

Abstract
Tau protein in a hyperphosphorylated state makes up the intracellular inclusions of several neurodegenerative diseases, including Alzheimer's disease and cases of frontotemporal dementia. Mutations in Tau cause familial forms of frontotemporal dementia, establishing that dysfunction of tau protein is sufficient to cause neurodegeneration and dementia. Transgenic mice expressing human mutant tau in neurons exhibit the essential features of tauopathies, including neurodegeneration and abundant filaments composed of hyperphosphorylated tau. Here we show that a previously described mouse line transgenic for human P301S tau exhibits an age-related, layer-specific loss of superficial cortical neurons, similar to what has been observed in human frontotemporal dementias. We also show that focal neural precursor cell implantation, resulting in glial cell differentiation, leads to the sustained rescue of cortical neurons. Together with evidence indicating that astrocyte transplantation may be neuroprotective, our findings suggest a beneficial role for glial cell-based repair in neurodegenerative diseases.
Introduction
Age-related neurodegenerative diseases, such as Alzheimer's disease (AD) and frontotemporal dementias (FTDs), are increasingly prevalent and there are no disease-modifying or reparative treatments. The mechanisms underlying neurodegeneration also remain largely unknown, although the accumulation of intracellular neurofibrillary lesions composed of abnormally phosphorylated and aggregated tau protein is characteristic of many neurodegenerative disorders (Goedert and Spillantini, 2006). Tau is a microtubule-associated protein that plays a role in regulating microtubule assembly and stabilization, as well as in axonal transport (Lee et al., 2001; Magnani et al., 2007). Under pathological conditions tau becomes hyperphosphorylated, resulting in its dissociation from microtubules and assembly into abnormal filaments.
The identification of mutations in Tau in familial forms of FTD (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998) has led to the development of transgenic models of tauopathy (Götz and Ittner, 2008) that reproduce salient pathological features of the human diseases, such as tau-containing intracellular inclusions and nerve cell loss in some regions of the CNS. However, degeneration of cortical neurons and astrogliosis, central features of human FTD (Broe et al., 2004), have not been reported as consistent features of these experimental models.
Accumulating evidence has suggested a role for cell-based repair strategies in models of acute and chronic neuronal injury. Embryonic and adult-derived neural precursor cells (NPCs) or stem cells have been shown to exert beneficial effects through a range of mechanisms. Although graft-derived neuronal differentiation has been reported, only little evidence of functional neuronal replacement in models of neuronal injury has been obtained to date (Björklund and Lindvall, 2000; Cao et al., 2001; Hofstetter et al., 2005; White et al., 2008). Alternative neuroprotective pathways increasingly implicate mechanisms independent of neuronal differentiation (Martino and Pluchino, 2006; Lepore et al., 2008). In addition to NPCs, there is an increasing literature that implicates a direct glial cell-mediated neuroprotective effect in models of acute and progressive neuronal injury through modification of a hostile endogenous astroglial environment in the context of noncell-autonomous neurodegeneration (Boillee et al., 2006; Lepore et al., 2008; Yamanaka et al., 2008).
Against this background we examined a previously characterized mouse line transgenic for human P301S tau for evidence of cortical neuron loss and astrogliosis. This transgenic model has previously been shown to accumulate hyperphosphorylated tau in neurons within a number of CNS regions, including cerebral cortex, amygdala, brainstem, and spinal cord, along with a neuroinflammatory response (Allen et al., 2002; Bellucci et al., 2004; Delobel et al., 2008; Gasparini et al., 2009).
In the present study we demonstrate that P301S tau mice display progressive cortical neuron loss, especially in the superficial layers, along with associated astrogliosis, and that focal transplantation of NPCs that differentiate into glia or direct astrocyte implantation results in sustained neuroprotection.
Materials and Methods
Animals.
All procedures were performed in compliance with national and institutional guidelines (UK Animals Scientific Procedures Act 1986 and the University of Cambridge Animal Care Committees). For characterization of neuronal cell loss, female and male homozygous P301S tau mice were killed at the onset of symptoms (Allen et al., 2002; Bellucci et al., 2004; Delobel et al., 2008; Gasparini et al., 2009) between 4 and 6 months of age, along with age-matched C57BL/6 mice. The mice are only allowed to survive to 5–6 months of age because of increasing hindlimb disability and United Kingdom Home Office animal license regulations requiring humane killing. For subsequent transplantation experiments, both P301S tau and age-matched C57BL/6 male and female mice were killed at 3 and 5 months of age. For neuronal characterization, cresyl violet was used in n = 4 P301S tau and n = 4 C57BL/6 mice, while for neuronal nuclei (NeuN) analysis n = 8 P301S tau and n = 7 C57BL/6 mice, respectively, were used. For the time course of NeuN and AT8 analysis, the following mice were investigated: n = 5 P301S tau and n = 4 C57BL/6 mice at 2 months; n = 5 P301S tau and n = 3 C57BL/6 mice at 3 months; and n = 8 P301S tau and n = 7 C57BL/6 mice at 5 months of age.
Immunostaining and histology.
Sections for immunofluorescence were processed as described previously (Scott et al., 2005; Hampton et al., 2008). Primary antibodies used were as follows: monoclonal anti-NeuN (1:400, Millipore Bioscience Research Reagents); mouse monoclonal anti-phosphotau (AT8, 1:1000, Autogen Bioclear); monoclonal anti- GFAP (glial fibrillary acidic protein) clone GA5-Cy3 (1:500, Sigma); monoclonal anti-neurofilament NF200 (1:500, Sigma); polyclonal rabbit anti-NG2 (1:500, a kind gift from Prof. W. B. Stallcup, Burnham Institute, La Jolla, CA); polyclonal rabbit-anti-Olig-2 (1:200, Millipore Bioscience Research Reagents); polyclonal goat or rabbit anti-GFP (green fluorescent protein) (1:1000, Abcam and Invitrogen, respectively); biotinylated WFA (Wisteria floribunda agglutinin lectin) (1:200, Sigma); rabbit polyclonal anti-GDNF (glial-derived neurotrophic factor) (1:20, Abcam); rabbit polyclonal anti-BDNF (brain-derived neurotrophic factor) (1:100 Abcam); polyclonal anti-NGF (nerve growth factor) (1:100, Calbiochem); monoclonal anti-Reelin (1:250, Abcam); polyclonal rabbit anti-Cux1 (1:250, Calbiochem); and rabbit polyclonal anti-GABA (1:500, Sigma). Secondary antibodies used were Alexa 488, Alexa 555, and streptavidin 555 (1:500, Invitrogen) in conjunction with bisbenzamide (1:5000, Sigma) to identify cell nuclei. Cresyl violet (Fisher Scientific) staining was performed after serial dehydration in alcohol, followed by brief immersion into diluted cresyl violet (0.5%) to identify neuronal cell bodies.
Quantitative analysis.
All sections were identified as being approximately equivalent to bregma, and then 0.9, 1.8 and 2.7 mm rostral of bregma (Paxinos and Franklin, 2001). All counts and analyses were performed throughout the cortex, and the cortical layers were defined as follows: layers I and II, surface to a depth of 200 μm; layer III, 200–350 μm; layer IV, 350–550 μm; layer V, 550–750 μm; and layer VI, 750–onwards (Rockel et al., 1980; Hill et al., 2000). We refer to the superficial cortex throughout, and this is defined as extending from the surface to a depth of 200 μm, equivalent to layers I and II. Cresyl violet counts were performed throughout the frontal cortex, two counts in either hemisphere under a 64 × 160 μm grid. For each animal, a minimum of three sections per area were counted. GFP double-labeled cells were also counted with three slides from each animal used and a minimum of three sections per area counted, with three 64 × 160 μm grids counted per section.
AT8 counts were performed under 100 × 500 μm grids for each cortical layer using three slides for each animal. Again, areas corresponding to bregma, as well as areas 0.9, 1.8, and 2.7 mm rostral of bregma, were identified and counts were performed such that two areas were counted per hemisphere (lesioned side versus unlesioned contralateral side), with a minimum of three sections per area. GABA and GFAP counts were performed exclusively in cortical layers I and II. Counts were performed at the same four locations relative to bregma as described above (n = 2–4 sections per area).
For threshold analysis, images were captured using Lucia or Leica software (Nikon) via a digital camera or a Zeiss AxioScope using AxioVision software and SigmaScan Pro (SPSS) was used for subsequent quantitative densitometric measurements of NeuN, bisbenzamide, and NF200, as detailed previously (Scott et al. 2005; Hampton et al. 2008). For NeuN and bisbenzamide, a percentage area of the cortex containing antibody signal was generated by dividing the number of positive readings by the total number of potential readings, multiplied by 100. Between four and six sections per animal were analyzed, from which a mean percentage area of the cortical layer containing a positive NeuN signal could be calculated. For NF200, once images had been set to a threshold the density of immunopositive objects was measured independently of their individual intensities. This allowed us to make comparisons between P301S tau, C57BL/6, and transplanted mice.
For confocal analysis, stacks of images were captured on a Leica TCS-NT-UV confocal laser scanning microscope using a 63× 1.25 numerical aperture Leica lens or a Zeiss 720 confocal laser scanning microscope using ZEN 2009 software. Typical stacks were composed of 10–20 optical sections of 1 μm thickness taken at 0.8 μm intervals. Sequential acquisitions were performed in the different channels to avoid any misinterpretation of the results caused by signal cross talk.
All data were quantified using statistical analysis, including one-way ANOVA (with the Holm-Sidak post hoc analysis) or t test, and significance was only assumed if p < 0.001.
Neural precursor cell culture.
Cell culture reagents were obtained from Invitrogen unless otherwise specified. Neonatal mice ubiquitously expressing enhanced green fluorescent protein (eGFP), a kind gift from Dr. M. Okabe, Osaka University, Osaka, Japan (Okabe et al., 1997) were anesthetized with ice and decapitated. Following removal of the meninges, the cerebral cortex was dissociated and a single cell suspension was generated, seeded at 1 × 106 per T25 flask with DMEM/F12 (Invitrogen) containing 2% B27, 1% N2, 10 ng/ml fibroblast growth factor (FGF)-2, and 10 ng/ml epidermal growth factor (EGF) (expansion media) and expanded until the spheres became confluent, as described previously (Vescovi et al., 1993; Nagahara et al., 2009). Confluent spheres were incubated with accutase for 3 min, triturated to return them to a single cell suspension, and reseeded into uncoated 25 cm2 flasks with expansion media. Spheres were passaged once before being used for transplantation or in vitro characterization. GFP+ astrocytes were generated from eGFP-NPCs treated with basal medium supplemented with bone morphogenetic protein 4 (BMP4) (10 ng/ml) for 96 h in the absence of EGF and FGF-2 before harvesting for transplantation (Gross et al., 1996; Mabie et al., 1997).
Paired samples of dissociated eGFP-NPCs were plated onto poly-d-lysine (1 μg/ml)/laminin (10 μg/ml)-coated borosilicate coverslips (∼5 × 104 cells/coverslip) corresponding to the samples used for transplantation. Twenty-four hours following plating, cells were fixed with 4% paraformaldehyde. The following antibodies were used to stain the cells: anti-A2B5 (1:5 supernatant, mouse IgM); anti-O4 (1:5 supernatant, mouse IgM); anti-galactocerebroside (1:5 supernatant, mouse IgG3), anti-NG2 (1:250, rabbit, kind gift of Prof. W. B. Stallcup); anti-microtubule-associated protein 2 (1:500, Sigma, clone-AP-20); Tuj1 (1:500, Sigma, β-III tubulin), and anti-GFAP (1:4000, rabbit, DakoCytomation). Cell nuclei were visualized with bisbenzamide stain (Hoechst 33323, Sigma). Coverslips were mounted on glass slides using FluroSave (Calbiochem) and viewed under a Leica DM6000 fluorescence microscope (Leica Microsystems) with appropriate filters for cell identification and counting. For each coverslip, five consecutive random fields were examined using a grid with all cells counted. At least four coverslips were analyzed for each immunostain. The number of GFP+ colabeled cells was expressed as a mean ± SEM. for each experiment (n = 4).
Transplantation.
eGFP-NPCs or eGFP-astrocytes were injected atraumatically into P301S tau and C57BL/6 male and female mice. Each animal received two injections of eGFP-NPCs (80,000 cells per injection of 0.8 μl) into the cortical gray matter (a depth of 0.4 mm, relative to the dura) at the following coordinates from bregma: 1.0 mm mediolateral and 1.0 mm or 1.8 mm anterioposterior (Franklin and Paxinos, 1997). In experiments parallel to those of the live cell transplants, dead cells and vehicle consisting of the control media were also injected. Dead cells were obtained by rapidly freeze-thawing eGFP-NPCs several times before transplantation. Injections were performed on 8-week-old animals, which were killed either 4 weeks (n = 4–10 and 5–7 for P301S tau and C57BL/6 mice, respectively) or 12 weeks after injection (n = 7–10 and 4–10 for P301S tau and C57BL/6, respectively). Mice were either perfused with 4% paraformaldehyde or their brains were snap frozen. Perfused brains were cryoprotected in 25% sucrose, frozen, cut at 25 μm on a cryostat, and stored at −80°C. Snap-frozen tissues were stored at −80°C and used for quantitative PCR (qPCR).
Stereology.
For stereological counting of surviving eGFP-NPCs, brain sections were stained with anti-GFP antibody. Briefly, sections were quenched in methanol/H2O2 and washed in PBS before blocking of nonspecific background. Sections were incubated overnight with rabbit anti-GFP (1:1000, Millipore Bioscience Research Reagents). They were then washed and incubated with biotinylated anti-rabbit antibody (1:500, Jackson ImmunoResearch Laboratories) before incubation with avidin-biotin (Vector Laboratories). GFP labeling was revealed using 3,3′-diaminobenzidine tetrahydrochloride (DAB) and coverslips were applied using DPX mounting medium. Stereology was performed using an Olympus microscope with CAST-Grid program.
The GFP+ cells were identified anterior to bregma. The whole brain section was first highlighted at ×4 objective before the areas of interest, including the cortical layers, were defined. Sample counting windows were identified using the computer software. The number of GFP-positive cells in each window was counted and the fractionator method was used to scale and approximate the number of GFP-positive cells in each transplanted adult mouse. The following formula was used:
 where number of objects counted is the total number of GFP-positive cells in all sections from one animal, Freq is the frequency of sections used, the XY step area is the x step length times the y step length, and the area of window is the area of the counting window at ×60 magnification.
where number of objects counted is the total number of GFP-positive cells in all sections from one animal, Freq is the frequency of sections used, the XY step area is the x step length times the y step length, and the area of window is the area of the counting window at ×60 magnification.
Quantitative real-time PCR.
Total RNA was extracted from tissues microdissected around the injection site using TRIzol (Invitrogen) and PureLink RNA mini kit (Invitrogen) according to the manufacturer's protocol. Total RNA (2 μg) was treated with RNase-free DNase I (New England Biolabs) and reverse transcribed in 100 μl with random hexamers using Moloney murine leukemia virus reverse transcriptase (Invitrogen). PCR was performed in 96-well plates using 1 μl of synthesized cDNA and forward and reverse primers [mouse (m)Reelin forward: 5′-CGAGTGGGTGAGGTGTAT-3′; mReelin reverse: 5′-AGCTATGCTTGACCGTTGCTC-3′; mCux2 forward: 5′-GCGGCGTTCCTGAGTGTTTAT-3′; mCux2 reverse: 5′-CTGGCAGGTGGTTACCGTT-3′; hypoxanthine phosphoribosyltransferase (HPRT) forward: 5′-TCAGTCAACGGGGGACATAAA-3′; HPRT reverse: 5′-GGGGCTGTACTGCTTAACCAG-3′]) with DyNAmo Flash SYBR Green qPCR kit (New England Biolabs) according to the manufacturer's protocol. Real-time PCR was performed on a Bio-Rad iCycler as follows: 95°C for 10 min and 40 cycles of 95°C for 10 s (denaturation) and 58°C for 45 s (annealing/extension), followed by a standard melting curve program. A minimum of three different samples for each condition was quantified in triplicate, and a negative control without cDNA template was run with every assay to assess overall specificity. Quantitative analysis of the data was performed using the iCycler analysis software. Expression levels were normalized against HPRT with PCR efficiency correction.
Laser capture microdissection and quantitative PCR.
Snap-frozen tissue was cut using a cryostat (Leica) at 20 μm and collected onto membrane slides (P.A.L.M Microlaser Technologies). Tissue around GFP-positive transplanted cells or the control region was microdissected using a Zeiss PALM laser capture microdissection (LCM) microscope, and total RNA was extracted using an RNAqueous-Micro LCM kit from Ambion according to the manufacturer's protocol. Total RNA (20–30 ng) was reverse transcribed using QuantiTect reverse-transcription kit from Qiagen. GFP expression was confirmed in transplanted tissues with standard PCR (GFP forward: 5′-CGACCACTACCAGCAGAACA-3′; GFP reverse: 5′-GAACTCCAGCAGGACCATGT-3′). cDNA from the above reaction was used in real-time PCR without dilution. The primers were as follows: mBDNF forward: 5′-TCATACTTCGGTTGCATGAAGG-3′; mBDNF reverse: 5′-AGACCTCTCGAACCTGCCC-3′; mGDNF forward: 5′-CACTCCTGGATTTGCTGATGT-3′; mGDNF reverse: 5′-AGTGTGCGGTACTTGGTGC-3′; mNGF forward: 5′-CCAAGGACGCAGCTTTCTATAC-3′; mNGF reverse: 5′-CTGCCTGTACGCCGATCAAAA-3′; mCNTF forward: 5′-TAGAGCGGCTACAGAGGTCC-3′; mCNTF reverse: 5′-CAAACCAGCTCACTTGTTTCC-3′.
Results
Progressive superficial cortical neuron loss in human mutant P301S tau mice
To determine whether there was layer specificity of cortical neurodegeneration, we undertook detailed quantitative neuronal and total cell count analysis on cortices from transgenic P301S tau and C57BL/6 control mice. Cortical neurodegeneration was quantified using cresyl violet cell counts in both hemispheres of brains from 5-month-old transgenic mice and age-matched controls. This is the latest time point at which the mice can be studied because of the need for humane killing on account of severe disability. A significant decrease in cresyl violet-positive cells was observed in the superficial cortex, defined as from the surface to a depth of 150 μm, in P301S tau-transgenic mice (1622 ± 125 cells per mm2) compared with C57BL/6 controls (2425 ± 495 cells per mm2, p < 0.001) (Fig. 1 A,B,E). To confirm superficial cortical neuron loss, quantification of NeuN and immunolabeling for GABA were undertaken (Snyder et al., 1997; Park et al., 2006). A significant reduction in the percentage area of the superficial cortex containing NeuN-positive cortical neurons was observed in P301S tau mice (10.6 ± 6.2%), compared with controls (33.6 ± 12.9%, p < 0.001) (Fig. 1 C,D,F) along with a reduction in the number of GABAergic cells (73.9 ± 20.8 in P301S tau mice versus 339.3 ± 44.1 in controls, p < 0.001) (Fig. 2 B–D). No significant changes were observed in deeper cortical layers (Figs. 1 E,F, 2 D). Quantitative PCR and immunohistochemistry of layer-specific markers also showed a significant loss in transgenic mice of Reelin, a marker of predominantly superficial layers (D'Arcangelo et al., 1995; Shen et al., 2006; Lein et al., 2007; Molyneaux et al., 2007), when compared with age-matched controls (Fig. 2 A,E,F), while Cux1 in deeper cortical layers displayed no significant change. Together, these findings demonstrate significant neuronal cell loss restricted to the superficial cortex in human P301S tau transgenic mice at 5 months of age.

Neuronal cell loss in the superficial cerebral cortex of mice transgenic for human mutant P301S tau. A , B , Cresyl violet counts were performed on cerebral cortex of 5-month-old C57BL/6 ( A ) and P301S tau ( B ) mice (dashed lines demarcate cortical layers). C–F , Significant neuronal loss was confined to the superficial cortex (rostral of bregma) of the P301S tau mice ( E , p < 0.001). Image-based threshold analysis of NeuN (Scott et al., 2005; Hampton et al., 2008) confirmed the significant loss of layer I and layer II neurons in 5-month-old P301S tau mice ( C , D , F , p < 0.001). Scale bar: (in A ) A–D , 200 μm.

Neuronal subtype loss and reduction in molecular marker of superficial cortical neurons in mice transgenic for human mutant P301S tau. A , Analysis of expression of mRNAs encoding layer-specific markers Reelin, Cux2, and ER81 by quantitative PCR showed a significant reduction in Reelin mRNA expression in transgenic mice compared with controls. B–D , Quantification of GABAergic cells revealed a significant reduction in both 3- and 5-month-old transgenic mice compared with C57BL/6 controls (p < 0.001, arrows indicate GABA-positive cells). E–H , Immunohistochemistry for the layer–specific marker Reelin revealed superficial nerve cell loss in the P301S tau mice ( F , arrowheads indicate Reelin-positive cells) compared with C57BL/6 controls ( E ), while no significant change in neuronal cell number was observed in deeper layers using immunostaining for either Cux1 ( G , H ) or TBR1 (T box brain 1) (data not shown). Scale bars: B (for B , C ), E (for E–H ), 100 μm.
To determine the temporal progression of neurodegeneration, quantitative analysis of the superficial cortex of P301S tau transgenic and C57BL/6 control mice was undertaken at 2 and 3 months of age. NeuN analysis revealed no significant loss at 2 months (Fig. 3 A,B,E, 18.4 ± 20.9%). However, by 3 months (45.3 ± 12.4%, p < 0.001) and 5 months (76.9 ± 16.6%, p < 0.001) of age there was a progressive and significant loss of neurons in the superficial cortex of P301S tau mice compared with age-matched controls (Fig. 3 C–E, supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Temporal characterization of staining with the phosphorylation-dependent anti-tau antibody AT8 was also performed. We saw the staining change from a ring-like appearance at 2 months, through an increase in cell body and dendritic staining at 3 months, to multiple, abundant tau inclusions at 5 months (Fig. 3 K–M). This was not exclusive to the superficial layers, and AT8 changes were observed in deeper layers.

Progressive neuron loss, astrocytosis, and accumulation of hyperphosphorylated tau in the cerebral cortex of mice transgenic for human mutant P301S tau. A–E , Serial quantitative NeuN analysis of the superficial cortex of P301S tau and C57BL/6 mice at 2, 3, and 5 months of age showed progressive nerve cell loss (p < 0.001). F–J , GFAP staining showed a progressive increase in P301S tau mice compared with C57BL/6 controls. K–M , Immunohistochemistry using anti-tau antibody AT8 demonstrated the progressive accumulation of hyperphosphorylated tau in the superficial cortex at 2, 3, and 5 months of age (dashed line demarcates cortical layers I and II, taken 1 mm lateral of the midline and rostral to bregma). AT8 staining went from a ring-like accumulation at 2 months ( K ), through an increase in cell bodies and dendrites at 3 months ( L ), to the presence of abundant inclusions at 5 months ( M ). Scale bars: A (for A–D ), F (for F–I ), K (for K–M ),100 μm.
In addition to progressive neuron loss, a significant increase in the number of GFAP-positive astrocytes was observed within the superficial frontal cortex of P301S tau mice compared with C57BL/6 controls from 2 to 5 months of age (Fig. 3 F–J, 23.3 ± 12.0 vs 8.3 ± 6.1, 109.2 ± 33.8 vs 13.1 ± 8.9, and 156.7 ± 33.7 v 11.7 ± 9.3 respectively; supplemental Fig. 1B–D, available at www.jneurosci.org as supplemental material). Serial comparisons of brain volume and total bisbenzamide-positive cell nuclei throughout all layers of the frontal cortex between P301S and control brains revealed no differences and, thus, confirmed progressive neuron cell loss specific to the superficial layers (supplemental Fig. 2A–D, available at www.jneurosci.org as supplemental material).
Together, these findings reveal that P301S tau mice display progressive, superficial cortical neuron loss with associated astrogliosis. Furthermore, at least a proportion of neurons containing the ring-like tau deposits die before the formation of abundant filamentous tau inclusions.
Transplanted neural precursors and astrocytes are neuroprotective
To test whether implantation of NPCs may influence the progressive superficial neuronal loss, cortices from neonatal C57BL/6 mice ubiquitously expressing eGFP (Okabe et al., 1997) were cultured under substrate-free conditions in the presence of EGF and FGF-2 (Vescovi et al., 1993). In vitro characterization confirmed neuronal and glial phenotypic potential of eGFP-NPCs upon plating and withdrawal of mitogens (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). At 2 months of age, P301S tau mice and age-matched controls received either live eGFP-NPCs (8 × 105/μl), dead eGFP-NPCs at an equal concentration, or vehicle unilaterally into the cortex at 2 sites, both under 1 mm lateral of the midline, the first 0.5 mm and the second 1.8 mm rostral of bregma. Brains were then analyzed at 1 month (3 months of age) and 3 months (5 months of age) after transplantation.
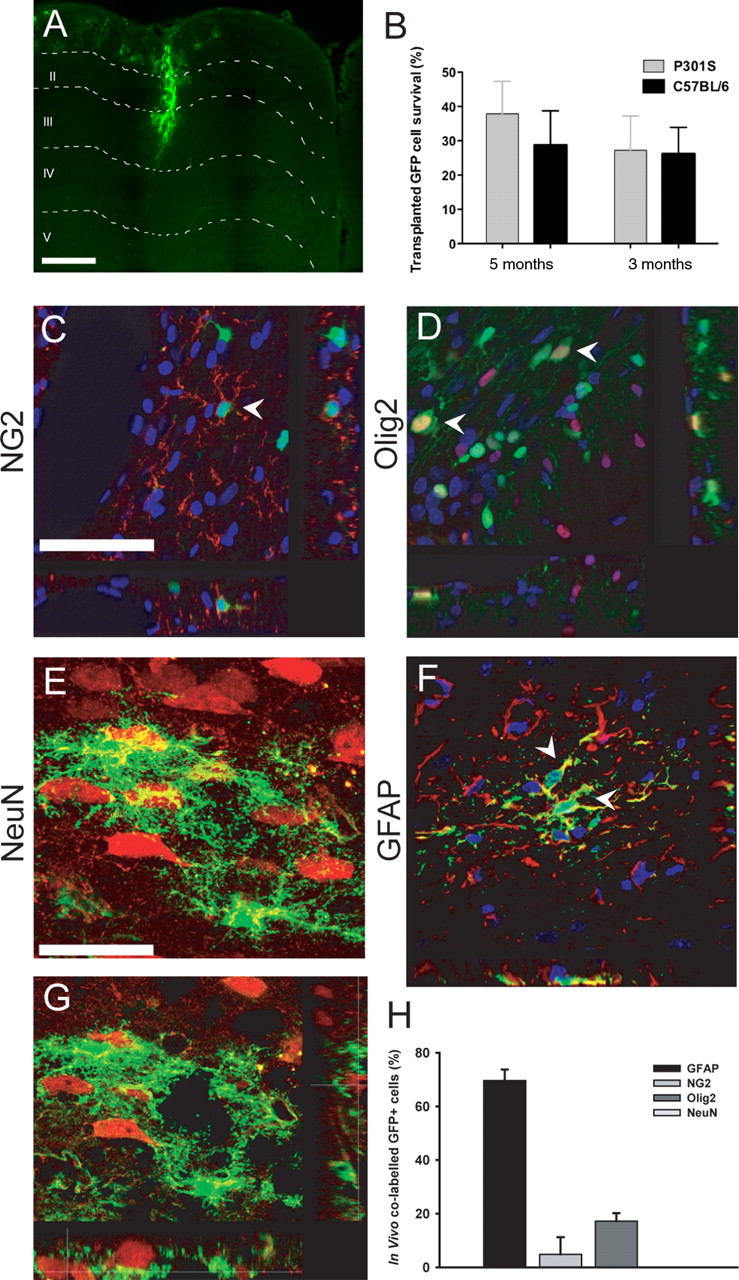
Immunohistochemistry revealed GFP+ cells in the cortical gray matter of P301S tau mice (Fig. 4A), while stereological analysis revealed that ∼30% of transplanted cells had survived and integrated into host tissue, as visualized by GFP-positive cell processes “wrapping” around host neurons up to 3 months after transplantation (Fig. 4 B) in both P301S tau and C57BL/6 mice. Phenotypic differentiation of transplanted eGFP-NPCs was examined by immunostaining. Although GFP+ cells were observed in close proximity to endogenous neurons and occasionally extended processes around them, there was no evidence of graft-derived neuronal differentiation, as assessed by GFP costaining with NeuN or doublecortin, at either 1 month (Fig. 4 E,G) or 3 months after transplantation. The majority of GFP+ cells costained for GFAP, consistent with an astroglial identity (Fig. 4 F, 69.6 ± 4.0%), with the remainder costaining with NG2 (Fig. 4 C, 4.8 ± 6.3%) and Olig2 (Fig. 4 D, 17.2 ± 2.9%). There was no evidence of staining for markers of mature oligodendrocytes, such as O4 or MBP.

eGFP-NPCs survive, integrate, and differentiate in the cerebral cortex of mice transgenic for human mutant P301S tau. A , B , Significant numbers of eGFP-NPCs were identified after transplantation. GFP+ cells were present in cortical gray and white matter, migrating along corpus callosum and cingulum ( A ). Stereological analysis revealed that ∼30% of cells had survived and integrated 1 and 3 months after transplantation ( B ). C–G , Following the labeling of brain sections with several phenotypic markers, confocal microscopy was used to examine the fate commitment of transplanted eGFP-NPCs. It was found that GFP-NPCs colabeled with NG2 ( C ) and Olig2 ( D ), markers of the oligodendrocyte lineage, and with GFAP ( F ), a marker of astrocytes (arrowheads indicate colabeled cells). Although GFP+ cells were found to surround NeuN+ neurons and to extend processes around host NeuN+ cells, no evidence of colabeled NeuN-GFP cells at either 1 month ( E , and an orthogonal projection image from this series, G ) or 3 months (data not shown) after transplantation was observed. Quantitative analysis of differentiation following transplantation showed that the majority of cells were GFAP positive (69.6 ± 4.0%), while immature oligodendrocyte differentiation was characteristic of a smaller number of cells (4.8 ± 6.3%) and (17.2 ± 2.9%) for NG2 and Olig2, respectively ( H ). Scale bars: (in A and C ) A , C , D , F , 250 μm; (in E ) E , G , 25 μm.
Having established the exclusive glial differentiation of focally transplanted NPCs, NeuN analysis of the superficial cortex of both hemispheres of mice that had received unilateral delivery of live GFP-NPCs, dead GFP-NPCs, or vehicle at 2 months of age was undertaken. A significant increase in cortical neuronal numbers was observed 1 month after transplantation in the hemisphere that had received GFP-NPCs compared with the contralateral, nontransplanted hemisphere in P301S tau mice, with no effect in C57BL/6 mice (Fig. 5 A–C, p < 0.001). Analysis of both vehicle alone (Fig. 5 C) and dead GFP-NPC cell transplants (data not shown) showed that neither had a beneficial effect in P301S tau mice (Fig. 5 C). Transplantation of live GFP-NPCs or dead GFP-NPCs into C57BL/6 mice had no detrimental effect on cortical neuronal cell numbers (Fig. 5 C). Following transplantation of dead GFP-NPCs, NeuN analysis revealed that only 16.1 ± 4.0% of the area of cortical layers I and II contained a NeuN signal, which was comparable to what was observed in the contralateral hemisphere of P301S tau mice transplanted with live GFP-NPCs (16.2 ± 3.6%), the vehicle-injected hemisphere, and its contralateral noninjected hemisphere (16.4 ± 6.0% and 14.4 ± 6.6%, respectively). The same was also true of the nontransplanted contralateral hemisphere of the GFP-NPC-transplanted P301S tau mice (15.5 ± 2.9%). A significant increase in neuronal survival was also observed in animals killed 3 months after transplantation (Fig. 5 D–F).
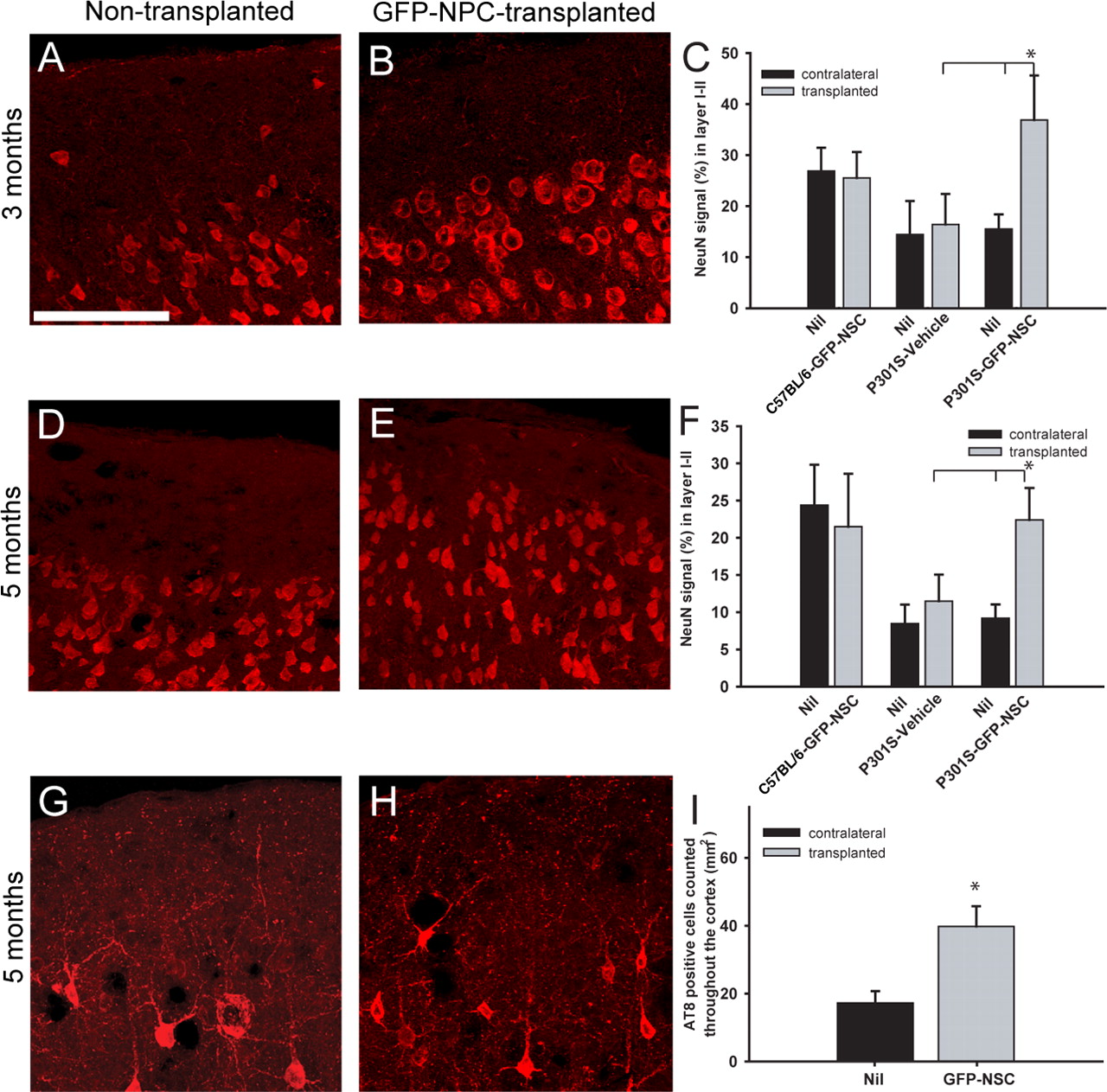
Transplantation of eGFP-NPCs reduces neuron loss in the superficial cortex of mice transgenic for human mutant P301S tau. A–C , One month after transplantation (3 months of age), a significant increase in the number of neurons in the superficial cortex was observed in the transplanted hemisphere when compared with the contralateral hemisphere, vehicle control-injected mice, and dead cell transplants (data not shown) (p < 0.001). D–F , Similarly, 3 months after transplantation, neuron cell numbers were significantly higher in layers I and II of the injected hemisphere when compared with the contralateral side and with vehicle controls (p < 0.001). G–I , Transplantation of eGFP-NPCs resulted in a significant increase in the number of AT8-positive cells in the superficial cortex 3 months after transplantation when compared with the contralateral hemisphere (p < 0.001). Scale bar: (in A ) A , B , D , E , G , H , 100 μm.
We next examined staining for phosphorylated tau following focal transplantation of GFP-NPCs. No cells were labeled for both AT8 and GFP. Significantly, more AT8-positive cells were present in the superficial cortical layers I and II of hemispheres injected with eGFP-NPCs than in the contralateral hemisphere or the hemispheres of vehicle controls (Fig. 5 G–I, p < 0.001). To confirm the neuroprotective effect of NPC transplantation, immunolabeling for NF200, WFA, and GABA was carried out. WFA colocalizes with GABAergic interneurons (Härtig et al., 1992; Brückner et al., 1993). Examination of NF200 density revealed a significant loss of neurofilaments in P301S tau mice compared with age-matched controls (Fig. 6 A,B, D). Densitometric analysis of NF200 3 months after transplantation of GFP-NPCs into P301S tau mice showed a significant increase in density, like that in C57BL/6 controls (Fig. 6 C,D). A similar pattern was also observed for WFA. (Fig. 6 E,F,H). Following transplantation, WFA-positive nets were significantly increased, equivalent to the levels in C57BL/6 mice (Fig. 6 G,H). Furthermore, when GABAergic neurons were examined in brain sections from GFP-NPC-transplanted and vehicle controls, a significant increase in the number of GABAergic cells was observed in P301S tau mice 1 and 3 months after transplantation (78.0 ± 26.3 to 220.5 ± 45.8 and 68.2 ± 24.4 to 229.8 ± 23.9, respectively, p < 0.001) (Fig. 7 A–E). Quantitative PCR of cortical layer-specific markers revealed a significant increase in Reelin mRNA expression following transplantation of GFP-NPCs (Fig. 7 F). Together, these findings confirm a neuroprotective effect of focal NPC implantation on the age-dependent loss of superficial cortical neurons in P301S tau transgenic mice.

Immunohistochemistry for NF200 and WFA shows superficial cortical neuroaxonal loss and protection following transplantation of eGFP-NPCs. A–C , Confocal maximum projection images of NF200-stained sections from 5-month-old C57BL/6 mice ( A ), P301S tau mice ( B ), and P301S tau mice following eGFP-NPC transplantation ( C ). D , Quantification of NF200 using threshold analysis demonstrated a significant decrease in density, which was significantly ameliorated following transplantation of eGFP-NPCs. E–H , WFA staining of cerebral cortex of C57BL/6 mice ( E ) with arrows highlighting examples of perineuronal nets that were lost in age-matched P301S tau transgenic mice ( F ). Following transplantation of eGFP-NPCs, WFA nets were significantly increased in number (p < 0.001) ( G ), equivalent to what was observed in C57BL/6 mice ( H ). Scale bars: A (for A–C ), E (for E–G ),50 μm.

A–E , Transplantation of eGFP-NPCs results in neuroprotection within the superficial cerebral cortex of mice transgenic for human P301S tau. At 1 and 3 months after transplantation, a significant increase in the number of GABAergic neurons was observed in the transplanted hemispheres compared with vehicle controls (p < 0.001). F , Transplantation of eGFP-NPCs also resulted in a significant increase in Reelin mRNA expression (p < 0.05). Scale bar: (in A ) A–C , 100 μm.
To examine whether cell transplantation resulted in a local increase in neurotrophic factors as suggested by the concept of the bystander neuroprotective effect (Ourednik et al., 2002; Pluchino et al., 2005), we undertook LCM and candidate growth factor qPCR analysis. Real-time (RT) PCR analysis confirmed that GFP mRNA was only expressed in the LCM-captured, GFP-NPC transplanted tissue and not in vehicle control samples (Fig. 8 A). Although expression of both CNTF and GDNF mRNAs was increased in areas containing GFP+ cells, only GDNF mRNA was significantly elevated relative to controls (p < 0.05) (Fig. 8 B). There was no increase observed in BDNF mRNA following transplantation. The observed increase in GDNF mRNA was accompanied by immunohistochemistry and confocal microscopy, revealing strong GDNF immunostaining restricted to the GFP+ transplant (Fig. 8 C,D). Interestingly, BDNF and NGF immunohistochemistry also appeared elevated in the GFP-NPC transplanted mice (supplemental 4A-D, available at www.jneurosci.org as supplemental material). These observed increases were restricted to regions of close proximity to the GFP+ transplant.

Quantification of expression of GDNF following transplantation of eGFP-NPCs. A , B , LCM and semiquantitative RT-PCR were used to identify GFP+ cells following transplantation. GFP mRNA was only detected in LCM samples from eGFP-NPC-transplanted P301S tau mice ( A ). HPRT was used as a loading control. Transplantation of eGFP-NPCs resulted in a significant increase in GDNF mRNA (p < 0.05) compared with control samples, as measured by quantitative PCR following LCM ( B ). C , D , GDNF immunohistochemistry showing a maximum projection image of GDNF, GFP and bisbenzamide following transplantation of eGFP-NPCs ( C ) and an orthogonal projection image ( D ) from this series showing GFP-positive processes located around an area of increased GDNF staining. Scale bars: C , D , 40 μm.
To address whether the protective effect was NPC dependent or glial dependent, we undertook unilateral focal transplantation of ex vivo differentiated eGFP-astrocytes derived from treatment of NPCs with BMP4 (Gross et al., 1996; Mabie et al., 1997). In vitro characterization showed that 96.2 ± 3.7% of cells were GFAP positive following 96 h of BMP4 treatment (Fig. 9 A). eGFP-astrocytes were transplanted unilaterally into the cortex of 2-month-old P301S tau mice using coordinates identical to those used earlier for the NPC transplants. Analysis at 1 month following transplantation revealed GFP+ cells located in the gray matter of transgenic mice (Fig. 9 B). These cells were immunopositive for GFAP and negative for NG2, NeuN, and AT8 (Fig. 9 B,C). Having established that eGFP-astrocytes survived and integrated into the host gray matter and did not differentiate into neurons, NeuN analysis of the superficial cortex was undertaken. A significant increase in the area containing NeuN signal was found in the transplanted hemisphere compared with the contralateral, nontransplanted hemisphere and the vehicle-injected hemisphere (Fig. 9 D,E, p < 0.001).

Transplantation of eGFP-astrocytes reduces neuron loss in the superficial cortex of mice transgenic for human P301S tau. A , GFP-NPCs were derived and differentiated into astrocytes using BMP4, followed by transplantation into P301S tau mice. B , C , Following unilateral transplantation, GFP+ astrocytes were identified within the gray matter of P301S tau mice and found to express the astrocytic marker GFAP (arrowheads indicate GFP-positive astrocytes). D–F , One month after transplantation, a significant increase in nerve cell numbers in the superficial cortex was observed in the transplanted hemisphere, when compared with the contralateral hemisphere and to vehicle controls (p < 0.001). Scale bars: (in A , C , D ) A–E , 100 μm.
Discussion
Our findings demonstrate a focal neuroprotective effect of transplanted NPCs and astrocytes in a mouse model of age-related, layer-specific cortical neurodegeneration. Although the mechanisms underlying neurodegeneration are unknown, the accumulation of hyperphosphorylated filamentous tau protein is characteristic of several human neurodegenerative diseases, including AD and cases of FTD. Mutations in Tau cause familial forms of FTD, establishing that dysfunction of tau protein is sufficient to cause neurodegeneration and dementia (Goedert and Spillantini, 2006).
Transgenic mice expressing human mutant tau in neurons exhibit the essential features of tauopathies, including neurodegeneration and abundant filaments made of hyperphosphorylated tau protein. Our results show a significant, progressive reduction of neurons in the superficial frontal cortex (layers I and II) of P301S tau mice compared with age-matched controls by using a range of quantitative morphological, histological, and molecular outcomes. Similar to other parts of the CNS (Allen et al., 2002; Delobel et al., 2008), human mutant tau accumulated over time in layers I and II, became hyperphosphorylated, and formed neuronal inclusions. Neuron loss, which was maximal at 5 months of age, the latest time point that could be studied, was already significant in 3-month-old mice and preceded the formation of abundant tau inclusions. Neuronal loss has been observed in this transgenic model within a number of CNS regions, including amygdala, brainstem, and spinal cord (Allen et al., 2002; Bellucci et al., 2004; Delobel et al., 2008; Gasparini et al., 2009).
In parallel with neuron loss, we also observed an increase in GFAP-positive astrocytes throughout the superficial cortex. This is particularly interesting, since endogenous, reactive astrocytes in conjunction with tau may play an important role in the progression of FTD (Broe et al., 2004; Kersaitis et al., 2004; Tan et al., 2005). Indeed, in human cases, there is an association between the extent of astrocytosis and the severity of disease (Broe et al., 2004). Furthermore, cortical astrogliosis appears to precede neuronal loss, suggesting an important role for astroglia in FTD (Broe et al., 2004; Kersaitis et al., 2004).
These findings reveal for the first time that P301S tau mice display progressive neuron loss with associated astrogliosis in the superficial layers of the cerebral cortex. This is similar to human FTD, where postmortem examination has shown significant neuronal loss and gliosis in the superficial frontal cortex (Rosso et al., 2001; Broe et al., 2004; Leverenz et al., 2007; Mackenzie et al., 2008).
Accumulating evidence has implicated cell-mediated neuroprotection through a variety of mechanisms ranging from cell replacement to growth factor-mediated functional recovery (Martino and Pluchino, 2006; Lepore et al., 2008, Barhum et al., 2010). Previous studies using a tetracycline-inducible promoter to focally ablate neurons in the hippocampus have demonstrated a significant reduction in NeuN+ cells in the CA1 region with evidence of functional improvement following focal transplantation of NPCs (Yamasaki et al., 2007). Furthermore, a recent study in a transgenic mouse model of AD exhibiting hippocampal synaptic dysfunction revealed functional improvement following focal NPC transplantation, accompanied by increased synaptic density mediated by increased production of BDNF (Blurton-Jones et al., 2009). This is of interest given that in the current study we observed an increase in neurotrophins, particularly GDNF, following transplantation of eGFP-NPCs. To examine whether focal implantation of neural populations may influence neuron loss in our model, we undertook focal transplantation studies. In vitro characterization confirmed the trilineage neuronal and glial phenotypic potential of eGFP+ stem cells (Vescovi et al., 1993; Webber et al., 2007). Following transplantation of eGFP-NPCs, we observed glial differentiation without graft-derived neuronal differentiation at 1 and 3 months after transplantation, consistent with previous studies using injury paradigms (Cao et al., 2001, 2002; Hofstetter et al., 2005; Webber et al., 2007; White and Jakeman, 2008). We demonstrate that following unilateral focal transplantation of eGFP-NPCs, superficial cortical neurons are protected from neurodegeneration, with transplanted NPCs being able to counteract the neurotoxic effects of misfolded tau. This was not a consequence of cell replacement, as no evidence of neuronal differentiation was obtained. We present several lines of evidence to demonstrate a neuroprotective effect of NPCs within the superficial cortex, including multiple nerve cell counts and analyses at both 1 and 3 months after transplantation. In addition, the finding that after 3 months P301S tau mice had significantly more AT8-positive cells per mm2 in the superficial cortex of hemispheres injected with eGFP-NPCs suggests that transplanted NPCs exert a neuroprotective effect independently of the progressive accumulation of hyperphosphorylated tau. The exclusively glial, predominantly astrocytic differentiation of grafted NPCs suggests that the neuroprotective effect of NPCs is glial dependent. To address this hypothesis, astrocyte transplantation was undertaken. Focal transplantation of ex vivo NPC-derived astrocytes that integrate and differentiate only into GFAP+ cells also resulted in a neuroprotective effect of superficial cortical neurons, a finding consistent with a glial-mediated neuroprotective effect of cell transplantation.
The precise mechanisms underlying the neuroprotection observed in our study are unknown. The comparable effects of transplantation of NPCs that differentiate into astrocytes and of direct astrocyte transplantation suggest that graft-derived astrocytes mediate the protective effect in P301S tau mice. This finding is consistent with recent studies that have reported an astroglia-mediated neuroprotective effect in different models of neuronal injury (Boucherie et al., 2009; Davies et al., 2006, 2008; Lepore et al., 2008). In view of the accumulating evidence suggesting a role for astrocytes in noncell-autonomous neurodegeneration (Boillee et al., 2006; Yamanaka et al., 2008), one interpretation is that exogenous astrocytes are improving a hostile glial environment. The concept of astrocytes being injurious or protective depending on context is supported by a growing literature (Davies et al., 2008; Lepore et al., 2008). This idea has gained support from a study showing that focal astrocyte transplantation exerted a neuroprotective effect with functional improvement in transgenic rats expressing human mutant superoxide dismutase-1 (SODG93A) (Lepore et al., 2008). The beneficial effect was, in part, mediated by the primary astrocyte glutamate transporter GLT1. Additional putative glia-mediated neuroprotective mechanisms suggested from spinal cord injury transplantation studies include immune modulation, suppression of proteoglycan inhibition, and secretion of neurotrophic growth factors (Davies et al., 2006; Boucherie et al., 2009). In this regard, our finding of an increase in neurotrophins in regions containing GFP+ transplanted cells, similar to the “bystander” effect observed in previous studies of neural stem cell populations, is of interest (Ourednik et al., 2002; Pluchino et al., 2005; Martino and Pluchino, 2006; Nagahara et al., 2009). The observed increase in neurotrophins following transplantation suggests local graft-mediated growth factor delivery as a potential explanation for the observed neuroprotection. Indeed, direct administration of BDNF in a rodent model of AD exerts substantial protective effects, leading to reduction in synaptic loss and restoration of learning and memory (Nagahara et al., 2009). Another potential mechanism for the neuroprotective effects of both the glial-derived NPC transplant and the direct astrocytes may be through secretion of activity-dependent neuroprotective protein (ADNP). Recent literature suggests this protein is secreted by astrocytes, while intranasal delivery of a peptide fragment derived from ADNP, termed NAP, has been demonstrated to increase soluble tau, reduce hyperphosphorylation, and reduce cognitive decline observed in tau transgenic mice (Furman et al., 2004; Shiryaev et al., 2009). This mechanism may be an additional effect that the exogenous transplanted astrocytes have on the endogenous populations and thus promotes neuroprotection.
In conclusion, the present study reports that exogenous NPCs and astrocytes exert a powerful neuroprotective effect in a novel model of progressive age-related, layer-specific, cortical neurodegeneration caused by dysfunction of tau protein. Our findings provide an important platform for exploring the mechanisms of glia-mediated neuroprotection and have significant implications for the development of future treatment strategies for neurodegenerative disorders, such as AD and FTD.
Footnotes
-
This work was supported by grants from the UK Medical Research Council and the Multiple Sclerosis Society.
- Correspondence should be addressed to either of the following: Siddharthan Chandran, Euan MacDonald Centre for Motor Neurone Disease Research, Centre for Regenerative Medicine, University of Edinburgh, Chancellor's Building, 49 Little France Crescent, Edinburgh EH16 4SB, UK, siddharthan.chandran{at}ed.ac.uk; or Maria Grazia Spillantini, Centre for Brain Repair, Department of Clinical Neurosciences, University of Cambridge, Robinson Way, Cambridge CB2 2PY, UK, mgs11{at}cam.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}