of ions and small metabolites between the cytoplasm and the extracellular space (Heiser et al., 2000; Volles et al., 2001; Volles and Lansbury, 2003). In support of this idea, in a number of studies pre-formed α-synuclein protofibrils generated from recombinant α-synuclein have been shown to cause neurotoxicity when added to the culture medium of healthy cells (El-Agnaf et al., 1998; Du et al., 2003; Zhang et al., 2005).
In conclusion, we demonstrate here that α-synuclein is secreted from neuronal cells in a calcium-dependent manner by exosomes and that, at physiological concentrations, it can impact the viability of neighboring neurons. Oligomeric secreted α-synuclein species are in part responsible for these effects. These findings provide an understanding of the mechanism of spread of pathology in PD and offer the opportunity to modulate this process at either the level of secretion of α-synuclein or the level of its impact on neighboring cells.
Footnotes
-
This work was supported in part by National Institutes of Health Grant R21 NS0556 (L.S., K.V.) and by a Rapid Response Grant from the Michael J. Fox Foundation (E.E.).
-
The authors have nothing to disclose.
- Correspondence should be addressed to Kostas Vekrellis, Division of Basic Neurosciences, Biomedical Research Foundation of the Academy of Athens, 4, Soranou Efessiou Street, Athens 11527, Greece. vekrellis{at}bioacademy.gr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}