
Abstract
Homeostatic control of synaptic efficacy is often mediated by dynamic regulation of excitatory synaptic receptors. Here, we report a novel form of homeostatic synaptic plasticity based on regulation of shunt currents that control dendritosomatic information transfer. In cortical pyramidal neurons from wild-type mice, HCN1 channels underlie a dendritic hyperpolarization-activated cationic current (I h) that serves to limit temporal summation of synaptic inputs. In HCN1 knock-out mice, as expected, I h is reduced in pyramidal neurons and its effects on synaptic summation are strongly diminished. Unexpectedly, we found a markedly enhanced bicuculline- and L-655,708-sensitive background GABAA current in these cells that could be attributed to selective upregulation of GABAA α5 subunit expression in the cortex of HCN1 knock-out mice. Strikingly, despite diminished I h, baseline sublinear summation of evoked EPSPs was unchanged in pyramidal neurons from HCN1 knock-out mice; however, blocking tonic GABAA currents with bicuculline enhanced synaptic summation more strongly in pyramidal cells from HCN1 knock-out mice than in those cells from wild-type mice. Increasing tonic GABAA receptor conductance in the context of reduced I h, using computational or pharmacological approaches, restored normal baseline synaptic summation, as observed in neurons from HCN1 knock-out mice. These data indicate that upregulation of α5 subunit-mediated GABAA receptor tonic current compensates quantitatively for loss of dendritic I h in cortical pyramidal neurons from HCN1 knock-out mice to maintain normal synaptic summation; they further imply that dendritosomatic synaptic efficacy is a controlled variable for homeostatic regulation of cortical neuron excitability in vivo.
Introduction
Hyperpolarization-activated mixed cationic currents (I h) represent a unique class of neuronal currents (for review, see Pape, 1996; Biel et al., 2009). Unlike most other voltage-dependent currents, I h activates when the membrane is hyperpolarized from resting potentials, providing increasing inward (i.e., depolarizing) current with membrane hyperpolarization. Conversely, its deactivation upon depolarization decreases net inward current to provide a hyperpolarizing bias to membrane potential. This combination of properties yields a current that stabilizes resting membrane potential (Pape, 1996; Biel et al., 2009). In hippocampal and cortical pyramidal neurons, in which I h has a predominant dendritic localization, these properties are best appreciated for their influence on dendritosomatic synaptic integration (for review, see Magee, 2000). In this context, the membrane-stabilizing actions of dendritic I h diminish depolarizing effects of EPSCs and limit synaptic efficacy (Magee, 2000).
The cloning of the HCN channels that underlie I h (Ludwig et al., 1998; Santoro and Tibbs, 1999) has provided the opportunity to identify the molecular basis for native neuronal currents (Santoro et al., 2000) and to manipulate their expression in genetically modified mice. In pyramidal neurons of hippocampus and cortex, HCN1 is the predominant subunit and genetic deletion of this subunit greatly reduces I h in those cells (Nolan et al., 2004; Chen et al., 2009b). In hippocampus, the reduction in I h is associated with enhanced long-term potentiation at the perforant path input to distal dendrites of CA1 pyramidal neurons and, correspondingly, with improved hippocampal-dependent learning and memory (Nolan et al., 2004). In addition, HCN1 gene deletion decreases I h in small, cold-sensitive sensory neurons and diminishes behavioral responses to cooling (Orio et al., 2009); it also reduces I h in cerebellar Purkinje neurons and disrupts motor learning (Nolan et al., 2003). Although these studies with knock-out mice show clear consequences of genetic deletion of HCN1 in defined behavioral contexts, the overall disposition of these animals is remarkably normal. This is somewhat surprising given prevailing ideas regarding the enhanced neural network activation expected to accompany a decrease in I h. For example, acute inhibition of I h with ZD-7288 enhances recurrent cortical network activity (Wang et al., 2007) and decreased expression of HCN1 is implicated as a potential causative factor in various rodent epilepsy models (Shah et al., 2004; Jung et al., 2007; Kole et al., 2007; Huang et al., 2009). However, such aberrant cortical activity has not been reported in HCN1 knock-out mice, suggesting that compensatory mechanisms may limit the excessive excitability predicted to result from diminished I h.
In this paper, we report a compensatory upregulation of tonic GABAA current mediated by α5-containing receptors in neocortical pyramidal neurons of HCN1 knock-out mice. We further show that this enhanced tonic GABAA conductance effectively blunts synaptic summation in cortical pyramidal neurons, apparently by providing an alternative current shunt to offset the loss of I h. These results indicate that cortical pyramidal neurons can engage distinct mechanisms to maintain normal synaptic summation properties. Further, they suggest that dendritosomatic synaptic integration is an important variable that is monitored for purposes of homeostatic control of cortical neuron excitability.
Materials and Methods
HCN1 knock-out mice.
All animal use was in accordance with guidelines approved by the University of Virginia Animal Care and Use Committee. Mice used in this work were age-, sex-, and weight-matched HCN1 knock-out and B6129SF2/J mice; both represent hybrids of the parental C57BL/6 and 129Sv substrains (Jackson Laboratories; stock #005034 and #101045). Mice homozygous for the targeted mutation are viable, fertile, and normal in size and longevity and they do not display any gross physical or behavioral abnormalities (Nolan et al., 2003, 2004). For some experiments, we compared HCN1+/+ and HCN1−/− littermates; these animals were generated from heterozygous HCN1+/− mice obtained by crossing animals from the homozygous HCN1 knock-out line with C57BL/6J mice.
Drugs and reagents.
To block GABAA currents nonselectively we used bicuculline (30 μm; Sigma) or gabazine (SR-95531, 20 μm; Sigma); we used the partial agonist L-655,708 (20 μm; Sigma) to block α5 subunit-containing GABAA receptors, and two agonists to selectively activate GABAA receptors containing α1 subunits (zolpidem, 100 nm; Sigma) and δ subunits (THDOC, tetrahydrodeoxycorticosterone, 100 nm; Sigma). The benzodiazepine diazepam was also used to activate GABAA receptors (1 μm; Sigma). In some experiments, ZD-7288 was used to block I h (50 μm; Tocris Cookson) and tetrodotoxin (TTX, 0.5 μm; Alomone Labs) was used to block voltage-dependent Na channels and action potential-dependent synaptic release; where noted, glutamate receptors were blocked with CNQX (10 μm; Sigma) and APV (50 μm; Sigma). Isoflurane was added to the perfusate by bubbling through calibrated vaporizers (Ohmeda).
Recording from mouse cortical pyramidal neurons.
Mice of either sex (14–22 d old) were anesthetized (ketamine/xylazine: 200/14 mg/kg, i.m.) and transverse brain slices were prepared as described previously (Chen et al., 2009a). Slices were submerged in a recording chamber on a Zeiss Axioskop 2 FS Plus microscope and visualized with Nomarski optics; pyramidal cells located in layer 5 of somatosensory cortex or area CA1 of hippocampus were targeted for recording in slices based on characteristic size and shape.
Recordings were obtained using pCLAMP software and an Axopatch 200B amplifier interfaced via a Digidata 1322A digitizer at room temperature (∼24°C); bath solution was perfused at ∼2 ml/min and contained the following (in mm): 140 NaCl, 3 KCl, 10 HEPES, 2 CaCl2, 2 MgCl2, 10 glucose. For voltage-clamp recording of tonic and phasic GABAA currents, pipettes (2–4 MΩ) were filled with the following (in mm): 125 CsCl, 0.1 CaCl2, 1 EGTA, 10 HEPES (pH 7.3); for current clamp, pipette solution contained the following (in mm): 17.5 KCl, 122.5 potassium gluconate, 10 HEPES, 0.2 EGTA, 9 NaCl, 1 MgCl2, 3 MgATP, 0.3 GTP-Tris (pH 7.2). Evoked EPSPs were recorded with internal containing the following (in mm): 128 CsCH3O3S, 9 NaCl, 2 MgCl2, 10 HEPES, 10 EGTA (pH 7.3). For perforated-patch recordings, gramicidin (10 mg/ml in DMSO; Sigma) was sonicated, diluted (30–60 μg/ml), and added to pipettes containing the following (in mm): 120 KCH3O3S, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 10 HEPES, 10 EGTA, 3 MgATP, 0.3 GTP-Tris, pH 7.2. Cell capacitance and series resistance (R s, typically <20 MΩ) were compensated using amplifier circuits (∼70%); compensation was continuously monitored throughout the recordings to ensure stability and to correct small changes. Cell capacitance was not different between wild-type and HCN1 knock-out mice (13.3 ± 1.2 pF vs 11.7 ± 1.1 pF, n = 42 and 36, p > 0.3). All data were corrected for a measured liquid junction potential of 8 mV.
Tonic and phasic GABAA currents were recorded under voltage clamp at a holding potential of −70 mV in the presence of a mixture of glutamate receptor blockers (CNQX, 10 μm; APV 50 μm), with the total tonic current established during blockade with either bicuculline or gabazine. All-points histograms were obtained under steady-state conditions (for 30–60 s) and fitted with Gaussian functions to provide a measure of holding current under each experimental condition (Clampfit). To evoke EPSPs, a pipette connected to a stimulator (Grass S48) via a stimulus isolation unit (Grass SIU5) was filled with standard bath solution and placed in the superficial layers of the cortex. Synaptic responses were evoked by applying 40 Hz, 5–10 V pulses (corresponding to 2–10 μA); for quantification, summation was measured as the ratio of amplitudes of the last and first evoked EPSP in the train (EPSP5:EPSP1), always from a membrane potential of approximately −70 mV (adjusted with DC). We recorded spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs, in the presence of 0.5 μm TTX) and analyzed frequency and amplitude of events using the Mini Analysis Program (Synaptosoft). Computer simulations were performed using the NEURON software package (Hines and Carnevale, 1997) and a previously published pyramidal neuron model (Day et al., 2005), kindly provided by Drs. Joshua Held and Jim Surmeier (Northwestern University, Evanston, IL). The model was adjusted to include a tonic GABAA conductance by adding a standing somatodendritic leak current with E rev = −65 mV; for simulating wild-type pyramidal neurons, the tonic GABAA conductance was set to 1/10 of the HCN conductance. To model cells from HCN1 knock-out mice, the HCN conductance was decreased by 75% and the V 1/2 shifted by −10 mV (Chen et al., 2009b) and the tonic GABAA conductance was increased by 2.5-fold. Using these parameters for wild-type and HCN1 knock-out pyramidal neurons, we compared baseline EPSP summation properties and effects of eliminating extant HCN or GABAA conductances.
Quantitative real-time PCR.
Cortical GABAA receptor subunit expression was determined in wild-type and knock-out mice by using quantitative real-time PCR (qRT-PCR). The cortex and hippocampus were microdissected for RNA isolation from n ≥ 5 animals per genotype and qRT-PCR was performed from each sample in quadruplicate, using an ICycler (Bio-Rad); each animal contributed a single data point for a given transcript. The primer sets used are provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material) and, except where noted, PCR conditions were as follows: 3 min, 95°C; 40 cycles: 15 s, 95°C; 40 s, 60°C; 40 s, 72°C. For all primer pairs and PCR conditions, preliminary experiments were performed to ensure high-efficiency amplification (all ≥95%). The identity of PCR products was verified by agarose gel electrophoresis (which yielded amplicons of appropriate size) and by melt curve analysis (which yielded a single peak near the expected melting temperature). Cyclophilin served as an internal standard and control samples with no added template were included with every experiment. We analyzed qRT-PCR data by using a modification of the so-called ΔΔCt normalization procedure to obtain GABAA receptor subunit mRNA levels for each genotype, relative to cyclophilin (Pfaffl, 2001).
GABAA receptor α5 subunit histochemistry.
In situ hybridization was performed as described previously (Talley et al., 2001; Berg et al., 2007). Following rapid decapitation of deeply anesthetized wild-type and HCN1 knock-out mice, brains were removed and rapidly frozen on dry ice. Sections (10 μm) were cut in a cryostat (Reichert Jung CM3000), thaw mounted onto charged slides (Superfrost Plus, Fisher Scientific), and stored at −80°C until use. Sections were fixed briefly (5 min) in 4% PFA, rinsed repeatedly in PBS, treated successively with glycine (0.2% in PBS) and acetic anhydride (0.25% in 0.1 m triethanolamine, 0.9% saline, pH 8), and dehydrated in a graded series of ethanols and chloroform. Hybridization was performed overnight at 60°C in a buffer of 50% formamide, 4× SSC, 1× Denhardt's solution (0.02% each of Ficoll, polyvinylpyrrolidone, and bovine serum albumin), 10% dextran sulfate, 100 mm DTT, 250 μg/ml yeast tRNA, and 0.5 mg/ml salmon testes DNA. A template for the GABAA α5 receptor subunit (∼1.3 kb) was generated by PCR, incorporating T7 and SP6 polymerase binding sites at the 5′ and 3′ ends of the template. In vitro transcription was performed using α-[33P]UTP to yield probes in either sense or antisense orientations. Radiolabeled probes were purified by ethanol precipitation, quantified by scintillation counting and used for hybridization at 1.4 × 108 cpm/ml. After hybridization, slides were washed through two changes of 4× SSC (30 min each), treated with RNase A at 37°C (50 μg/ml, 30 min), washed again through two changes each of 2× SSC and 0.5× SSC (20 min each), and finally subjected to a high-stringency wash of 0.1× SSC (30 min). All washes were performed at 37°C and included 10 mm sodium thiosulphate, with the exception of the high-stringency wash, which was performed at 62.5°C without sodium thiosulphate. Slides were exposed to film (GE Healthcare Hyperfilm β-Max; GE Healthcare) for 5–7 d and images obtained on a Zeiss Discovery V.20 microscope with a QImaging Retiga 1300 camera (QImaging) and IPLab software (BioVision).
To quantify results from in situ hybridization experiments, we determined optical density over the cortex and hippocampus (usually from three different sites) from at least three sections for each wild-type and HCN1 knock-out mouse; this was repeated across all experiments (n = 5) and the averaged value for each animal was treated as a single data point in subsequent analysis. To account for differences in film exposure times and allow pooling of results from different experiments, we normalized background-subtracted optical density measures in the cortex to those obtained from the hippocampus, which were virtually identical between wild-type and HCN1 knock-out mice within each experiment (i.e., values from hippocampus in HCNI knock-out mice were 101.3 ± 1.4% of those values in wild-type mice). Thus, differences in cortex:hippocampus optical density served as a measure of the relative change in levels of GABAA α5 subunit transcripts.
Immunohistochemical analysis of GABAA receptor α5 subunit was performed on coronal brainstem sections from HCN1 knock-out and wild-type mice, processed concurrently (n = 2 for each). Animals were anesthetized with urethane (4 mg/kg) and perfused through the ascending aorta with 0.9% NaCl, followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB, pH 7.4). The brains were removed and postfixed in the same fixative for 1 h at 4°C. After overnight incubation in 30% sucrose in 0.1 m PB, the brains were frozen by immersion in −70°C isopentane and sectioned on a cryostat at 40 μm. Following three washes in 0.1 m PB, sections were preincubated for 1 h in blocking solution [5% normal goat serum (NGS), 0.1% Triton X-100 in 0.1 m PBS, pH 7.4] and then incubated with two primary antibodies (rabbit anti-α5; 5 μg/ml, gift from Dr. W. Sieghart, University of Vienna, Vienna, Austria; mouse anti-NeuN, diluted at 1:200, Millipore) at 4°C for 72 h on a shaker in antibody diluent (2% NGS, 0.2% BSA in PBS). Subsequently, sections were incubated with Alexa fluor 594-conjugated goat anti-rabbit and Alexa fluor 488-conjugated goat anti-mouse secondary antibodies (both at 5 μg/ml in antibody diluent) for 1 h on a shaker at room temperature in the dark and then mounted on slides with FLUORO-GEL (Electron Microscopy Sciences). The α5-subunit antibody has previously been characterized (Pirker et al., 2000); controls in which primary antibodies were omitted provided weak nonspecific staining. Fluorescent images of immunostained tissue sections were captured on Zeiss 510 confocal-multiphoton-spectral imaging system.
All images of histological material were prepared for presentation using Adobe Photoshop. Adjustments of contrast and brightness were applied identically to images from wild-type and HCN1 knock-out animals.
Data analysis.
Results are presented as mean ± SEM. Data were analyzed statistically using one-way and two-way ANOVA or Student's t test; post hoc pairwise comparisons used Bonferroni's correction of the t test (Excel and/or SigmaStat). Differences in mean values were considered significant if p < 0.05.
Results
Tonic GABAA receptor current is enhanced in cortical pyramidal neurons from HCN1 knock-out mice
In the course of examining effects of inhalational anesthetics on layer V pyramidal neurons in brain slices from somatosensory cortex (see Chen et al., 2009b), we noticed that isoflurane evoked a maintained tonic inward current that was substantially larger in cells from HCN1 knock-out mice, by comparison to wild-type mice; representative records of tonic currents activated by 0.6 mm isoflurane at a holding potential of −70 mV are shown in Figure 1 A. We suspected that the tonic current might be mediated by anesthetic-sensitive GABAA receptors and, consistent with this, we found that the isoflurane-activated tonic current was fully inhibited by bicuculline (Fig. 1 A) and gabazine (SR-95531, 20 μm; data not shown). We fitted Gaussian functions to all-points histograms of membrane current under control conditions and in the presence of the drugs to provide a measure of holding current; averaged data reveal >2-fold higher isoflurane-activated, bicuculline-sensitive current density in pyramidal neurons from HCN1 knock-out mice than in cells from wild-type mice (−3.2 ± 1.3 pA/pF vs −8.4 ± 1.3 pA/pF, n = 5 for both, p < 0.05).

Tonic GABAA receptor currents are increased in cortical pyramidal neurons from HCN1 knock-out mice. A , Tonic and synaptic GABAA receptor currents in cortical pyramidal neurons from wild-type and HCN1 knock-out (KO) mice were recorded at a holding potential of −70 mV during application of isoflurane (0.6 mm) and bicuculline (30 μm). All-points histograms, shown at the left of each current trace, were obtained for 30–60 s epochs and fitted with Gaussian functions to obtain tonic current levels under different conditions. In this and other experiments, recordings of inward GABAA currents were obtained in the presence of glutamate receptor blockers (CNQX, 10 μm; APV 50 μm) and using pipettes with elevated intracellular Cl. B , Total tonic GABAA current was determined as the bicuculline-inhibited current, and quantified from Gaussian fits to all-points histograms. Averaged values of isoflurane-activated and bicuculline-inhibited currents were ∼2-fold larger in cells from HCN1 knock-out mice (see text).
The tonic GABAA (i.e., bicuculline-sensitive) current was also evident in pyramidal neurons in the absence of isoflurane, as depicted in Figure 1 B; again, the bicuculline-sensitive current density was ∼2-fold larger in cells from HCN1 knock-out mice (3.0 ± 0.5 pA/pF vs 5.8 ± 0.5 pA/pF, n = 11 and 14, p < 0.005). We examined properties of spontaneous and miniature IPSCs (sIPSC and mIPSCs) recorded in pyramidal neurons from wild-type and HCN1 knock-out mice; neither the amplitude nor the frequency of sIPSCs or mIPSCs was different in these mice (see supplemental Fig. S1, available at www.jneurosci.org as supplemental material). These data indicate that properties of synaptic GABAA currents are relatively preserved in layer V pyramidal neurons from HCN1 knock-out mice but these cells present with a strongly upregulated tonic GABAA current.
GABAA receptor α5 subunit expression is upregulated in cortex of HCN1 knock-out mice
The GABAA receptor is a heteropentamer, usually including two α subunits (of 6 possible α variants), two β subunits (of 3 possible β variants) and one additional subunit, often γ2 (of three possible γ variants) but occasionally δ (Farrant and Nusser, 2005). Of myriad possible subunit combinations, the specific receptor conformations that support tonic GABAA currents typically contain the δ subunit (with either α4 or α6 subunits) or they include the α5 subunit (Semyanov et al., 2004; Farrant and Nusser, 2005; Glykys and Mody, 2007; Jacob et al., 2008).
We performed qRT-PCR on cortical tissue from wild-type and HCN1 knock-out mice to determine which, if any, GABAA receptor subunits were upregulated to account for the enhanced tonic GABAA receptor currents. As depicted in Figure 2 A, we were able to detect expression of all subunits examined (although additional PCR cycles were required to detect α3 and α6 subunits, which are expressed at very low levels in cortex). Among these, only the α5 subunit showed higher cortical expression in HCN1 knock-out mice; in this case, the expression of α5 was ∼2-fold greater in HCN1 knock-outs. There was no difference between wild-type and HCN1 knock-out mice in expression of the δ subunit, which also can mediate tonic GABAA currents, or in expression of the other α subunits that often pair with δ in those extrasynaptic receptors (i.e., α4 and α6) (Semyanov et al., 2004; Farrant and Nusser, 2005; Glykys and Mody, 2007; Jacob et al., 2008).

GABAA receptor α5 subunit expression and α5 receptor-mediated tonic currents are upregulated in cortical pyramidal neurons from HCN1 knock-out mice. A , By qRT-PCR of cortical RNA, we found selective upregulation of GABAA α5 subunit transcripts in HCN1 knock-out (KO) mice (*p < 0.05 vs WT); expression of α1–α4, α6, β1–β3, γ1–γ3, and δ subunits was unaltered. B , Coronal forebrain sections from wild-type (WT) and HCN1 knock-out mice were hybridized with 33P-labeled cRNA probes for the GABAA α5 subunit; expression was higher in cortex of HCN1 knock-out mice (arrow). Note that hippocampal expression of α5 subunit mRNA is comparatively high and not obviously altered in HCN1 knock-out mice. C , Coronal brain sections of cortex from wild-type and HCN1 knock-out mice were immunostained with antibodies to GABAA α5 subunit (red; left) and NeuN (green; middle) and images were overlaid (right); note that α5 subunit immunoreactivity is higher in cortical layer 5 of the HCN1 knock-out than in that region of wild-type mice (arrows). D , High-power confocal images of cortical layer 5 (from C ) illustrating the enhanced GABAA α5 subunit immunofluorescence; note that immunoreactivity is not typically associated with cell somata but is present mostly in the neuropil. E , Voltage-clamp recordings of tonic GABAergic currents under control conditions and with 20 μm L-655,708 and 30 μm bicuculline in cortical pyramidal neurons from wild-type and HCN1 knock-out mice. F , Averaged L-655,708-inhibited currents and fractional contribution of α5 subunit currents to total (i.e., bicuculline-inhibited) tonic GABAA current in control and HCN1 knock-out cells (*p < 0.05 vs WT).
The elevated α5 transcript expression in HCN1 knock-out mice was also verified by using histochemical approaches. By in situ hybridization, we observed higher levels of α5 expression in cortex of HCN1 knock-out mice than of wild-type mice, as evident in film autoradiograms of coronal forebrain sections hybridized with a 33P-labeled cRNA probe for the GABAA α5 subunit (Fig. 2 B). Similar results were obtained in at least 5 sets of material from wild-type and knock-out mice processed concurrently, and there was little to no background hybridization in corresponding sections exposed to 33P-labeled sense strand control probes (data not shown). Also, consistent with the known distribution of α5 subunits (Wisden et al., 1992; Sperk et al., 1997; Pirker et al., 2000), note the uniformly high expression throughout the hippocampus that was not different between wild-type and HCN1 knock-out mice (also see below). To quantify these data, we compared the ratio of optical density over the cortex and hippocampus from each animal (see Materials and Methods); this cortex:hippocampus ratio was ∼0.4 in wild-type animals and ∼0.6 in HCN1 knock-out mice (0.40 ± 0.06 vs 0.62 ± 0.07, n = 5, p < 0.05), representing an increase in signal intensity of ∼60% in the HCN1 knock-out animals (58.5 ± 6.2%). This value is somewhat less than that obtained by qRT-PCR, but nevertheless consistent with increased expression of GABAA α5 receptor subunits in cortex of HCN1 knock-out mice.
We also examined cortical distribution of α5-containing GABAA receptors by immunohistochemistry in wild-type and HCN1 knock-out mice. As seen in Figure 2 C, staining for α5 subunits was stronger in cortex from HCN1 knock-out mice, especially in layer 5 (see arrows, compare with NeuN staining); images obtained at high magnification highlight the stronger immunostaining in layer 5 of the HCN1 knock-out mice and illustrate that α5 staining is evident in the neuropil and not obviously associated with NeuN-stained cell somata (Fig. 2 D). Together, these data indicate that expression of the α5 subunits that contribute to tonic GABAA currents is upregulated in cortex of HCN1 knock-out mice.
GABAA receptor containing α5 subunits contribute to upregulated tonic currents in cortical pyramidal neurons
We took advantage of differential receptor pharmacology to examine subunit contributions to the upregulated GABAA tonic currents in pyramidal neurons from HCN1 knock-out mice. In particular, we were interested in determining whether the greater α5 subunit expression in HCN1 knock-out mice was associated with elevated α5-mediated GABAA current. To this end, we evaluated tonic currents attributed to α5 subunit-containing receptors by using the α5 subunit-selective partial inverse agonist, L-655,708 (Quirk et al., 1996; Caraiscos et al., 2004; Bonin et al., 2007). We used L-655,708 at a concentration (20 μm) shown to depolarize hippocampal pyramidal neurons from wild-type mice but to have no effect on those same cells from α5 subunit knock-out mice (Bonin et al., 2007). The mice used to evaluate α5 subunit-mediated currents were from a restricted age range (P14-P16) to obviate concerns with developmental changes in GABAA subunit expression that occur over this early postnatal period (Laurie et al., 1992).
As depicted in the representative records of Figure 2 E, the tonic current sensitive to L-655,708 was much greater in cells from HCN1 knock-out mice; averaged data reveal an ∼3-fold greater L-655,708-sensitive current by comparison to cells from wild-type mice (Fig. 2 F) (1.4 ± 0.3 pA/pF vs 3.8 ± 0.7 pA/pF, n = 5 and 5, p < 0.05). For these cells tested with both L-655,708 and bicuculline, the fraction of total tonic GABAA current (3.3 ± 0.7 pA/pF vs 6.4 ± 1.0 pA/pF) blocked by L-655,708 averaged 43.5 ± 3.3% in wild-type mice and 58.9 ± 4.6% in HCN1 knock-out mice (Fig. 2 F), and the increase in L-655,708-sensitive-current accounted for most of the upregulated tonic GABAA current (77.4%).
The data presented to this point were obtained from two separate lines of mice that are both hybrids of the parental C57BL/6 and 129Sv substrains, one wild-type at the HCN1 locus and the other deleted for HCN1. To verify that the enhanced tonic GABAA current is due to HCN1 gene deletion and independent of other differences associated with these two mouse lines, we also examined homozygous HCN1+/+ and HCN1−/− littermates derived from crossing a single line of HCN1+/− heterozygous mice. Again, we found a marked upregulation of α5 subunit-mediated tonic GABAA receptor current in cortical pyramidal neurons; the L-655,708-sensitive current was 2.1 ± 1.0 pA/pF in HCN1+/+ and 5.9 ± 1.0 pA/pF in HCN1−/− mice (n = 5 and 8, p < 0.05; data not shown). Thus, the increase in α5 subunit expression and tonic GABAA current is a robust phenomenon, independent of background strain and clearly associated with deletion of HCN1 subunits.
We examined whether the elevated tonic GABAA current in pyramidal neurons from HCN1 knock-out mice could be attributed to other subunits that can also generate extrasynaptic GABAA receptors. In this respect, zolpidem-sensitive GABAA α1 subunits normally contribute a minor component of tonic current in layer V cortical pyramidal cells (Yamada et al., 2007) and δ subunit-containing GABAA receptors generate tonic currents in other cell types (Stell et al., 2003; Glykys et al., 2008). However, we found no increase in tonic currents activated by the α1 agonist zolpidem (100 nm; −2.2 ± 0.6 pA/pF vs −2.7 ± 0.4 pA/pF, n = 6 and 7, p > 0.55) or by the δ subunit-selective neurosteroid agonist, THDOC (100 nm; −2.2 ± 0.8 pA/pF vs −2.5 ± 0.5 pA/pF, n = 5 and 7, p > 0.75) in cells from HCN1 knock-out mice (Fig. 3). These results are consistent with our qRT-PCR data indicating no change in α1 or δ subunit expression in HCN1 knock-out mice, and they support the conclusion that enhanced tonic GABAA currents in cortical pyramidal neurons from HCN1 knock-out mice are due, in large measure, to increased activity of α5 subunit-containing GABAA receptors.

Tonic GABAA currents mediated by α1 and δ subunits are not increased in cortical pyramidal cells from HCN1 knock-out (KO) mice. A , B , Effects of zolpidem ( A , 100 nm) and 5-THDOC ( B , 100 nm) on tonic currents in cortical pyramidal neurons from wild-type and HCN1 knock-out mice. The drug-activated currents were quantified from Gaussian fits to all-points histograms, and averaged values were not different between genotypes (see text).
In HCN1 knock-out mice, tonic α5-mediated GABAA currents remain elevated after blocking GABA uptake
The increased α5-mediated tonic GABAA currents in HCN1 knock-out mice could reflect elevated GABA levels in the slices, in addition to increased receptor expression, perhaps as a result of diminished GABA transporter activity. To test a role for decreased transporter activity in the elevated tonic GABAA currents, we performed experiments with an inhibitor of GABA uptake, NO-711 (Suzdak et al., 1992). The uptake inhibitor caused an increase in tonic GABAA current in cells from both wild-type and HCN1 knock-out mice (Fig. 4 A,B). However, the effect of NO-711 on tonic current was greater in the knock-outs and, as observed under standard conditions in the absence of the uptake blocker, the total tonic current remained elevated in HCN1 knock-out mice (2.5 ± 0.7 pA/pF vs 5.6 ± 1.2 pA/pF, n = 7 for both, p < 0.05). The stronger effect of NO-711 in neurons from HCN1 knock-out mice indicates that decreased GABA transporter activity is not primarily responsible for elevated tonic GABAA current in the HCN1 knock-out mice.

GABAA α5 subunit-mediated tonic currents are enhanced in cortical pyramidal cells from HCN1 knock-out mice under conditions of reduced GABA transporter activity with saturating GABA concentrations. A , Effect of the GABA transport inhibitor NO-711 (5 μm) on GABA currents in cortical neurons from wild-type (WT) and HCN1 knock-out (KO) mice. B , Summary data illustrating that averaged NO-711-activated currents (left) and the total bicuculline-inhibited currents during NO-711 application (right) are enhanced in cortical pyramidal neurons from HCN1 knock-out mice. (p < 0.05, n = 7). C , Effect of a fixed concentration of GABA (5 μm) in the presence of the GABA transport inhibitor NO-711 (5 μm) on tonic GABA currents in cortical neurons from wild-type and HCN1 knock-out mice. D , Averaged data illustrating that both the α5 subunit-mediated component of maximal tonic GABAA current (left) and the total tonic GABAA current (right) were larger in cortical pyramidal neurons from HCN1 knock-out mice. (p < 0.05, n = 9 and 6).
The larger tonic GABAA current in the presence of NO-711 might reflect higher endogenous GABA levels in the slices from HCN1 knock-out mice, rather than differences in GABAA receptor activity. To examine this possibility, we performed experiments in which excess GABA was included in the bath solution at a fixed concentration (5 μm), together with NO-711. We found that tonic bicuculline- and L-655,708-sensitive currents were much bigger in cells from both wild-type and HCN1 knock-out mice, by comparison to currents with NO-711 alone (Fig. 4, compare B and D). This indicates that GABAA receptors were not saturated by endogenous GABA in mice of either genotype. In the combined presence of NO-711 and GABA, the α5-subunit-mediated and total tonic GABAA receptor currents remained greater in cells from HCN1 knock-outs (Fig. 4 C,D); bicuculline-sensitive currents were 9.8 ± 1.4 pA/pF vs 16.4 ± 1.5 pA/pF and L-655,708-sensitive currents were 5.1 ± 1.2 pA/pF vs 10.0 ± 1.7 pA/pF (n = 9 and n = 6, p < 0.05). Under these conditions, when one might expect a greater contribution from other lower affinity GABAA receptor subunits to the total current, the α5 subunit-mediated current again accounted for most of the increase in tonic current (∼75%). So, enhanced α5 subunit-mediated current is retained in cortical pyramidal neurons after blocking GABA transporter activity and in the presence of fixed elevated GABA concentrations. These data indicate that increased endogenous GABA levels are not required for the upregulated tonic GABAA current mediated by α5 subunits in pyramidal cells from HCN1 knock-out mice.
Enhanced tonic GABAA currents provide a background shunt conductance that normalizes synaptic summation in pyramidal neurons from HCN1 knock-out mice
We considered potential functional consequences of the upregulated tonic GABAA current on cortical neuron excitability, focusing primarily on how such a current might compensate for the loss of I h in cortical pyramidal neurons from HCN1 knock-out mice. In this respect, it is now well known that HCN1 subunits contribute primarily to dendritic I h in pyramidal cells, providing an electrotonic shunt leading to sublinear EPSP summation (Magee, 2000). Therefore, we hypothesized that the upregulated tonic GABAA currents in cortical pyramidal neurons from HCN1 knock-out mice might replace the shunt typically provided by I h— and thereby restore its synaptic dampening function.
We first compared input resistance (R N) in pyramidal cells from wild-type and HCN1 knock-out mice under current-clamp conditions to verify that the enhanced GABAA tonic current contributes to normalizing cell conductance at the resting membrane potential (−66.0 ± 3.3 mV and −73.1 ± 3.4 mV; n = 7 and 5). Indeed, under control conditions, R N was unaffected by deletion of HCN1 subunits (182 ± 20.7 MΩ vs 193.4 ± 36.3 MΩ in neurons from wild-type and HCN1 knock-out mice, n = 7 and n = 5, p = 0.75). Moreover, consistent with the idea that R N was maintained in the HCN1 knock-out mice by virtue of upregulated tonic GABAA currents, we found that gabazine had a more pronounced effect on R N in cells from the HCN1 knock-out mice (∼5% increase in control vs ∼40% increase in HCN1 knock-out mice). Note, also, that the absolute values of R N we found in the presence of gabazine in this dataset (190.8 ± 25.3 MΩ and 268.8 ± 56.6 MΩ) are very similar to those we found after GABAA receptor blockade with bicuculline in a different group of pyramidal neurons from control and HCN1 knock-out mice (∼200 MΩ and ∼300 MΩ; see Chen et al., 2009b). We also measured input conductance (G N) from instantaneous I–V relationships obtained under voltage-clamp conditions, all from the same holding potential of −60 mV. Again, there was no significant difference between neurons from wild-type and HCN1 knock-out mice under control conditions (G N: 4.5 ± 0.2 nS vs 3.6 ± 0.3 nS; n = 5 and 6) and gabazine significantly decreased G N only in HCN1 knock-out cells (by ∼23%; to 2.8 ± 0.2 nS, p < 0.05). Thus, despite the substantial reduction of I h in cortical pyramidal neurons from HCN1 knock-out mice (Chen et al., 2009b), baseline conductance properties are relatively preserved by increased tonic GABAA receptor currents.
To test directly the synaptic summation properties in cortical pyramidal neurons from HCN1 knock-out mice we recorded excitatory synaptic responses evoked by high-frequency (40 Hz) extracellular stimulation of the superficial cortex (Fig. 5 A, schematic). As shown for the representative pyramidal neuron from a wild-type animal, EPSP summation was clearly sublinear under control conditions (Fig. 5 B, top); the amplitude of the final evoked EPSP (EPSP5) relative to the first (EPSP1) averaged only ∼2 (Fig. 5 C) (2.1 ± 0.1, n = 6). In these cells from wild-type mice, exposure to bicuculline caused a membrane hyperpolarization and only a modest effect on EPSP summation properties (Fig. 5 B,C, aligned traces and averaged data, respectively) (EPSP5:EPSP1 ratio to 2.4 ± 0.1, p = 0.12 vs control), whereas blocking I h with ZD-7288 (50 μm) caused an additional hyperpolarization and greatly enhanced synaptic summation (Fig. 5 B,C) (EPSP5:EPSP1 ratio to 4.0 ± 0.3, p < 0.05 vs control). As shown, the enhanced EPSP summation was observed in the presence of bicuculline or ZD-7288 despite membrane hyperpolarization (Fig. 5 B, sample traces) but for quantitative comparisons of EPSP summation we used DC injection to set membrane potential to approximately −70 mV under all conditions (Fig. 5 C,D). In sum, these data are as expected for cells from wild-type mice. That is, they confirm that blocking I h acutely with ZD-7288 enhances synaptic function, consistent with the idea that I h normally dampens dendritosomatic coupling (Magee, 2000).
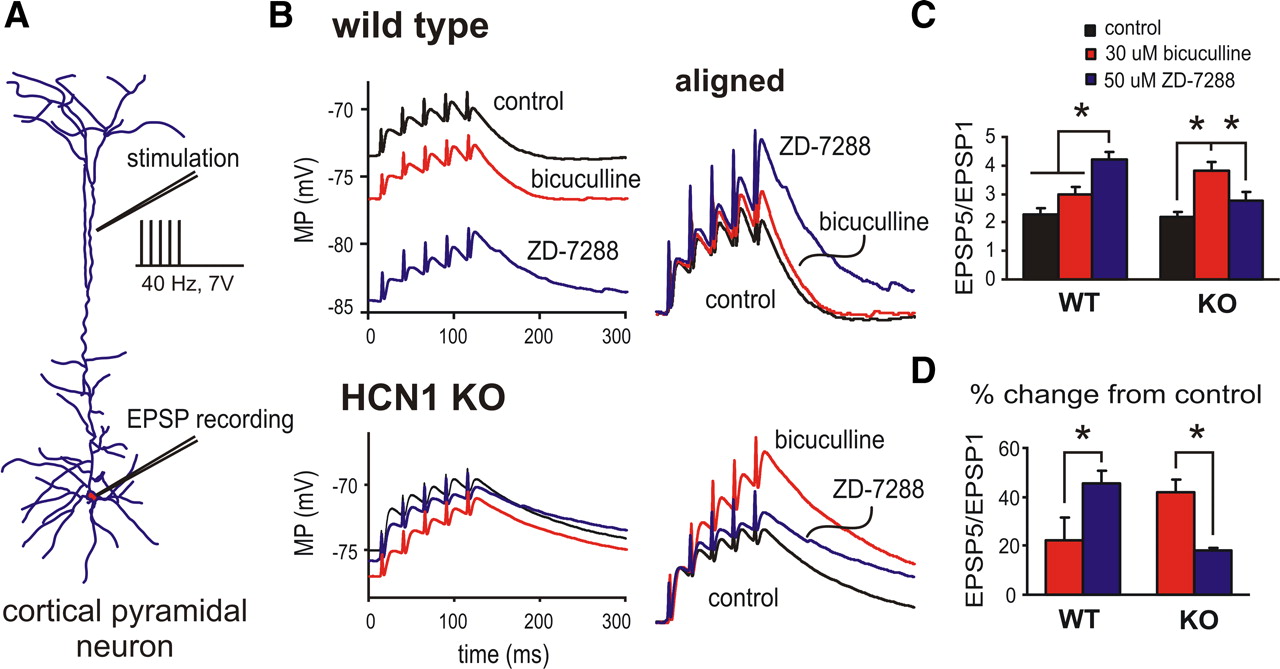
Increased tonic GABAA currents maintain normal EPSP summation properties in cortical pyramidal neurons from HCN1 knock-out mice. A , Reconstruction of a biocytin-filled cortical pyramidal neuron illustrating schematically the stimulating and recording configuration. B , Sample voltage traces of EPSPs obtained in cortical pyramidal neurons from wild-type (top) and HCN1 knock-out (bottom) mice in response to 40 Hz, 7 V stimulation under control conditions, after incubation in bicuculline (30 μm) or ZD-7288 (50 μm). EPSPs are aligned to initial membrane potential to highlight enhanced temporal summation of EPSPs associated with bicuculline or ZD-7288. C , Summation properties of evoked EPSPs (EPSP5/EPSP1 ratio) under control conditions and in the presence of 30 μm bicuculline or 50 μm ZD-7288 in wild-type (WT) or HCN1 knock-out (KO) mice. D , Enhancement of EPSP summation by bicuculline and ZD-7288 (percentage of control) in cells from wild-type and HCN1 knock-out mice. Note that whereas blocking HCN channels with ZD-7288 had a prominent effect on synaptic summation in cortical neurons from WT mice, EPSP summation was most strongly enhanced by blocking GABAA currents in cells from HCN1 knock-out mice. (*p < 0.05 vs control).
In pyramidal neurons from HCN1 knock-out mice, however, despite the loss of HCN1 channels and a markedly reduced I h (Chen et al., 2009b), we found that synaptic summation properties under control conditions were virtually identical to those from wild-type mice. As presented in Figure 5 B (bottom), control EPSPs summated sublinearly with an EPSP5:EPSP1 ratio close to 2 (Fig. 5 C) (2.1 ± 0.1, n = 5, p = 0.25 vs wild-type control). So, unlike the situation obtained with acute pharmacological inhibition of I h, genetic deletion of HCN1 channels did not enhance synaptic summation in cortical pyramidal neurons, suggesting engagement of a compensatory mechanism. In this respect, there was a striking difference between wild-type and HCN1 knock-out mice in effects of I h and GABAA blockers on EPSP summation. In cells from HCN1 knock-out mice, as expected, there was little effect of ZD-7288 on synaptic summation (Fig. 5 B,C) (EPSP5:EPSP1 ratio to 2.6 ± 0.2, p < 0.05 vs control). On the other hand, blocking GABAA receptors with bicuculline caused a marked enhancement of EPSP summation in these cells (Fig. 5 B,C) (EPSP5:EPSP1 ratio to 3.5 ± 0.4, p < 0.05 vs control). Thus, it appears that upregulation of tonic GABAA receptor currents can serve to maintain essentially normal dendritosomatic coupling in pyramidal neurons from HCN1 knock-out mice, despite the loss of I h (Fig. 5 C, compare black bars). Moreover, this compensation of synaptic integration in the HCN1 knock-out mice is quantitatively precise, as highlighted by examining the relative contributions of I h and tonic GABAA currents to EPSP summation (Fig. 5 D). In cells from wild-type mice, blocking GABAA currents enhanced EPSP summation by 12.9 ± 5.7% and blocking I h enhanced summation by 47.1 ± 4.1%; the situation was reversed in neurons from HCN1 knock-out mice, wherein bicuculline increased summation by 37.3 ± 5.8% while ZD-7288 increased summation by only 15.8 ± 1.5%.
As expected from our data on GABAA receptor subunit expression and pharmacology we found that selective inhibition of α5 subunit-containing GABAA receptors with L-655,708 increased the EPSP5:EPSP1 ratio in HCN1 knock-out mice (by ∼20%; from 1.9 ± 0.1 to 2.3 ± 0.2, n = 4, p < 0.05), whereas it had essentially no effect on EPSP summation in control mice (by <2%, n = 5; data not shown). In addition, to ensure that contributions of tonic GABAA currents to EPSP summation were not dependent on altered intracellular Cl concentration ([Cl]i) resulting from whole-cell recordings, we repeated these experiments using gramicidin perforated-patch recordings, which do not disrupt [Cl]i. Importantly, as shown in Figure 6, bicuculline again enhanced EPSP summation more strongly in pyramidal neurons from HCN1 knock-out mice (by ∼27%; from 2.7 ± 0.4 to 3.7 ± 0.4; n = 3), than in cells from control mice (by only ∼10%; from 2.3 ± 0.6 to 2.6 ± 0.7; n = 3). We were also able to establish ECl in these gramicidin recordings of cortical pyramidal neurons from wild-type and HCN1 knock-out mice (i.e., by determining the reversal potential of gabazine-sensitive current). For both genotypes, ECl was approximately −65 mV (−66.0 ± 3.4 mV vs −68.1 ± 3.8 mV; n = 5), indicating that genotype-dependent differences in tonic GABAA currents and their effects on EPSP summation properties could not be explained by altered Cl− gradients.

Effects of upregulated GABAA currents on EPSP summation are observed in cortical pyramidal neurons from HCN1 knock-out mice with unperturbed intracellular Cl−. A , Cortical pyramidal neurons from wild-type (top) and HCN1 knock-out (KO) mice (bottom) were recorded using gramicidin perforated patch to preserve [Cl−]i; aligned voltage traces of evoked EPSPs obtained in response to 40 Hz, 7 V stimulation under control conditions, after incubation in bicuculline (30 μm) or ZD-7288 (50 μm). B , Averaged EPSP5/EPSP1 summation ratio (top) and percentage enhancement (bottom) in cells from wild-type (WT) and HCN1 knock-out mice under control conditions and in the presence of 30 μm bicuculline or 50 μm ZD-7288. As with whole-cell recordings, blocking HCN channels with ZD-7288 had a prominent effect on synaptic summation in cortical neurons from WT mice, whereas EPSP summation was most strongly enhanced by blocking GABAA currents in cells from HCN1 knock-out mice. (*p < 0.05 vs control).
Computer simulations and pharmacological manipulations recapitulate synaptic summation properties in pyramidal neurons from HCN1 knock-out mice
There are obvious differences in the properties of tonic GABAA currents and neuronal I h, notably the absence of voltage and time dependence and a more hyperpolarized reversal potential for the tonic currents; in addition to a simple current shunt effect, it is believed that the additional voltage-dependent properties of I h also contribute to limiting temporal summation of EPSPs (Magee, 2000). Therefore, we used a computer simulation in an existing cortical pyramidal neuron model (Day et al., 2005) to test whether simply substituting a tonic GABAA current for I h could yield normal sublinear summation, as our experimental results suggest. For this modeling, we added a tonic GABAA conductance by including a standing somatodendritic leak conductance with a reversal potential at −65 mV. To simulate wild-type pyramidal neurons, the tonic GABAA conductance was set at 1/10 of the HCN conductance, as determined from measured values of I h and tonic GABAA current in those cells. To model pyramidal neurons from HCN1 knock-out mice, we decreased the HCN conductance by 75% and shifted V 1/2 by −10 mV (Chen et al., 2009b), and the tonic GABAA current was increased 2.5-fold. Again, this approximated measured changes in these currents (this work and see Chen et al., 2009b). Other parameters from the original model were unchanged, although we set the existing Kir conductance to zero to match our experimental situation, in which recordings were performed in the presence of 200 μm barium. We also used simulated DC injection to set the model neuron membrane potential to approximately −70 mV for analysis of EPSP summation, reproducing conditions we used for measuring evoked EPSPs.
As illustrated in Figure 7, there was clear sublinear EPSP summation in the simulation mimicking wild-type cortical pyramidal cells under control conditions (i.e., with dendritic I h and a relatively small tonic GABAA current); eliminating the tonic GABAA current to simulate effects of bicuculline had only a minor effect on summation properties, whereas eliminating I h to mimic effects of ZD-7288 enhanced the EPSP5:EPSP1 ratio (from ∼2 to ∼3). These relative changes in EPSP properties were similar to those obtained experimentally in cells from wild-type mice. In the model of the pyramidal cell from an HCN1 knock-out mouse (i.e., smaller I h with more hyperpolarized V 1/2 and increased tonic GABAA current), we again found a sublinear EPSP summation under initial conditions. In this case, however, removing the residual I h to mimic effects of ZD-7288 had little effect on summation properties while removing the elevated tonic GABAA current strongly increased EPSP summation. We obtained qualitatively similar results if the upregulated tonic GABAA current was modeled exclusively within the somatic or the dendritic compartments, although compensation was slightly more effective when GABAA conductance was increased only in dendrites than when it was inserted only on the soma (data not shown). Overall, the results of this simple simulation match our experimental data remarkably well.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Computer simulation and pharmacological manipulation recapitulates effects of increased tonic GABAA currents on EPSP summation properties in cortical pyramidal neuron model. A , B , Simulation of EPSP summation properties (40 Hz) was obtained using an existing NEURON model of a cortical pyramidal cell (from Day et al., 2005), with Kir conductance set to zero and the membrane potential adjusted to −70 mV in all conditions using simulated DC. The existing model for the wild-type (WT) pyramidal neuron ( A ) was modified to include a small tonic GABAA-like conductance (1/10 the size of HCN conductance); for the HCN1 knock-out (KO) cell simulation ( B ), the HCN conductance was decreased by 75% with a −10 mV shift in V 1/2 and the tonic GABAA conductance was increased by a factor of 2.5. The HCN and GABAA conductances were eliminated to mimic effects of ZD-7288 and bicuculline. C , Values obtained for EPSP5:EPSP1 (top) and percentage change in EPSP summation ratio (bottom). Note that effects of eliminating I h and tonic GABAA currents on EPSP summation were reversed in the simulations of wild-type and HCN1 cortical pyramidal neurons, reproducing results obtained experimentally. D , In pyramidal cells from wild-type mice, diazepam (1 μm) approximately doubled the tonic GABAA current. E , EPSPs were evoked in wild-type cortical pyramidal neurons under control conditions (C), after blocking I h with ZD-7288 (Z, 50 μm) and after activating tonic GABAA current with diazepam (D, 1 μm) in the continued presence of ZD-7288. Top, Aligned voltage traces of evoked EPSPs under the indicated conditions. Bottom, Values obtained for EPSP5:EPSP1 (left) and percentage change in EPSP summation ratio (right). Note that the enhanced EPSP summation associated with block of I h in wild-type neurons was reversed by pharmacologically activating tonic GABAA currents.
To further test the idea that enhanced tonic GABAA currents can substitute for diminished I h in regulating EPSP summation properties, we used a pharmacological approach designed to mimic acutely in wild-type cells the situation obtained in pyramidal neurons from HCN1 knock-out mice. For these experiments, neuronal I h was eliminated with ZD-7288 and diazepam was used to activate existing GABAA channels on wild-type cortical pyramidal cells. Diazepam was applied at a concentration (1 μm) that caused approximately a doubling of bicuculline-sensitive tonic GABAA current (Fig. 7 D) (averaged fold increase of 2.1 ± 0.0, n = 3), similar to the increase measured in pyramidal neurons from HCN1 knock-out mice. As shown in Figure 7 E, blocking I h increased EPSP summation (from 1.9 ± 0.3 to 3.2 ± 0.5) and this enhanced summation was restored toward control levels in the presence of diazepam (to 2.3 ± 0.5). It is important to note that diazepam can activate many different types of GABAA receptor, in addition to those containing α5 subunits. In addition, activation of these existing receptors may not faithfully represent the situation obtained with upregulated α5 subunit-containing GABAA receptors in pyramidal neurons from knock-out mice (e.g., those new receptors may be localized in distinct subcellular domains). Nevertheless, the ability of diazepam to reverse the enhanced summation following block of I h provides further support for the concept that upregulated tonic GABAA currents can indeed substitute for the loss of I h in regulating synaptic summation properties, despite obvious differences in the properties of these currents.
GABAA a5 subunit expression and tonic GABAA currents are unchanged in hippocampal pyramidal neurons from HCN1 knock-out mice
There is strong expression of both HCN1 and GABAA α5 subunits in pyramidal neurons of the hippocampus (Wisden et al., 1992; Sperk et al., 1997; Santoro et al., 2000; Pirker et al., 2000), in which the currents they generate play similar roles as in the neocortex: HCN1 is localized to dendrites in which it depresses synaptic integration (Magee, 2000) and α5 subunit-containing GABAA receptors are found extrasynaptically where they mediate tonic currents (Brünig et al., 2002; Caraiscos et al., 2004; Glykys et al., 2008). We therefore examined whether tonic GABAA currents are upregulated in hippocampal pyramidal neurons, as we found for neocortical pyramidal neurons.
As mentioned above, our in situ hybridization experiments did not reveal any obvious difference in α5 expression between wild-type and HCN1 knock-out mice (Fig. 2 B). To assess this quantitatively, we performed qRT-PCR for α5 subunit mRNA on hippocampal tissue; α5 subunit transcript levels were much higher in hippocampus than in cortex (by ∼4-fold), but we found no difference in expression between wild-type and HCN1 knock-out mice (2−ΔCt values: 1.9 ± 0.5 vs 2.2 ± 0.1, n = 5, p = 0.18). We also looked for functional evidence of an upregulated tonic GABAA current in hippocampal pyramidal neurons, but we found that bicuculline-sensitive tonic currents were not different in those neurons from wild-type and HCN1 knock-out mice (see supplemental Fig. S2, available at www.jneurosci.org as supplemental material) (2.5 ± 0.5 pA/pF vs 2.4 ± 0.6 pA/pF, n = 5 and 6, p = 0.97). Thus, it appears that the compensatory upregulation of α5 subunit expression and the enhanced tonic GABAA currents that result from HCN1 deletion in neocortical pyramidal neurons may not be present in other cell types, even those in which the ablated HCN1 channels are known to mediate very similar functions.
Discussion
In this work, we uncovered a homeostatic interaction between two manifestly different types of ion channel; specifically, in cortical pyramidal neurons, we found that genetic deletion of HCN1 channels that underlie dendritic I h leads to increased expression of α5 subunit-containing GABAA receptors that mediate a tonic background current. We further demonstrated that upregulation of tonic GABAA receptor currents provides an important functional compensation for the loss of I h in HCN1 knock-out mice; the sublinear synaptic summation that depends on I h in cortical pyramidal neurons of wild-type mice was preserved in HCN1 knock-out mice by the enhanced tonic GABAA receptor current. Strikingly, the compensation was quantitatively precise. Despite the loss of I h, baseline EPSP summation properties were essentially identical in cortical pyramidal neurons from wild-type and HCN1 knock-out mice; moreover, the fractional contributions of I h and tonic GABAA current to synaptic summation properties were reversed in the HCN1 knock-out mice. These data reveal a novel homeostatic mechanism that serves to restrict excitability in cortical pyramidal neurons and they imply that dendritosomatic synaptic integration is a carefully monitored variable subject to strict regulatory control.
In the absence of Ih, tonic GABAA currents serve to constrain synaptic integration
Homeostatic compensation of neuronal excitability can be accomplished in myriad and surprising ways (Destexhe and Marder, 2004; Turrigiano, 2008). In this respect, it is perhaps not immediately obvious how such seemingly distinct channel types— one that underlies a voltage-dependent mixed cationic current and the other that supports agonist-gated tonic Cl currents—could substitute for each other. However, our results indicate that these currents influence dendritosomatic integration in similar ways: both dampen temporal summation of synaptic inputs. We suggest that this reflects the shared property of both currents to minimize deviations in membrane potential. In the case of I h, voltage- and time-dependent deactivation of this cationic current during a depolarizing EPSP provides a relative outward current that decreases synaptically mediated membrane depolarization (Magee, 2000). In addition, the persistent nature of the noninactivating I h at resting membrane potential decreases resting input resistance to provide a current shunt that limits synaptic summation (Magee, 2000). Similarly, tonic GABAA currents provide a shunting inhibition (Farrant and Nusser, 2005; Bonin et al., 2007), and our experimental and computational data indicate that addition of this simple conductance can substitute for the loss of I h in constraining synaptic summation. So, although the basic properties of these channels are very different, especially in terms of ion selectivity and mechanisms of gating, they can both diminish EPSP summation and thereby constrain neuronal excitation.
Other reported instances of homeostatic compensation involving I h and tonic GABAA currents engage different ion channels that also generate subthreshold currents responsible for controlling neuronal excitability. For example, genetic deletion of the GABAA receptor α6 subunits that mediate a tonic hyperpolarizing current in cerebellar granule neurons results in upregulation of TASK-1, a two-pore-domain “leak” potassium channel (Brickley et al., 2001). In Kv4.2 knock-out mice, loss of the associated dendritic transient K+ current in hippocampal pyramidal neurons leads to a compensatory upregulation of tonic GABAA current, resulting in relatively normal neuronal excitability (Andrásfalvy et al., 2008). Also in hippocampal pyramidal neurons, activity-dependent changes in synaptic strength evoke counterbalancing effects on general excitability that are mediated by dynamic upregulation and downregulation of I h (Narayanan and Johnston, 2007). Overexpression of a transient K+ current in lobster stomatogastric ganglion neurons causes upregulation of I h, which normalizes rhythmic firing behavior in those cells. This latter example is particularly interesting since the increase in I h expression was obtained even when a nonconducting version of the same transient K+ channel was expressed, indicating that channel upregulation occurred without any perturbation of normal neuronal activity (MacLean et al., 2003). Thus, the mechanisms underlying homeostatic compensation can be diverse and the regulated neural output can be obscure (Destexhe and Marder, 2004).
In light of these complexities, it is worth considering the present results in the context of recent reports that have also combined experimental and computational analyses to examine the role of I h in regulation of membrane potential and synaptic responses in CA1 pyramidal neurons (Dyhrfjeld-Johnsen et al., 2009; George et al., 2009). In that work, it was shown that the influence of I h, when considered in isolation, is always excitatory; its direct depolarizing actions outweigh inhibitory effects of the associated shunt conductance such that peak membrane potential during EPSPs is greater when I h is elevated (Dyhrfjeld-Johnsen et al., 2009; George et al., 2009). Given this, one might think that homeostatic changes compensating for the loss of I h in HCN1 knock-out mice would tend to enhance, rather than diminish, excitability. However, the relative influence of I h is determined by interactions with various other currents that together determine the membrane potential (George et al., 2009). In neocortical pyramidal neurons in vivo, multiple ionic mechanisms conspire to shift membrane potential between preferred depolarized and hyperpolarized levels, with synaptic activity superimposed on those so-called up states and down states (Destexhe et al., 2003). Thus, we suggest that the enhanced GABAA tonic currents we observed in HCN1 knock-out mice can be best understood as a homeostatic response specifically to loss of the inhibitory aspect of I h on synaptic integration, with prevailing cortical neuron membrane potential in those animals governed primarily by the multiple additional currents that control sojourns between up and down states.
Upregulated α5 subunit expression accounts for enhanced tonic GABAA currents in cortical pyramidal neurons
It is known that GABAA receptors containing α5 subunits primarily account for tonic currents in cortical pyramidal neurons (Yamada et al., 2007) and accordingly, we found that the enhanced tonic GABAA currents in cortical pyramidal neurons from HCN1 knock-out mice could be attributed to elevated cortical expression of α5 subunits. We did not find evidence for upregulation of other GABAA receptor subunits that typically associate with α5 (e.g., β3, γ2; Sur et al., 1998; Farrant and Nusser, 2005), suggesting that those associated subunits may be present in excess under normal conditions in cortex of wild-type mice. The mechanism by which a decrease in HCN1 expression is selectively coupled to upregulation of α5 expression remains to be determined. We did not observe any obvious increase in α5 subunit transcript levels or immunoreactivity in neuronal somata in cortex of HCN1 knock-out mice, but our analysis did not include ultrastructural assessment of subcellular localization of α5 subunits. Other work has shown a predominant extrasynaptic, and even dendritic, distribution of α5-containing GABAA receptors (Sperk et al., 1997; Brünig et al., 2002). If a similar pattern were obtained for the upregulated tonic GABAA receptors in HCN1 knock-out mice, the channels might be especially well placed to substitute for the loss of dendritic HCN1 channels.
Tonic GABAA currents are not enhanced in hippocampal pyramidal neurons from HCN1 knock-out mice
We also examined effects of HCN1 deletion on tonic GABAA currents in hippocampal pyramidal neurons, in which expression of HCN1 and the GABAA receptor α5 subunit are prominent. In slices from wild-type mice, tonic GABAA current amplitude was similar to that reported by others (Glykys et al., 2008) and not greater than in cortical pyramidal neurons, despite the fourfold higher α5 subunit expression in hippocampus. Thus, it appears that α5 transcript expression is not strictly correlated with tonic current amplitude in hippocampus (Glykys et al., 2008). Importantly, and in contrast to neocortical pyramidal neurons, we found no increase in tonic GABAA current in hippocampal pyramidal neurons from HCN1 knock-out mice, and no enhanced expression of α5 subunits in hippocampus. The absence of this compensatory mechanism in hippocampal pyramidal neurons was somewhat unexpected since dendritic HCN1 channels subserve a similar function in both types of pyramidal neuron (Stuart and Spruston, 1998; Magee, 2000). The reasons for these differences between neocortical and hippocampal pyramidal neurons are unknown, but may reflect higher basal expression of α5 subunits in the hippocampus (Wisden et al., 1992), and thus a more limited capacity for compensatory upregulation of α5 expression. However, this does not seem to reflect a general inability of hippocampal pyramidal neurons to homeostatically regulate tonic GABAA currents. As mentioned, upregulated tonic GABAA current preserves relatively normal dendritic excitability in hippocampal pyramidal neurons of mice deleted for Kv4.2 (Andrásfalvy et al., 2008). The molecular basis for that upregulated tonic GABAA current in Kv4.2 knock-out mice was not determined (Andrásfalvy et al., 2008), but it could involve other receptor subunits that carry tonic currents in hippocampal neurons, and which may have greater compensatory reserve (e.g., δ subunits). In any case, our results with hippocampal neurons from HCN1 knock-out mice are consistent with previous reports that demonstrated clear functional consequences of HCN1 deletion on dendritic excitability in hippocampal pyramidal neurons, suggesting little (or at least incomplete) compensation in those cells (Nolan et al., 2004).
Functional implications of GABAA receptor compensation for decreased HCN1 channels
The importance of I h in regulating synaptic integration and excitability in cortical pyramidal neurons is well recognized (Stuart and Spruston, 1998; Magee, 2000). It is becoming increasingly clear that levels of HCN1 channel expression are not invariant, but are adjusted under different physiological or pathophysiological conditions. For example, HCN channel expression is dynamically regulated by coexpression of different isoforms of the auxiliary channel protein, TRIP8b (Lewis et al., 2009; Santoro et al., 2009). Also, HCN1 channel downregulation may contribute to development of epilepsy in various rodent models (Shah et al., 2004; Jung et al., 2007; Kole et al., 2007; Huang et al., 2009). Our data suggest that under conditions of chronically downregulated HCN1 channel expression, enhanced expression of tonic GABAA receptor currents may provide a countervailing current that can compensate for the loss of I h—and thereby mitigate attendant changes in excitability.
Footnotes
- Received August 3, 2009.
- Revision received December 18, 2009.
- Accepted December 30, 2009.
-
This work was supported by American Heart Association Beginning Grant-in-Aid 0665349U to X.C. and National Institutes of Health Grants NS44370 to J.K. and GM66181 to D.A.B. We thank Dr. Werner Sieghart (Medical University of Vienna) for the GABAA α5 subunit antibody; we also thank Drs. Daniel Johnston and Payne Chang (UT-Austin) for sharing preliminary data from hippocampal pyramidal neurons and Dr. Johnston for helpful comments on this manuscript.
- Correspondence should be addressed to Douglas A. Bayliss, Department of Pharmacology, University of Virginia Health System, P.O. Box 800735, 1300 Jefferson Park Avenue, Charlottesville, VA 22908-0735. dab3y{at}virginia.edu
- Copyright © 2010 the authors 0270-6474/10/302611-12$15.00/0