

Abstract
Oligomannosidic glycans play important roles in nervous system development and function. By performing a phage display screening with oligomannose-specific antibodies, we identified an oligomannose-mimicking peptide that was functionally active in modulating neurite outgrowth and neuron–astrocyte adhesion. Using the oligomannose-mimicking peptide in crosslinking experiments, synapsin I was identified as a novel oligomannose-binding protein in mouse brain. Further analyses not only verified that synapsin I is an oligomannose-binding lectin, but also indicated that it is a glycoprotein carrying oligomannose and Lewisx. We also found that synapsin I is expressed in glia-enriched cultures and is released from glial cells via exosomes. Incubation of glial-derived exosomes in the presence of high KCl concentrations or subjecting glial cell cultures to either oxygen/glucose deprivation or hydrogen peroxide resulted in release of synapsin I from exosomes. Application of synapsin I promoted neurite outgrowth from hippocampal neurons and increased survival of cortical neurons upon hydrogen peroxide treatment or oxygen/glucose deprivation. Coculture experiments using wild-type hippocampal neurons and wild-type or synapsin-deficient glial cells showed enhanced neurite outgrowth when synapsin was expressed by glial cells. Synapsin-induced neurite outgrowth was dependent on oligomannose on synapsin I and the neural cell adhesion molecule NCAM at the neuronal cell surface. The data indicate that, under conditions of high neuronal activity and/or oxidative stress, synapsin can be released from glial-derived exosomes and promotes neurite outgrowth and neuronal survival by modulating the interactions between glia and neurons.
Introduction
N-linked oligomannosidic glycans play important roles in modulating nervous system development and function (for review, see Kleene and Schachner, 2004). Oligomannosidic glycans are particularly abundant in the brain, are carried by adhesion molecules such as L1, NCAM, and AMOG (adhesion molecule on glia), are present on both NMDA and AMPA subtypes of glutamate receptors, and are concentrated at presynaptic and postsynaptic membranes of glutamatergic synapses of adult brains (for review, see Kleene and Schachner, 2004). Analysis of oligomannoses isolated from synaptosomes showed that the amount of the two major glycans containing five (man5) and eight (man8) mannoses increases during development with man5 being the predominant form in adult brain (Fu and Gurd, 1983). Oligomannoses also play a role in long-term potentiation of mouse hippocampal neurons (Lüthl et al., 1994) and in regeneration of retinotectal projections in goldfish (Schmidt and Schachner, 1998).
Proteins that bind these oligomannoses as oligomannose-binding lectin-like proteins are the cell adhesion molecules NCAM and basigin (Horstkorte et al., 1993; Heller et al., 2003). Basigin, which is an important player in neuron–glia interactions and in the formation and/or maintenance of the blood–brain barrier, promotes outgrowth of astrocytic processes in an oligomannose-dependent manner (Heller et al., 2003). The interaction of NCAM with L1 depends on oligomannoses on L1, and disturbance of this oligomannose-dependent interaction reduces L1-induced neurite outgrowth (Horstkorte et al., 1993).
Due to their importance in the nervous system, we further investigated the role of oligomannoses in mediating or modulating neural cell adhesion with the intention to identify novel oligomannose-carrying and oligomannose-recognizing proteins from mouse brain. Because of the limited sources of natural oligomannosidic glycans and difficulty in chemical synthesis, it was necessary to use surrogates that structurally and functionally mimic the oligosaccharides. Here, an oligomannose-mimicking peptide was identified by screening a phage display peptide library using two monoclonal oligomannose-specific antibodies (Kücherer et al., 1987; Fahrig et al., 1990). This peptide was then used to identify oligomannose-binding proteins, and synapsin I (hereafter called synapsin) was identified by this approach.
Synapsin is expressed in nervous tissue of vertebrates and invertebrates (Kao et al., 1999; Candiani et al., 2010) and is a neuron-specific, synaptic vesicle-associated protein implicated in neural development (Fornasiero et al., 2010) and synaptic transmission (Cesca et al., 2010). However, some degree of synapsin expression was also found in cultured astrocytes (Maienschein et al., 1999), epithelial cells (Bustos et al., 2001), pancreatic β cells (Krueger et al., 1999), and several cell lines of neural and endocrine origin (Romano et al., 1987; Tooze et al., 1989; Matsumoto et al., 1995).
Here, we show that synapsin is a glycoprotein and an oligomannose-binding lectin that modulates neurite outgrowth in an oligomannose- and NCAM-dependent manner. Furthermore, we provide evidence that glial cells express synapsin and release it via exosomes. Synapsin released from exosomes under distinct conditions such as neuronal activity, oxidative stress, and/or ischemia can modulate neuronal outgrowth and neuron–glia interactions.
Materials and Methods
Antibodies and reagents.
Monoclonal antibodies L3 and L4 recognizing oligomannosidic glycans were prepared as described previously (Fahrig et al., 1990). The antibodies recognizing Lewisx (L5), polysialic acid (735), or HNK-1 (412, HNK-1) have been described previously (for references, see Kleene and Schachner, 2004). The commercially available antibodies directed to the following proteins were used: Alix, flotillin, and PDI from BD Transduction Laboratories; GAPDH and myelin basic protein from Millipore; calreticulin, hsc70, and 14-3-3 from Santa Cruz Biotechnology; synapsin and synaptophysin from Synaptic Systems; phosphorylated neurofilament heavy chain (SMI-310R) from Covance; Iba-1 from Wako; neuronal class III β-tubulin (T2200) and actin from Sigma-Aldrich. Synthetic peptides were obtained from Schafer-N and RNase B from Sigma-Aldrich. Synapsin was purified from calf brain under non-denaturing conditions and antibodies against synapsin were prepared as previously described (Bähler and Greengard, 1987). AMOG was prepared from mouse brain by affinity chromatography using AMOG-specific monoclonal antibodies as described previously (Antonicek and Schachner, 1988). The extracellular domain of NCAM fused to the Fc fragment of human IgG (NCAM-Fc) and the extracellular domain of the close homolog of L1 (CHL1) fused to the Fc fragment (CHL1-Fc) were prepared and purified as described previously (Chen et al., 1999; Schmid et al., 1999). Endoglycosidase H (EndoH) and peptidyl-N-glycosidase F (PNGase F) were purchased from Roche Diagnostics. NCAM-deficient mice (Cremer et al., 1994) and C57BL/6 mice were bred and maintained at the animal facility of the Universitätsklinikum Hamburg-Eppendorf. Mice of either sex were used.
Screening of phage display peptide library.
The antibodies L3 and L4 (10 μg) were immobilized on 0.5 mg of magnetic Dynabeads M 270 (Dynal Biotech), followed by blocking with 0.5% bovine serum albumin (BSA) in 0.1 m sodium phosphate buffer, pH 7.4. The Ph.D.-12 Phage Display Peptide Library Kit (New England Biolabs) was used to express linear random 12-mer peptides fused to the N terminus of M13 coat protein PIII. Three rounds of panning were performed as described previously (Simon-Haldi et al., 2002). Briefly, 4 × 1010 phages were incubated with beads carrying L3 and L4 antibodies for 30 min at room temperature, and beads were washed 10 times with 1 ml of Tris-buffered saline (TBS; pH 7.4) containing 0.1%, 0.3%, and 0.5% (v/v) Tween 20 in the first, second, and third rounds of panning, respectively. In each round of panning, phages were eluted using either L3 or L4 antibodies (10 μg in 100 μl of TBS) or oligomannoses (5 μg in 100 μl of TBS) isolated from RNase B after EndoH treatment. Eluted phages were amplified and 2 × 1011 amplified phages were applied for panning in the second and third rounds. After three rounds of panning, individual phage clones were isolated and single-stranded phage DNA was purified and subjected to DNA sequencing to determine the peptide sequences expressed by the phages.
Conjugation of synthetic peptides to catalase.
Synthetic peptides bearing a cysteine residue at the C terminus were coupled to catalase (Sigma-Aldrich) using the crosslinker m-maleimido-benzoyl-N-hydroxy-succinimidyl ester (MBS; Pierce Biotechnology). Catalase (0.1 mm) dissolved in 0.9 ml of conjugation buffer (5 mm EDTA in PBS, pH 7.2) was mixed with 1 mm MBS freshly dissolved in 0.1 ml of dimethyl sulfoxide (DMSO). After 30 min incubation at room temperature, unbound MBS was removed by gel filtration through a PD-10 column (GE Healthcare). The flow-through was incubated with 1 mm peptides freshly dissolved in 0.2 ml of 20% DMSO in PBS. This mixture was incubated at room temperature for 30 min, dialyzed overnight at 4°C against 4 L of TBS, and concentrated by Vivaspin concentrators (Vivaspin 20, Vivascience) with 30 kDa molecular weight cutoff. For quantifying the amount of conjugated peptide, free sulfhydryl groups before and after coupling were determined by Ellman's test (Pierce).
ELISA.
To determine binding of phage clones to L3 and L4 antibodies, L3 or L4 antibodies (10 μg/ml in PBS, 50 μl per well) were applied to MaxiSorp Immuno Modules (Nunc) and incubated overnight at 4°C. Wells were blocked with 100 μl of blocking solution per well (1% BSA in PBS, pH 7.4) for 1 h at room temperature, and 5 × 109 purified phages in 50 μl of blocking buffer were added to each well and incubated for 1 h at room temperature. Plates were washed 5 times with 100 μl of PBST (0.03% Tween 20 in PBS, pH 7.4) and incubated with 50 μl of horseradish peroxidase (HRP)-conjugated M13 antibodies in blocking solution. The plates were then washed 5 times with PBST, and finally 0.5% o-phenylenediamine dihydrochloride (OPD; Pierce) was added. The absorbance was determined at 490 nm in a microtiter plate reader (μQuant, BioTek).
To monitor the binding between catalase-coupled peptides and L3 or L4 antibodies, wells of 96-well microtiter plates were coated with serial dilutions of catalase-coupled peptides in CANDOR blocking solution (CANDOR Bioscience; 50 μl per well) overnight at 4°C and then wells were blocked with 100 μl of CANDOR blocking solution for 1 h at room temperature. L3 and L4 antibodies (10 μg/ml; 50 μl per well) in LowCross-Buffer (CANDOR Bioscience) were added to the wells and incubated for 1 h at room temperature. For competition ELISA, wells were coated with 40 μg/ml catalase-coupled peptides in blocking solution (50 μl per well), and L3 and L4 antibodies preincubated in 50 μl of LowCross-Buffer with different concentrations of RNase B or EndoH-treated RNase B for 30 min were added to the wells and incubated for 1.5 h at room temperature. After washing with PBST for 5 times, 50 μl of HRP-conjugated anti-rat secondary antibodies in LowCross-Buffer were added and incubated for 1 h at room temperature. The wells were then washed for 5 times with PBST, 0.5% OPD was added, and absorbance at 490 nm was determined.
To identify the oligomannose-specific interaction between AMOG and synapsin, 10 pmol (in 50 μl of PBS) of untreated or EndoH-treated AMOG was coated overnight at 4°C. After washing with PBS and blocking with blocking solution (1% BSA in PBS) for 1 h, 6.7 pmol (in 50 μl of blocking solution) of synapsin alone or preincubated for 30 min with 50 μg/ml (in 50 μl of blocking solution) oligomannoses isolated from AMOG upon EndoH-treatment was incubated for 1 h at room temperature. After washing, synapsin antibody was added, followed by the corresponding secondary antibody after washing with PBST. Wells were washed with PBST, OPD was added, and absorbance at 490 nm was determined. Alternatively, wells were coated with synapsin (6.7 pmol/well) overnight at 4°C and blocked for 1 h at room temperature, and increasing amounts of AMOG or EndoH-treated AMOG in 2% BSA were added and incubated overnight at 4°C. After washing with PBST, AMOG antibody in 2% BSA was applied for 1 h at room temperature, and after washing with PBST, HRP-coupled secondary antibody in 2% BSA was applied for 1 h at room temperature. After washing with PBST, 1% OPD was used for detection at 490 nm.
To investigate the interaction between synapsin and NCAM, 6.7 pmol (in 50 μl of PBS) of either untreated or EndoH-treated purified bovine synapsin was coated overnight at 4°C. After blocking with 100 μl of blocking solution (1% BSA in PBS) at room temperature, 6.5 pmol (in 50 μl of blocking solution) of NCAM-Fc or CHL1-Fc preincubated with either untreated or EndoH-treated AMOG (40 pmol) at room temperature for 30 min was incubated at 4°C overnight. Plates were washed with TBST (0.03% Tween 20 in PBS) and incubated with HRP-conjugated anti-human Fc antibody at room temperature for 1 h. Wells were washed with TBST, OPD was added, and absorbance at 490 nm was determined.
Neurite outgrowth assay.
Dissociated hippocampal neurons obtained from neonatal C57BL/6 wild-type and NCAM-deficient mice were used in neurite outgrowth experiments. Briefly, isolated hippocampi from mouse brains were placed in ice-cold dissection solution (4.16 mm NaHCO3, 10 mm HEPES, 34 mm d-glucose, 12 mm MgSO4, 2.88 mg/ml BSA, and 5 μg/ml gentamicin in HBSS) and then incubated in digestion solution (5 mg/ml trypsin, 1 mg/ml DNase I, 135 mm NaCl, 5 mm KCl, 7 mm Na2HPO4, 4.16 mm NaHCO3, and 25 mm HEPES, pH 7.3) for 15 min at room temperature. The tissue was washed three times with ice-cold dissection solution and incubated in dissociation solution containing 1 mg/ml DNase I using fire-polished Pasteur pipettes. Dissociated cells were resuspended in 10 ml of dissection solution, centrifuged at 800 × g for 10 min at 4°C, and resuspended in culture medium (Neurobasal A supplied with 2 mm l-glutamine, 2% B27, and 5 μg/ml gentamicin). Cells at a density of 2.5 × 104 were seeded on coverslips coated with 0.01% poly-l-lysine (PLL), PLL with catalase or catalase-conjugated peptides (100 μg/ml), PLL with untreated, heat-denatured RNase B (60°C for 10 min) or EndoH-treated RNase B (50 μg/ml), native synapsin, heat-denatured synapsin, or EndoH-treated synapsin (10 μg/ml). For substrate coating, molecules were diluted in HBSS and incubated on coverslips overnight at 4°C. Before seeding, coverslips were washed three times with ice-cold HBSS. After seeding, cells were cultured in the absence or presence of soluble synapsin (10 μg/ml) for 24 h in a humidified chamber at 37°C with 5% CO2. In some experiments, RNase B diluted in prewarmed culture medium was added to the culture 1 h after seeding. Cultures were fixed with 2.5% glutaraldehyde and stained with 1% toluidine blue in 1% borax. The length of neurites was determined from neurons that were not in contact with other cells and had at least one process that was as long as the diameter of the cell body. Measurements were performed with AxioVision 4.6 software (Zeiss Microimaging).
Cell adhesion assay.
Astrocytes were obtained from the cerebral cortex of newborn C57BL/6 mice (Won and Oh, 2000) and seeded as single-cell suspensions (5 × 105 cells/ml) in DMEM with 10% fetal calf serum (FCS), 2 mm l-glutamine, and 50 U/ml penicillin/streptomycin onto tissue culture Petri dishes. On the third day of culture, the culture medium was replaced with serum-free medium. After 7 d, freshly dissociated neurons obtained from the cerebellum of 7-d-old mice expressing green fluorescent protein as described previously (Husmann et al., 1992) were suspended in HBSS, adjusted to a cell density of 2 × 105 cells/ml, and seeded onto the monolayer of cultured astrocytes. Both astrocytes and neurons were independently preincubated with catalase or catalase-coupled peptides in Ca2+/Mg2+-free HBSS (CMF-HBSS) for 5 min on ice. Cocultures were incubated for 10 min at room temperature in a reciprocal shaker at 40 cycles/min. Nonbound neurons were removed from the astrocyte monolayer by four gentle washings with CMF-HBSS. Adhering neurons were counted in at least 50 microscopic fields at 100-fold magnification with an inverted fluorescence microscope (Zeiss Microimaging). Aggregated clusters of green cells were not counted.
Cross-linking assay.
Sulfo-succinimidyl-2-[6-(biotinamido)-2-(p-azido-benzamido) hexanoamido] ethyl-1,3′-dithiopropionate (Sulfo-SBED, 0.25 mg; Pierce Biotechnology) predissolved in 6 μl of DMSO was incubated with 200 μg of catalase-coupled peptides resuspended in 200 μl of PBS for 30 min in the dark under constant stirring at room temperature. Nonreacted crosslinker was removed by using Vivaspin concentrators with 10 kDa molecular weight cutoff. Brains from 2-week-old wild-type mice were homogenized in ice-cold homogenization buffer [50 mm Tris-HCl, pH 7.5, 2 mm EDTA, and EDTA-free protease inhibitor cocktail (Roche Diagnostics)]. Homogenates were centrifuged at 1000 × g for 10 min at 4°C. N-Octyl β-d-glucopyranoside (50 mm) was added to the supernatant, and this crude homogenate was incubated overnight at 4°C. The protein concentration was adjusted to 1 mg/ml with homogenization buffer and the crosslinker conjugates were added and incubated with 1 ml of the crude detergent-solubilized homogenate overnight at 4°C in the dark. The samples were then exposed to UV light (365 nm) for 15 min on ice to photoactivate the aryl azide group of the crosslinker and to covalently attach bound proteins. To enrich biotin-carrying proteins, 50 μl magnetic MagnaBind Streptavidin beads (Pierce Biotechnology) were incubated with the samples for 1 h at room temperature with mild rotation. Beads were washed 5 times with 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS, Sigma-Aldrich) in PBS. Beads were then boiled in sample buffer (60 mm Tris/HCl, pH 6.8, 2% SDS, 1% β-mercaptoethanol, 10% glycerol, and 0.02% bromophenol blue) and resolved by SDS-PAGE under reducing conditions, resulting in a transfer of the biotin residue from the crosslinker to the bound protein(s). Biotinylated proteins were detected by Western blot analysis using HRP-conjugated streptavidin.
Immunoaffinity chromatography.
Freeze-dried powder of CNBr-activated Sepharose 4B (120 mg; GE Healthcare) was incubated with 5 ml of swelling solution (1 mm HCl) for 15 min at 4°C. The resin was washed three times with swelling solution and twice with 5 ml of coupling solution (100 mm NaHCO3, 500 mm NaCl, pH 8.3) for 15 min at 4°C and then pelleted by centrifugation at 200 × g for 5 min. Either L3 or L4 antibodies (200 mg) were incubated with pretreated resin in coupling solution overnight at 4°C with end-over-end rotation. The resin was washed with 10 ml of coupling buffer and remaining active groups were blocked by incubation with 5 ml of blocking solution (200 mm glycine, 500 mm NaCl, pH 8.0) at 4°C with end-over-end rotation. After three cycles of washing with buffer A (100 mm sodium acetate and 500 mm NaCl, pH 4.0), buffer B (100 mm NaHCO3 and 500 mm NaCl, pH 8.3), and buffer C (PBS, pH 7.4), the resin was packed into a column. The column was washed with 20 ml of buffer D (25 mm Tris, 150 mm NaCl, and 5 mm EDTA, pH 7.4), and the brain extract was applied to the column overnight at 4°C. The column was then washed with 10 ml of buffer E (25 mm Tris, 150 mm NaCl, 1% Triton X-100, and 0.02% NaN3, pH 7.4) followed by 10 ml of buffer F (25 mm Tris/HCl, 500 mm NaCl, 0.1% Triton X-100, and 0.02% NaN3, pH 7.4). Proteins that bound to either L3 or L4 antibodies were eluted with elution buffer (50 mm ethanolamine, 150 mm NaCl, and 0.2% CHAPS, pH 11.5). The eluates were dialyzed against TBS and concentrated with Vivaspin concentrators with 5 kDa molecular weight cutoff.
Mass spectrometry.
After SDS-PAGE, the gel was stained with colloidal Coomassie Blue (Roth) and protein bands were cut out. After successive treatment with dithiothreitol and iodoacetamide, in-gel digestion of proteins by 5 ng/μl trypsin (Promega) in 50 mm NH4HCO3 was performed overnight at 37°C. Gel pieces were then repeatedly extracted with 50% acetonitrile/5% formic acid/45% distilled water, and the combined extracts were dried down in a vacuum concentrator, redissolved in 5% methanol/5% formic acid in water, desalted on a C18 μZipTip (Millipore), eluted with 1 μl of 60% methanol/5% formic acid/35% water, and analyzed by nano-electrospray mass spectrometry in a QTOF II instrument (Micromass). The MS/MS spectra obtained by collision-induced fragmentation of the peptides were evaluated both manually and by the Mascot MS/MS ion search algorithm (Matrix Sciences).
Deglycosylation.
For the EndoH-treatment, 10 μg of glycoprotein, 10 μl of 5× reaction buffer (250 mm sodium acetate, pH 5.5), and 2.5 μl of 20× denaturing buffer (2% SDS and 10% β-mercaptoethanol) were incubated in a total reaction volume of 45 μl at 100°C for 10 min. EndoH (25 mU in 5 μl) was added and incubated at 37°C for 3 h. For treatment with PNGase F, 10 μg of glycoprotein, 10 μl of 5× reaction buffer (0.5 m sodium phosphate, pH 7.4), and 5 μl of 10× denaturing buffer (2% SDS and 10% β-mercaptoethanol) were incubated in a total reaction volume of 45 μl at 100°C for 10 min. Triton X-100 (2.5 μl; 10%) and PNGase F (2.5 μl; 2.5 U) were added and incubated at 37°C for 3 h.
Western blot analysis and lectin staining.
Western blot analysis was performed as described previously (Kleene et al., 2007). For lectin staining, synapsin (0.5 μg) was subjected to SDS-PAGE and transferred onto nitrocellulose membranes. Glycans on synapsin were detected by DIG glycan differentiation kit (Roche Diagnostics) according to the manufacturer's protocol.
Isolation and treatment of exosomes from astrocytes.
Astrocyte-enriched cultures obtained from cortex or whole brain of 1- to 2-d-old mice were prepared as described previously (Won and Oh, 2000; Kleene et al., 2007) and cultured for 2 weeks or 1 week with serum-containing DMEM medium and then in serum-free DMEM medium for 12 h, respectively. For hydrogen peroxide treatment, astrocytes were cultured for 6 d in serum-containing DMEM medium and then in serum-free DMEM medium for 24 h in the absence or presence of 20 μm hydrogen peroxide. For oxygen/glucose deprivation, astrocytes obtained from whole brain were grown for 6 d in DMEM containing high glucose (4.5 g/L) and supplemented with 10% FCS. The medium was replaced by serum-free DMEM containing low glucose (1 g/L). For oxygen deprivation, cultures were placed into a humidified hypoxia chamber (Billups-Rothenberg) and flushed with 95% N2/ 5% CO2 for 10 min, and the sealed hypoxia chamber was incubated overnight at 37°C.
All following steps were performed at 4°C, unless stated otherwise. Cell culture supernatants were collected and centrifuged at 1000 × g for 10 min. The resulting 1000 × g supernatants were further centrifuged at 10,000 × g for 30 min followed by centrifugation of the resulting supernatants at 100,000 × g for 3 h. The resulting pellets were either taken as exosome-enriched fraction for further analysis or were subjected to further enrichment by sucrose gradient centrifugation. Therefore, the 100,000 × g pellet was resuspended in 2.5 m sucrose buffered with 20 mm HEPES, pH 7.4, and overlaid with a sucrose step gradient (2.25, 2.0, 1.75, 1.5, 1.25, 1.0, 0.75, 0.5, 0.25 m). The sucrose gradient was then centrifuged at 100,000 × g for 5 h and 10 fractions were collected from the top of the gradient and diluted with 20 mm HEPES, pH 7.4, and centrifuged at 100,000 × g for 1.5 h. Pellets from fractions enriched in synapsin and the exosomal marker proteins Alix and flotillin were combined and used for further analyses as indicated. For treatments of isolated exosomes, the pellet derived from the combined sucrose gradient fractions was resuspended in PBS and treated with for 5 min at 37°C under conditions as indicated. The samples were then centrifuged at 100,000 × g for 1 h. Cell culture supernatants were subjected to protein precipitation using 20% trichloroacetic acid. Proteins were collected by centrifugation at 20,000 × g for 30 min, and the resulting pellets were washed with acetone and resuspended in sample buffer. For preparation of cell lysates, astrocytes were collected in PBS using a cell scraper.
Electron microscopy.
Pellets containing exosomes were incubated in fixing solution (4% paraformaldehyde and 1% glutaraldehyde in 0.1 m sodium phosphate buffer, pH 7.3) for 60 min at room temperature. After washing with PBS, pH 7.3, for 10 min, exosomes were embedded in 2% low-melting agarose, and postfixed with 1% OsO4 in 0.1 m sodium phosphate buffer, pH 7.3, for 30 min on ice. Dehydration was performed by exposure to increasing concentrations of ethanol (30%, 50%, 70%, 80%, 90%) followed by two incubation steps with 100% ethanol for 15 min and a final incubation with propylenoxid for 15 min. Specimens were incubated with propylenoxid/epoxy resin (1:1) for 90 min and with propylenoxid/epoxy resin (1:2) for 2 h followed by embedding in neat epoxy resin. Ultrathin sections were cut and analyzed with a Zeiss 902 electron microscope.
Cell surface biotinylation.
Astrocyte-enriched cell cultures maintained for 7–9 d were washed twice with ice-cooled PBS2+ (PBS with 0.5 mm CaCl2 and 2 mm MgCl2) and incubated for 10 min on ice with sulfo-NHS-LC-biotin (Thermo Scientific) freshly prepared in ice-cooled PBS2+. After treating the cells with 20 mm glycine in PBS2+ for 5 min on ice, cells were washed twice with ice-cooled PBS2+ and lysed with RIPA buffer [50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1% NP40, and protease inhibitor cocktail (Roche)] for 30 min at 4°C with mild shaking. Lysed cells were collected using a rubber scraper, centrifuged for 10 min at 1000 × g and 4°C. Streptavidin-coupled magnetic beads (Pierce Biotechnology) were added to the lysate and incubated overnight at 4°C. After washing the beads twice with RIPA buffer and once with PBS2+, beads were boiled in sample buffer and the eluate resulting from centrifugation at 10,000 × g for 5 min was saved for further analyses.
Immunoprecipitation and double immunostaining.
For immunoprecipitation of synapsin, cell lysates or cell culture supernatants were incubated overnight with synapsin antibody at 4°C under constant rotation. Then, protein A/G agarose beads (Santa Cruz Biotechnology) were added and the incubation was continued for 3 h at 4°C. After sedimentation, beads were then washed twice with RIPA buffer and once with PBS and boiled in SDS sample buffer. The eluate from the beads was subjected to Western blot analysis.
For double immunostaining, astrocyte-enriched glial cell cultures were prepared from whole mouse brain and maintained for 6 d on PLL. All following steps were performed at room temperature. After removing the culture medium, cells were fixed by incubation with 4% formaldehyde for 10 min, washed twice with PBS, incubated for 1 h with 0.1% BSA/ 0.3% Triton X-100 followed by incubation with primary antibodies resuspended in PBS containing 0.1% BSA. After three washes with PBS, the corresponding secondary antibodies labeled with the fluorescent dye Cy2 or Cy3 were resuspended in PBS with 0.1% BSA and incubated for 1 h with fixed cells. After two washes with PBS, coverslips were mounted with Roti-Mount FluorCare DAPI (Roth). Immunopositive cells in five view fields were counted under the inverted fluorescence microscope at 20× magnification. The numbers relative to the number of DAPI-stained nuclei were calculated from two independent experiments.
Neuronal cell death assay.
Dissociated cortical neurons were obtained from E16–E17 mouse embryos. Briefly, pregnant mice were killed with CO2 inhalation, uterus and embryos were exposed, and cerebral cortices were removed, dissected, and incubated in digestion solution (0.025% trypsin in HBSS) for 15 min at 37°C. After addition of 1% BSA and 1% trypsin inhibitor in HBSS, the samples was centrifuged at 800 × g for 10 min and the pellet was resuspended in culture medium (Neurobasal A supplied with 2 mm l-glutamine, 2% B27, and 5 μg/ml gentamicin) and triturated with a fire-polished Pasteur pipette. Dissociated cells were seeded onto PLL- and synapsin (1.5 μg/well)-coated 48-well plates at a density of 2 × 105 cells in 250 μl of medium per well. After 24 h in a humidified chamber at 37°C with 5% CO2, 20 μm hydrogen peroxide was added to the cells, which were incubated under these conditions for 24 h. Cells were stained with calcein (1 μg/ml) and propidium iodide (2 μg/ml) for 30 min at 37°C in a humidified chamber and then washed with culture medium. Live cells stained with calcein and dead cells stained with propidium iodide in each corresponding view field were quantified under the inverted fluorescence microscope at 20× magnification. At least four view fields were quantified for each well using the AxioVision 4.6 software (Zeiss Microimaging). Cell survival was determined as live cell/total cell ratio, and survival in the nonstressed control group was set as 100%.
Oxygen/glucose deprivation.
Dissociated cortical neurons were obtained from E16 mouse embryos and seeded onto PLL- or synapsin-coated plastic plates at a density of with 5 × 105 cells/ml in Neurobasal A medium supplemented with B-27, 2 mm l-glutamine, and antibiotics. After 2 d, the conditioned cell culture medium was removed and saved. Cells were washed twice with prewarmed deoxygenated PBS (gassed for 30 min with N2) and incubated in deoxygenated PBS without or with 20 μg/ml synapsin. Cultures were then placed into a humidified hypoxia chamber (Billups-Rothenberg) and flushed with 100% N2 for 10 min. The sealed hypoxia chamber with the treated cell cultures was placed into an incubator at 37°C for 3 h. Reoxygenation was induced by removing PBS and adding the previous conditioned cell culture media and further maintained for 12 h in an incubator at 37°C. Cell survival was estimated as described above.
Coculture experiments.
Primary glial cultures were prepared from P0 wild-type or synapsin I/II/III triple knock-out (TKO) (Gitler et al., 2004) C57B/L6 mice (with modifications from Kaech and Banker, 2006). Briefly, cortices were dissected in ice-cold HBSS and incubated in 0.25% trypsin plus 1 mg/ml DNase for 30 min at 37°C. Cells were centrifuged, resuspended in glial culture medium (MEM, 10% horse serum, 33 mm glucose, and antibiotics) and plated onto PLL-coated 25 mm glass coverslips at low density (45,000 cells per coverslip). After 24 h, the medium was replaced with fresh glial culture medium, and cells were grown to confluence for 2 weeks. Hippocampal neurons obtained from E18 C57BL/6 wild-type embryos [with modifications from Kaech and Banker (2006)] were plated on mature glial cultures that had been starved for 48 h in neuronal medium (Neurobasal with B27 supplement, 2 mm glutamine, and antibiotics). Cocultures were grown for 24 h, fixed, and stained with antibody against neurofilament heavy chain (SMI-31) and against β-III tubulin antibody (T2200) to identify axons and the neurite network, respectively. Images were acquired with an Olympus BX51 upright microscope equipped with a Neurolucida XY stage and motorized Z movement, using a UPlanFL N 20×/0.50 objective, and the filter cubes Semrock CY5–4040A and Olympus FITC U-MNIBA3. Images were visualized with the Neurolucida Version 8 software (MBF-Bioscience). Sholl analysis was performed using the Sholl plugin of the ImageJ software. Axonal length and branching were measured using the NeuronJ plugin of the ImageJ software. Mean area staining intensity was determined by using the ImageJ software.
Results
Identification of an oligomannose-mimicking peptide
The aim of our project was to identify novel oligomannose-recognizing receptors in mouse brain. Because oligomannosidic glycans from natural sources are only available in limited amounts and also difficult to synthesize chemically, we had to use a surrogate to achieve our goal. Peptides have shown to structurally and functionally mimic oligosaccharides (Pirofski, 2001; Johnson et al., 2002; Monzavi-Karbassi et al., 2002) and thus are potential surrogates for carbohydrates. Therefore, we first attempted to isolate an oligomannose-mimicking peptide. The oligomannose-recognizing antibodies L3 and L4 (Fahrig et al., 1990) were used in a phage display screen to isolate oligomannose-mimicking peptides from a 12-mer peptide library. Phages that bound to L3 and L4 antibodies were eluted with antibody and/or sugar and further analyzed by ELISA using L3 or L4 antibodies as substrate coat. One phage obtained by antibody and sugar elution showed very strong binding to both antibodies (data not shown) and expressed the peptide sequence TISWWHLWPSPA. A synthetic peptide with this sequence was used for further analyses to verify that this peptide is indeed an oligomannose-specific glycomimetic. The synthetic peptide, which showed low solubility in aqueous buffers, was conjugated to the soluble carrier protein catalase to improve solubility and to increase the binding avidity of the peptide to its binding partners. First, the binding of the oligomannose-specific antibodies L3 and L4 to substrate-coated peptide coupled to catalase was analyzed. Both antibodies showed a concentration-dependent and saturable binding to the catalase-coupled peptide, whereas they showed only a very weak binding to catalase or to catalase-coupled control peptide with a scrambled sequence of the mimicking peptide (Fig. 1A).

Characterization of a synthetic oligomannose-mimicking peptide. A, Binding of L3 and L4 antibodies to increasing amounts of substrate-coated catalase (c), catalase-coupled putative oligomannose-mimicking peptide (c-pep), or catalase-coupled scrambled peptide (c-scr) was determined by ELISA. B, L3 and L4 antibodies were preincubated with various amounts of untreated RNase B (RB) or EndoH-treated, deglycosylated RNase B (de-RB) and added to the substrate-coated putative oligomannose-mimicking peptide. Binding of the antibodies was determined by ELISA. Mean values ± SEM from at least three independent experiments are shown.
To confirm that the peptide is an oligomannose-mimicking peptide, we tested whether the binding of the oligomannose-specific antibodies to the peptide was oligomannose dependent. Therefore, we performed a competition ELISA using oligomannose-bearing RNase B for competition and EndoH-treated oligomannose-lacking RNase B as control. In the presence of untreated RNase B, the binding of the antibodies to the substrate-coated catalase-coupled peptide decreased in a concentration-dependent manner, while EndoH-treated RNase B, being devoid of oligomannoses, showed no effect on the binding (Fig. 1B). This result confirms that the peptide indeed is an oligomannose-mimicking peptide, because it competes with the oligomannoses on RNase B for binding to the antibodies L3 and L4.
The oligomannose-mimicking peptide stimulates neurite outgrowth and disturbs adhesion between neurons and astrocytes
Next, we performed functional tests to investigate whether the oligomannose-mimicking peptide is functionally active. Since oligomannoses have been reported to promote neurite outgrowth (Chandrasekaran et al., 1991; Tanzer et al., 1993), we tested whether also the oligomannose-mimicking peptide had this effect. Hippocampal neurons were seeded onto substrate-coated catalase-coupled oligomannose-mimicking peptide. Oligomannose-carrying RNase B served as positive control and PLL, catalase, catalase-coupled scrambled peptide, and EndoH-treated oligomannose-lacking RNase B were used as negative controls. In comparison to PLL, neurite outgrowth of hippocampal neurons was promoted on substrate-coated catalase-coupled oligomannose-mimicking peptide and on oligomannose-carrying RNase B, while substrate-coated catalase, catalase-coupled scrambled peptide and oligomannose-lacking EndoH-treated RNase B were virtually ineffective (Fig. 2A).

The oligomannose-mimicking peptide promotes neurite outgrowth and disturbs adhesion of neurons to cortical astrocytes. A, Hippocampal neurons were grown on substrate-coated PLL alone or with catalase (c), catalase-coupled oligomannose-mimicking peptide (c-pep), catalase-coupled scrambled peptide (c-scr), RNase B (RB), or deglycosylated RNase B (de-RB). After 24 h in culture, cells were fixed and the length of the longest neurite per cell was determined. Mean length of the longest neurites obtained from PLL treatment was set to 100%. B, Freshly prepared GFP-expressing cerebellar granule neurons were preincubated with either solvent alone (control), catalase (c), catalase-coupled oligomannose-mimicking peptide (c-pep), or catalase-coupled scrambled peptide (c-scr) and subsequently added to a monolayer of cultured cortical astrocytes pretreated with solvent alone, catalase (c), catalase-coupled oligomannose-mimicking peptide (c-pep), or catalase-coupled scrambled peptide (c-scr). After removal of nonadherent cells, GFP-positive cells attached to the astrocyte monolayer were quantified. The mean number of adherent cells in the solvent-treated (PLL) control was set to 100%. Mean values ± SEM of three independent experiments are shown (**p < 0.01; one-way ANOVA followed by Dunnett's multiple-comparison test).
Since oligomannose-binding antibodies L3 and L4 have been shown to interfere with the adhesion between neurons and astrocytes (Fahrig et al., 1990), we also investigated whether the oligomannose-mimicking peptide affected the interaction between neurons and astrocytes. In a short-term adhesion assay, cerebellar neurons were plated onto cultured cortical astrocytes in the presence of catalase or catalase coupled to either oligomannose-mimicking peptide or scrambled peptide. In the presence of catalase-coupled oligomannose-mimicking peptide, adhesion between neurons and astrocytes decreased by 50% with respect to the control, while adhesion in the presence of catalase or scrambled peptide-coupled catalase was not or only slightly affected, respectively (Fig. 2B). These results confirm that the oligomannose-mimicking peptide is functionally active in stimulating neurite outgrowth and inhibiting adhesion of neurons to astrocytes.
Identification of synapsin as oligomannose-binding lectin
Next, we used the oligomannose-mimicking peptide in crosslinking experiments with the aim to identify novel oligomannose-binding proteins in mouse brain. Oligomannose-mimicking peptide and scrambled peptide coupled to catalase, and catalase alone, were conjugated to the trifunctional crosslinker sulfo-SBED, and these conjugates were used as baits to isolate bound target proteins from crude brain homogenate. By UV crosslinking, target proteins were covalently attached to the bait proteins and isolated via the biotin moiety present on the crosslinker. Reducing conditions during gel electrophoresis led to transfer of the biotin label from the bait to the bound target protein(s). Western blot analysis with streptavidin to detect biotinylated proteins revealed a very intense staining of a protein band with an apparent molecular weight of 75 kDa and a weak staining of several other protein bands, when catalase-coupled oligomannose-mimicking peptide was used as bait, whereas no biotinylated protein was detectable when catalase or catalase-coupled scrambled peptide was used as bait (Fig. 3A). Protein bands showing an apparent molecular weight of ∼75 kDa were cut out from a Coomassie Blue-stained gel (Fig. 3B), and mass spectrometry of these bands identified synapsin I (Table 1) (hereafter called synapsin). To confirm that synapsin was identical to the 75 kDa protein that predominantly bound to the oligomannose-mimicking peptide, Western blot analysis using synapsin-specific antibodies was carried out. A strong synapsin signal around 75 kDa was observed when the oligomannose-mimicking peptide was used as bait, whereas no signal or a very weak signal was seen when either catalase or catalase-coupled scrambled peptide was used as bait, respectively (Fig. 3C).

Synapsin is an oligomannose-specific lectin. Either catalase (c), catalase-coupled oligomannose-mimicking peptide (c-pep), or catalase-coupled scrambled peptide (c-scr) was conjugated to the biotin-carrying multifunctional and cleavable crosslinker sulfo-SBED. The conjugates were incubated with detergent extracts of crude brain homogenate. After UV crosslinking, proteins bound to the conjugates were isolated, separated by gel electrophoresis and subjected to Western blot analysis using HRP-coupled streptavidin (A), Coomassie Blue staining (CB) with the arrow indicating the position of the protein band identified as synapsin (B), or Western blot analysis with synapsin antibody (C). D, Synapsin preincubated in the absence (syn) or presence (syn+OM) of AMOG-derived oligomannose was incubated with substrate-coated untreated AMOG (AM) or EndoH-treated, deglycosylated AMOG (de-AM). The binding of synapsin was determined by ELISA using a synapsin antibody and background staining was subtracted. Mean values ± SEM of three independent experiments are indicated (**p < 0.01; one-way ANOVA followed by Dunnett's multiple-comparison test). E, Increasing amounts of AMOG (AM) or EndoH-treated, deglycosylated AMOG (de-AM) were incubated with substrate-coated synapsin. The binding of AMOG was determined by ELISA using an AMOG antibody and background staining was subtracted. Mean values ± SEM of three independent experiments are indicated.
Mass spectrometry of the 75 kDa protein band isolated by crosslinking with immobilized oligomannose-mimicking peptide revealed six peptide masses that match to mouse synapsin
To further confirm that synapsin acts as an oligomannose-specific lectin, the binding of purified synapsin or synapsin preincubated with AMOG-derived oligomannoses to substrate-coated oligomannose-bearing AMOG or to EndoH-treated, oligomannose-free AMOG was determined in ELISA experiments. Synapsin showed binding to untreated oligomannose-carrying AMOG, but a reduced binding to EndoH-treated oligomannose-lacking AMOG (Fig. 3D). The binding of synapsin to AMOG was also reduced when synapsin was preincubated with AMOG-derived oligomannose (Fig. 3D). An ELISA experiment performed by applying increasing amounts of AMOG or EndoH-treated AMOG to substrate-coated synapsin showed a concentration-dependent saturable binding of untreated oligomannose-carrying AMOG, but no binding of EndoH-treated oligomannose-lacking AMOG to synapsin (Fig. 3E). These results indicate that synapsin is indeed an oligomannose-binding lectin-like protein.
Synapsin is a glycoprotein
In an attempt to identify novel oligomannose-carrying proteins, immunoaffinity chromatography with L3, L4, or nonimmune control antibody using mouse brain extracts was performed. Western blot analysis using L3 or L4 antibody revealed a very prominent L3- and L4-positive band of ∼75 kDa in the eluates from the L3 and L4 antibody columns, but not in the eluate from the control antibody column (Fig. 4A,B). This result suggested that the 75 kDa protein is the most prominent oligomannose carrier. According to the molecular weight, we considered the possibility that the 75 kDa protein was synapsin and probed the eluates with synapsin antibodies in Western blot analysis. A strong signal at 75 kDa was observed in the eluates from to L3 and L4 columns, but no visible band was observed in the eluate from the control antibody column (Fig. 4C). These results imply that synapsin is a major oligomannose carrier. To further confirm that synapsin is a glycoprotein, purified synapsin was digested with EndoH to remove oligomannose, or with PNGase F to remove all N-glycans. Western blot analysis showed that synapsin deglycosylated by EndoH or PNGase F was no longer recognized by L3 and L4 antibodies, but it was still detected by the synapsin antibody (Fig. 4D). These results show that synapsin carries N-glycans and, thus, is a glycoprotein. Since synapsin has been reported to carry fucose in α1,2-linkage to galactose (Murrey et al., 2006) and we showed that it carries oligomannose, we further characterized the glycosylation of purified synapsin using carbohydrate-specific antibodies and lectins. No synapsin staining was observed with antibodies recognizing the HNK-1 epitope or polysialic acid, while L5 antibody, which recognizes Lewisx, bound to synapsin (Fig. 4E). The persistence of synapsin staining by the L5 antibody after treatment with PNGase F (data not shown) indicates that Lewisx is present on O-linked glycans of synapsin. All tested lectins showed binding to synapsin (Fig. 4F). These results indicate that synapsin is a glycoprotein carrying N-glycans and O-glycans.

Synapsin is an oligomannose-bearing glycoprotein. Eluates from L3, L4, or control (ctrl) antibody columns were probed with oligomannose reactive L3 (A) or L4 (B) antibody or with synapsin antibody (C). The band around 50 kDa (arrow) represents the heavy chain of antibodies leaking from the column. D, Purified bovine synapsin was incubated in the absence (syn) or in the presence of EndoH (syn/H) or PNGase F (syn/F) and subjected to Western blot analysis using L3, L4, or synapsin antibody. E, F, Purified bovine synapsin was either subjected to Western blot analysis with antibodies against synapsin (syn) (E, F), HNK1 (HNK, 412) (E), polysialic acid (PSA) (E), and Lewisx (Lex) (E) or tested for lectin staining using Galanthus nivalis agglutinin (GNA), Datura stramonium agglutinin (DSA), peanut agglutinin (PNA), Sambucus nigra agglutinin (SNA), and Maackia amurensis agglutinin (MAA) (F).
Synapsin is present in exosomes isolated from astrocyte-enriched cell cultures
Since synapsin was found to be both a glycoprotein and a glycan-binding protein, it was conceivable that it is released from the cell and functions as an extracellular protein. So far, synapsin is known to be a neuron-specific protein associated with the cytoplasmic side of synaptic vesicles and regulating synaptic vesicle trafficking. However, it has been reported that synapsin is also expressed by other cell types, including cultured astrocytes, albeit at much lower levels (Maienschein et al., 1999). Therefore, we first used cultured neurons and astrocytes for immunocytochemistry and cell surface biotinylation to check whether synapsin was detectable as an extracellular protein at the cell surface of these cells. Synapsin immunoreactivity was not detectable at the cell surface by immunostaining of live neurons of different origin, such as hippocampal, cerebellar, and cortical neurons, as well as astrocytes isolated from cortex or whole brain of neonatal mice (data not shown). Since cultured cortical astrocytes have been shown to express synapsin (Maienschein et al., 1999), we used this cell culture system for the following analyses. For comparison and because of higher yields, astrocyte-enriched cultures from whole brain, which we had used previously (Kleene et al., 2007), were used in most of the following experiments either in parallel to or instead of cortical astrocyte-enriched cultures. After cell surface biotinylation, isolation of biotinylated protein by streptavidin beads, and Western blot analysis of biotinylated proteins, synapsin immunoreactivity was detected in the samples obtained from astrocyte-enriched cultures derived from cortex and whole brain (Fig. 5A), but not in the sample derived from cerebellar neurons (data not shown). As seen before by Western blot analysis (Kleene et al., 2007), astrocytic marker proteins, such as GFAP or AMOG, but not neuron-specific markers, such as β-III tubulin or L1, were detected in our glial cell cultures (data not shown), indicating that our cell culture system is enriched in astrocytes. Densitometric analysis of the signal intensities obtained for synapsin in the eluate and for total synapsin indicated that ∼10 ± 5% of total synapsin was present in the eluate containing biotinylated proteins, suggesting that this amount of synapsin expressed by cultured astrocytes is exposed at the cell surface. As a control, Western blot analysis using a GAPDH antibody revealed that GAPDH was not present in the eluate containing biotinylated proteins (Fig. 5A). To confirm that synapsin was indeed biotinylated and did not merely coisolate with biotinylated transmembrane cell surface proteins, synapsin was immunoprecipitated after cell surface biotinylation of cultured cortical astrocytes and the immunoprecipitates were subjected to Western blot analysis using either HRP-conjugated streptavidin or the synapsin antibody. In comparison to a strong signal at 75 kDa seen for synapsin with the synapsin antibody, low, but clearly detectable, amounts of only one 75 kDa protein were observed with streptavidin labeling (Fig. 5B), indicating that a significant amount of immunoprecipitated synapsin was biotinylated and thus was exposed at the cell surface of astrocytes. No bands were observed when a nonimmune control antibody was used for immunoprecipitation (Fig. 5B). An approximate estimation of the signal intensities obtained for immunoprecipitated, biotinylated synapsin and total synapsin by densitometry revealed that 5 ± 4% of synapsin expressed by cultured cortical astrocytes is exposed at the cell surface.

Synapsin is localized at the cell surface of cultured glial cells and present in glia-derived exosomes. A, Cultured cortical or whole-brain astrocytes were subjected to cell surface biotinylation and biotinylated cell surface proteins were isolated using streptavidin-conjugated beads. Lysates and biotinylated proteins (surface) were subjected to Western blot analysis (WB) with synapsin (syn) or GAPDH antibody. B, Biotinylated proteins were used for immunoprecipitation (IP) with synapsin antibody (syn) or nonimmune control antibody (Ig) and the resulting immunoprecipitates were subjected to Western blot analysis using HRP-coupled streptavidin (strept) or synapsin antibody (syn). C, Lysates and crude exosomal fractions (exo) isolated from the cell culture supernatant of cultured mouse cortical astrocytes by serial centrifugation were probed in Western blot analysis with synapsin, Alix, hsc70, actin, GAPDH, PDI, synaptophysin (synapto), or 14-3-3 antibodies. D, Crude exosomes were subjected to sucrose gradient centrifugation. After centrifugation fractions were collected and probed in Western blot analysis using synapsin, Alix, and flotillin antibodies. E, Fractions of exosomes that contain synapsin were analyzed by electron microscopy (scale bar, 100 nm). F, Cell-free cell culture supernatant of cortical astrocytes (sup+exo) or the supernatant obtained after 100,000 × g centrifugation of the cell culture supernatant of cortical astrocytes (sup-exo) were subjected to immunoprecipitation (IP) with synapsin antibody (syn) or nonimmune control antibody (Ig). The immunoprecipitates were subjected to Western blot (WB) analysis using synapsin (syn) antibody. G, Exosomes isolated from astrocyte culture supernatant were incubated in the presence of 60, 75, or 80 mm KCl without (KCl) or with 1.5 mm MgCl2 and CaCl2 (KCl+MgCl2/CaCl2). After incubation for 5 min at 37°C, samples were centrifuged at 100,000 × g to isolate exosomes (pellet) and proteins released from the exosomes (sup) by subjecting the supernatant to protein precipitation. The pellet and supernatant fractions were subjected to Western blot analysis using synapsin, actin, or PDI antibodies.
In a next step, we tested the possibility that synapsin is released from cells via exosomes, which mediate the transport of cytosolic proteins from one cell to another cell or to the extracellular milieu [for review, see Simons and Raposo (2009)]. Exosomes are formed by invaginations at the membrane of multivesicular bodies and they pinch-off from these invaginations as intraluminal vesicles into the lumen of the multivesicular bodies, resulting in packaging of cytoplasmic proteins inside of these vesicles (for review, see Lakkaraju and Rodriguez-Boulan, 2008). The intraluminal vesicles are released as exosomes into the extracellular space upon fusion of multivesicular bodies with the plasma membrane. Via this pathway, the interior of exosomes contains cytoplasmic proteins, such as synapsin. Western blot analysis showed that synapsin was expressed in astrocyte-enriched cell cultures and that a significant amount of exosomes that contain synapsin and the exosomal marker protein Alix could be isolated by serial centrifugation from the cell culture supernatant of cultures enriched in cortical astrocytes (Fig. 5C). In addition, other soluble, cytoplasmic proteins, such as hsc70, actin, and GAPDH, which all have been found in exosomes (Simpson et al., 2008), and proteins of the endoplasmic reticulum, such as PDI, were also detected in cell lysates and in exosomes from cultures enriched in cortical astrocytes (Fig. 5C). However, synaptophysin and 14-3-3 were detectable in the cell lysates, but not in exosomes derived from astrocyte-enriched cell cultures (Fig. 5C). Although cerebellar neurons expressed large amounts of synapsin, only negligible amounts of exosomes were obtained from cultured neurons, such as cerebellar or cortical neurons (data not shown). Therefore, we used cultured cortical astrocytes to enrich exosomes from cell culture supernatants by serial centrifugation and sucrose gradient centrifugation. Analysis of the gradient fractions showed that synapsin was detectable in exosomal fractions enriched in the exosomal marker proteins Alix and flotillin (Fig. 5D). The analysis of the combined exosomal fractions by electron microscopy showed a high degree of homogeneity: ∼95% of the particles were vesicles with a diameter of ∼80 nm (Fig. 5E), which fits well with the reported size of exosomes (Simons and Raposo, 2009). These results imply that synapsin is released from cortical astrocytes via exosomes.
To investigate whether soluble synapsin is also present in the extracellular medium, cell-free cell culture supernatants from astrocyte-enriched cell cultures were used for immunoprecipitation either before or after removal of exosomes by ultracentrifugation. Western blot analysis of the immunoprecipitates with synapsin antibody showed that a proportion of synapsin could be immunoprecipitated from the cell culture supernatant both before and after removal of exosomes (Fig. 5F). However, the amount of immunoprecipitated synapsin was reduced by ∼70% when exosomes had been removed from the cell culture supernatant, indicating that synapsin was predominantly released from astrocytes via exosomes.
Synapsin is released from glial-derived exosomes and is neuroprotective under conditions of oxidative stress and ischemia
The presence of significant amounts of soluble synapsin in the cell culture supernatant of cultured glial cells raised the question of whether synapsin is directly released from glial cells as soluble protein or whether it is released from these cells as soluble protein via exosomes. To address the question whether synapsin is released from the lumen of exosomes under certain conditions, such as stress or high metabolic activity, we first exposed isolated exosomes under conditions that prevail at high neuronal activity, namely elevated K+ concentrations. Therefore, exosomes from cell cultures enriched in cortical astrocytes were resuspended in PBS and incubated in the absence or presence of increasing KCl concentrations. After ultracentrifugation, the pelleted exosomes and the supernatant fractions containing proteins released from exosomes were subjected to Western blot analysis. After incubation in PBS or in the presence of increasing concentrations from 40 to 70 mm KCl, synapsin was exclusively found in the pellet fraction, but not in the supernatant (Fig. 5G), indicating that it was not released from exosomes, while PDI was completely released from the exosomes already by 55 mm KCl (Fig. 5G). In the presence of 75 mm KCl, small amounts of synapsin were observed in the supernatant (Fig. 5G), whereas in the presence of 80 mm KCl, the vast majority of synapsin was present in the supernatant and only very small residual amounts were seen in the pellet fraction (Fig. 5G). Actin, which was also detectable in exosomes (Fig. 5C), or the soluble exosomal marker protein hsc70 (data not shown), was not released from the exosomes under these conditions (Fig. 5G). It is noteworthy that synapsin was also released from exosomes by addition of 80 mm LiCl, but not at lower concentrations, whereas addition of 80 mm NaCl did not promote release of synapsin (data not shown). Synapsin was also not released at lower pH values, namely pH 6.0, or in the presence of 0.23 m sucrose or 1.5 mm MgCl2/1.5 mm CaCl2 (data not shown). However, in the presence of 1.5 mm MgCl2/1.5 mm CaCl2, the release of synapsin by 80 mm KCl was inhibited (Fig. 5G). These results indicate that elevated concentrations of KCl or LiCl, but not of NaCl, result in a release of synapsin from isolated glial-derived exosomes and that the release at elevated KCl concentrations is inhibited in the presence of physiological concentrations of Ca2+ and Mg2+ ions, suggesting that the release of synapsin from exosomes is not due to an nonspecific effect of high ionic strength.
KCl concentrations up to 80 mm can be reached during propagation of spreading depression (SD) waves (for review, see Irwin and Walz, 1999) in the normal and the diseased or injured brain under pathological oxidative stress conditions, such as ischemia/stroke (for review, see Lauritzen et al., 2011). SD waves lead to an activation of microglia and astrocytes, which have neuroprotective functions (for review, see Irwin and Walz, 1999). These observations led to the idea that synapsin may be released from glial-derived exosomes under oxidative stress conditions to protect neurons against oxidative stress. To test this idea, we treated astrocyte-enriched cell cultures isolated from cortex or from whole brain of neonatal mice with concentrations of hydrogen peroxide that cause oxidative stress and determined the amounts of synapsin levels in exosomes and in the cell culture supernatant after removal of exosomes. In the presence of hydrogen peroxide, the amount of exosome-associated synapsin decreased and the amount of soluble synapsin in the cell culture supernatant increased in comparison to that observed in the absence of hydrogen peroxide (Fig. 6A). The amounts of other exosome-associated and/or cytoplasmic proteins, such as Alix or GAPDH, in exosomes and cell culture supernatants as well as the expression of synapsin in astrocytes were not affected by hydrogen peroxide (Fig. 6A).

Synapsin released from exosomes of cultured glia cells is neuroprotective under oxidative stress conditions. A, Cell culture supernatants from cortical and whole-brain glial cells cultured in the absence or presence of 20 μm hydrogen peroxide (H2O2) were subjected to serial centrifugation. After centrifugation at 100,000 × g, the resulting pellets containing exosomes and supernatants (medium) containing soluble proteins were subjected to Western blot analysis with synapsin and Alix or GAPDH antibodies. The cells were also lysed and probed with synapsin and GAPDH antibodies in Western blot analysis. B, Cortical neurons were grown on substrate-coated PLL or synapsin in the absence or presence of 20 μm hydrogen peroxide (H2O2). After 24 h, neurons were stained with calcein and propidium iodide to determine the number of live and dead cells. Cell survival (ratio between number of live cells and total cell number) was calculated and survival of control cells was set to 100%. C, Cell culture supernatants from whole-brain glial cells subjected to glucose deprivation and either hypoxia (OGD) or hydrogen peroxide (H2O2) treatment were applied to serial centrifugation. After centrifugation at 100,000 × g, the resulting supernatants (medium) containing soluble proteins and pellets containing exosomes were subjected to Western blot analysis with synapsin, Alix, GAPDH, or calreticulin (CRT) antibodies. Cell lysates were probed with synapsin, Alix, calreticulin, and GAPDH antibodies. D, Cortical neurons grown on substrate-coated PLL or synapsin were subjected to oxygen/glucose deprivation (OGD) in the absence or presence of soluble synapsin. After 3 h, neurons were stained with calcein and propidium iodide to determine the number of live and dead cells. Cell survival was analyzed as described above (B). A, C, Representative blots out of at least two blots from at least three independent experiments are shown. C, Western blots of the cell culture supernatants probed with synapsin antibody from three independent experiments are shown. B, D, Mean values ± SEM of three independent experiments are shown (**p < 0.01 obtained by one-way ANOVA with Turkey's multiple-comparison test).
Similarly, determination of cell death showed that survival of cultured astrocytes was not significantly affected by hydrogen peroxide treatment: only 5.2 ± 2.9% of all cells were found to be dead. Densitometric quantification indicates that hydrogen peroxide resulted in a 2.6 ± 0.7-fold increase of synapsin levels in the cell culture supernatant (n = 8; p < 0.005) and a concomitant 6.5 ± 0.2-fold decrease of synapsin in exosomes (n = 6; p < 0.005). Interestingly, treatment of isolated exosomes with hydrogen peroxide did not lead to a significant release of synapsin. In summary, the results indicate that synapsin is released from glia-derived exosomes under oxidative stress conditions.
Subsequently, we addressed the question whether extracellular synapsin protects neurons from oxidative stress. Survival of cultured cortical neurons grown on substrate-coated synapsin or on control substrate PLL and in the absence or presence of hydrogen peroxide was determined. In the absence of hydrogen peroxide, survival of neurons on substrate-coated synapsin was similar to that on PLL. In the presence of hydrogen peroxide, neuronal cell death increased on both substrates, but survival of neurons was increased on the synapsin coat relative to that determined on the control substrate PLL (Fig. 6B). This result indicates that extracellular synapsin is able to protect neurons from oxidative stress.
We further examined the release of synapsin under oxidative stress conditions by subjecting astrocyte-enriched cell cultures from whole brain to ischemic conditions induced by oxygen/glucose deprivation or glucose deprivation in the presence of hydrogen peroxide. After 7–9 d in culture, cells were incubated in medium with low glucose in the absence or presence of hydrogen peroxide under normoxic or hypoxic conditions. In comparison to normoxic control conditions and in the absence of hydrogen peroxide, hydrogen peroxide treatment as well as hypoxia in combination with glucose deprivation led to a 2.6 ± 0.4-fold increase of synapsin in the cell culture supernatant (n = 4; p < 0.05) and to a 6.7 ± 0.4-fold decrease of synapsin in exosomes (n = 4; p < 0.05), while the amount of synapsin in cell lysates was not altered (Fig. 6C). The amounts of GAPDH, Alix, and calreticulin in the cell culture supernatants and cell lysates as well as the amounts of Alix in exosomes were not altered by either treatment (Fig. 6C).
To investigate whether extracellular synapsin protects neurons from oxygen/glucose deprivation, cultured cortical neurons grown on substrate-coated synapsin or on the control substrate PLL were subjected to oxygen/glucose deprivation in the absence or presence of soluble synapsin. In comparison to the control conditions without oxygen/glucose deprivation, a dramatic increase of neuronal death was observed upon oxygen/glucose deprivation. Neurons grown on substrate-coated synapsin and/or in the presence of soluble synapsin showed a higher survival rate upon oxygen/glucose deprivation than neurons grown on PLL and, thus, in the absence of synapsin (Fig. 6D). These results indicate that extracellular synapsin protects neurons from ischemia and cell death.
Synapsin promotes neurite outgrowth in an oligomannose-dependent manner
So far, synapsin has been considered as a protein extrinsically associated with the cytoplasmic surface of small synaptic vesicles. Here, we have demonstrated that cultured astrocytes release synapsin via synapsin-containing exosomes and that synapsin can be released from exosomes into the extracellular medium by elevated KCl concentrations or oxidative stress or ischemic conditions.
To investigate whether extracellular synapsin may affect neuronal development, we first analyzed whether neurite outgrowth is affected when neurons are exposed to exogenous substrate-coated or soluble synapsin mimicking cell-associated or soluble extracellular synapsin, respectively. In comparison to neurons maintained on PLL, a pronounced increase in neurite lengths was observed both when hippocampal neurons were maintained on substrate-coated synapsin and when they were maintained in the presence of soluble synapsin (Fig. 7A). Neurite outgrowth in the concomitant presence of substrate-coated and soluble synapsin was not significantly changed relative to that observed in the presence of soluble synapsin or of substrate-coated synapsin alone (Fig. 7A). In summary, the results suggest that that extracellular synapsin released by astrocytes promotes neurite outgrowth. To evaluate the physiological role of glia-derived synapsin proteins in mediating neuronal growth, we plated wild-type hippocampal neurons on glial cultures derived from either wild-type or synapsin I/II/III TKO mice. We chose to use synapsin TKO animals rather than synapsin I knock-out to avoid the possibility that other members of the synapsin family might compensate for the lack of synapsin I in the system. Hippocampal neurons were maintained on glial cultures for 24 h, fixed, and stained with antibodies against neurofilament heavy chain and β-III tubulin to identify axons and the neurite network, respectively (Fig. 7B). Sholl analysis performed on neuronal profiles showed that wild-type hippocampal neurons grown on synapsin TKO cells have a simpler neurite network than neurons grown in parallel on wild-type glia (Fig. 7C). Quantification of axonal growth and branching also revealed that wild-type neurons maintained on synapsin TKO glia display shorter and less branched axons than neurons maintained on wild-type glia (Fig. 7D). Altogether, these results support the idea that synapsin proteins secreted from glial cells contributes to the optimal growth of neurons.

Glia-derived synapsin promotes neurite outgrowth in an oligomannose-dependent manner. A, Hippocampal neurons were grown on substrate-coated PLL or synapsin (syn) in the absence (−) or presence of soluble synapsin. After fixation, the length of the longest neurite per cell was determined and value obtained with PLL coat in the absence of soluble synapsin was set to 100%. B–D, Wild-type (WT) hippocampal neurons were plated on mature wild-type or synapsin TKO glial cultures, maintained for 24 h, fixed, and stained with antibodies against neurofilament heavy chain and β-III tubulin to identify axons and the neurite network, respectively. B, Representative images of wild-type hippocampal neurons maintained on wild-type (top) or synapsin TKO glia (bottom). Scale bar, 10 μm. C, WT neurons grown on TKO glia show a simpler neurite network than neurons grown in parallel on WT glia. Sholl analysis was performed on n = 225 cells per genotype, from three independent experiments. The number of intersections (means ± SEM) is plotted as function of the distance from the cell body (***p < 0.001, Mann–Whitney U test). D, The average axonal length and the average number of axonal branching of wild-type (WT) hippocampal neurons maintained on either WT or synapsin TKO glia was determined and compared. Axons were identified as processes that were positive for neurofilament heavy chain immunostaining, and manually measured. n = 60 cells per genotype, from two independent experiments (**p < 0.01, ***p < 0.001, Mann–Whitney U test). E, Hippocampal neurons were grown on substrate-coated PLL alone (−), synapsin (syn), or EndoH-treated synapsin (de-syn) in the absence (−) or presence of untreated RNase B (RB), deglycosylated RNase B (de-RB), or catalase-coupled oligomannose-mimicking peptide (c-pep). The length of the longest neurite per cell was determined and the mean length of the longest neurites obtained with PLL coat was set to 100%. A, E, Mean values ± SEM of three independent experiments are shown (*p < 0.05; one-way ANOVA followed by Dunnett's multiple-comparison test).
We next analyzed whether extracellularly localized synapsin affects neuritogenesis as an oligomannose-carrying and -binding protein. Neurite outgrowth on substrate-coated native synapsin was very similar to that on denatured synapsin (data not shown), and it was higher than that on control substrate PLL, while neurite outgrowth on substrate-coated EndoH-treated denatured synapsin was similar to that on PLL (Fig. 7E). To further analyze whether the neurite-promoting effect of synapsin depends on oligomannose, oligomannose-bearing RNase B, EndoH-treated oligomannose-lacking RNase B, or oligomannose-mimicking peptide coupled to catalase was added to the medium to block the binding of synapsin to oligomannoses at the cell surface and/or to inhibit the recognition of oligomannoses on synapsin by cell surface receptors. In the presence of oligomannose-carrying untreated RNase B or catalase-coupled oligomannose-mimicking peptide, the synapsin-induced promotion of neurite outgrowth was virtually abolished, while in the presence of EndoH-treated RNase B devoid of oligomannose, the synapsin-induced neurite outgrowth was not altered (Fig. 7E). These results indicate that the enhancement of neurite outgrowth by extracellular synapsin is oligomannose dependent.
Synapsin binds to the extracellular domain of NCAM in an oligomannose-dependent manner and promotes neurite outgrowth in an NCAM-dependent manner
Glia, such as astrocytes, is involved in functional modulation of neuronal properties, such as neurite outgrowth. Since synapsin is released from astrocytes via exosomes and promotes neurite outgrowth in an oligomannose-dependent manner, it is likely that the binding of the extracellular glial-derived oligomannose-bearing synapsin to neuronal oligomannose-binding cell surface receptors may trigger neurite outgrowth. NCAM is an oligomannose-binding neuronal protein involved in neurite outgrowth (Horstkorte et al., 1993; Heiland et al., 1998) and thus is a prime candidate to interact with synapsin in an oligomannose-dependent manner. First, we investigated by ELISA whether NCAM binds to synapsin using NCAM-Fc, and CHL1-Fc as negative control. The binding of NCAM-Fc to substrate-coated denatured synapsin, but not to EndoH-treated denatured synapsin, increased in a concentration-dependent and saturable manner, while CHL1-Fc did not show any binding to substrate-coated synapsin (Fig. 8A). In a similar ELISA experiment using increasing amounts of NCAM-Fc and a constant amount of denatured or EndoH-treated denatured synapsin as substrate coat, a concentration-dependent and saturable binding of NCAM-Fc to substrate-coated synapsin, but not to EndoH-treated synapsin, was observed (Fig. 8B). To confirm that NCAM binds to oligomannoses on synapsin, we performed a competition ELISA with NCAM-Fc using either untreated or EndoH-treated synapsin as substrate coat in the presence of untreated oligomannose-bearing AMOG or EndoH-treated AMOG as competitors. NCAM-Fc bound to synapsin, but not to deglycosylated synapsin (Fig. 8C). When NCAM-Fc was preincubated with oligomannose-bearing AMOG, binding of NCAM-Fc to synapsin decreased to levels obtained with deglycosylated synapsin (Fig. 8C). In contrast, preincubation with EndoH-treated oligomannose-lacking AMOG had no effect on binding of NCAM-Fc to synapsin (Fig. 8C). These results demonstrate an oligomannose-dependent binding of NCAM to synapsin.

Synapsin binds to the extracellular domain of NCAM in an oligomannose-dependent manner and promotes neurite outgrowth in an NCAM-dependent manner. A, Increasing amounts of either untreated (syn) or EndoH-treated (de-syn) synapsin were substrate coated and incubated with either NCAM-Fc or CHL1-Fc. B, Substrate-coated untreated (syn) or EndoH-treated (de-syn) synapsin was incubated with increasing amounts of NCAM-Fc. C, Synapsin (syn) or synapsin treated with EndoH (de-syn) was substrate coated and incubated in the absence or presence of NCAM-Fc (NCAM), NCAM-Fc preincubated with AMOG (NCAM+AM), or NCAM preincubated with EndoH-deglycosylated AMOG (NCAM+de-AM). A–C, The binding of NCAM-Fc or CHL1 was determined by ELISA using a human Fc antibody. Mean values ± SEM of three independent experiments are shown (*p < 0.05, **p < 0.01, ***p < 0.001 one-way ANOVA followed by Dunnett's multiple-comparison test). D, Hippocampal neurons of NCAM-deficient (NCAM−/−) and wild-type (NCAM+/+) mice were grown on substrate-coated untreated (syn) or EndoH-treated (de-syn) synapsin. After 24 h in culture, cells were fixed and stained, and the length of the longest neurite per cell was determined. Mean length of the longest neurites obtained on PLL was set to 100%. Mean values ± SEM of two independent experiments are shown (**p < 0.01 one-way ANOVA followed by Dunnett's multiple-comparison test).
Since synapsin promotes neurite outgrowth and interacts with NCAM in an oligomannose-dependent manner, it is reasonable to assume that the effects of synapsin on neurite outgrowth are mediated by its interaction with NCAM. Thus, we measured neurite outgrowth using NCAM-deficient hippocampal neurons and found that synapsin did not enhance neurite outgrowth in these neurons (Fig. 8D). This result indicates that promotion of neurite outgrowth by extracellular synapsin is mediated by its oligomannose-dependent interaction with NCAM at the cell surface of neurons.
Synapsin is expressed by glial cells
To verify the expression of synapsin in primary astrocytes, the astrocyte-enriched cell culture isolated from whole brain was characterized by coimmunostaining with antibodies against synapsin and the neuronal marker β-III tubulin or the astrocytic marker GFAP. As expected, the vast majority of cells (61.5 ± 11.2%) showed the typical flattened morphology of protoplasmic astrocytes and were intensely stained by GFAP (Fig. 9A). GFAP-positive astrocytes displayed a relatively weak synapsin staining, while a significant number of GFAP-negative cells (21.4 ± 8.3%) showed a strong synapsin staining (Fig. 9A). A very small number of cells (2.2 ± 1.1%) displayed neuronal features and were strongly positive for both β-III tubulin and synapsin (Fig. 9B). Quantification of the synapsin staining intensity in neurons and glial cells (Fig. 9C) revealed that synapsin immunoreactivity in neurons was fourfold to fivefold higher than that of glial cells. The contribution of autofluorescence or nonspecific background staining was ruled out by omitting primary antibodies and by using a Cy3-labeled instead of a Cy2-labeled secondary antibody for the detection of synapsin (data not shown). In addition, astrocyte-enriched cultures from wild-type and TKO mice were stained with GFAP and synapsin antibodies. A weak synapsin immunoreactivity was seen in GFAP-positive wild-type astrocytes, but not in TKO astrocytes (Fig. 9D). These results demonstrate that the staining by the synapsin antibody was specific. Interestingly, a number of cells (14.3 ± 4.1%) not resembling neurons or protoplasmic astrocytes showed no detectable staining of β-III tubulin, but a pronounced synapsin staining (Fig. 9B). To analyze whether these β-III tubulin- and GFAP-negative cells were microglia, oligodendrocytes, or NG2-glia, which represent a glial cell type distinct from astrocytes, microglia, and oligodendrocytes [for review, see Trotter et al. (2010), Wigley and Butt (2009), and Bergles et al. (2010)], we performed double immunostaining with an antibody to synapsin and antibodies to the microglial marker Iba-1, the oligodendrocyte marker myelin basic protein, and the NG2-glia marker NG2. Cells with a specific Iba-1 or myelin basic protein immunoreactivity were not observed in our cell culture (data not shown), while a remarkable number of cells (10.6 ± 2.2%) showed strong immunoreactivity for both synapsin and NG2 (Fig. 9E). In an astrocyte-enriched cell culture from cortex, a similar percentage of total cells (11.3 ± 3.3%) were positive for NG2 staining and the NG2-positive cell showed a pronounced synapsin staining in comparison to the weak synapsin staining of NG2-negative cells (Fig. 9F). The mean area intensity of NG2-positive cells was determined to be twofold to threefold higher than that measured for NG2-negative cells. These results indicate that NG2-positive glial cells express synapsin, while GFAP-positive astrocytes express much smaller amounts of synapsin. Thus, it is possible that synapsin is released only by NG2-positive glia and not by astrocytes. On the other hand, it is also possible that astrocytes release synapsin and thus, show only a weak staining for synapsin, while NG2-glia express but do not release synapsin.

Synapsin is expressed by glial cells. A–E, Upon fixation and permeabilization, astrocyte-enriched glial cell cultures from whole brain of wild-type mice (A–C, E) or cortex of wild-type (D, F) or TKO (D) mice were subjected to double immunostaining with a mouse synapsin antibody and rabbit GFAP (A, D), β-III tubulin (B), or NG2 (E, F) antibodies. Secondary anti-mouse antibodies were labeled with the fluorescent dye Cy2 (A–C, E) or Cy3 (D, F), and secondary anti-rabbit antibodies were labeled with Cy3 (A–C, E) or Cy2 (D, F). Representative images of GFAP+/synapsin+ and GFAP−/synapsin+ glial cells (A), a tubulin+/synapsin+ neuron-like cell (B, top) and tubulin−/synapsin+ cells (B, bottom), synapsin+ neurons (C, arrow) and surrounding synapsin+ glial cells (C, arrowhead), GFAP+/synapsin+ glial cells (D, arrowhead), and synapsin+/NG2+ (E, F) and synapsin+/NG2− (F) glial cells are shown. D, Synapsin immunoreactivity (red) is shown in a GFAP+ wild-type (WT) astrocyte (arrowhead) as well as in a neuronal cell body (arrow) and along neurites (left), while no synapsin immunoreactivity is detectable in a GFAP+ TKO astrocytes (right). A–F, Scale bars, 50 μm.
Discussion
Synapsin is an oligomannose-bearing glycoprotein and oligomannose-recognizing lectin
To gain further insights into the functions of oligomannoses in the nervous system, we tried to identify new oligomannose-carrying proteins and oligomannose-binding lectin-like proteins and identified synapsin as an oligomannose-binding and oligomannose-carrying protein. Both synapsin Ia and synapsin Ib, the two splice variants of the synapsin I gene, have been shown to carry fucose-α1,2-galactose on O-glycans (Murrey et al., 2006). Here, we showed that synapsin I carries Lewisx also on O-glycans and oligomannoses on N-glycans. Synapsin Ia carries an N-linked glycosylation site at the very beginning of the E domain, which plays a fundamental role in synapsin function (Hilfiker et al., 1998; Fassio et al., 2006), and it is thus possible that only this synapsin isoform carries the N-linked oligomannoses.
Since synapsin occurs as a glycoprotein and since glycosylation is a characteristic posttranslational modification of extracellular proteins, our findings are consistent with the idea that synapsin may be secreted as extracellular glycoprotein. The route by which cytoplasmic synapsin is glycosylated and the secretion pathway of synapsin in glia cells are not known. The observation of an interaction between synapsin and the glycosyl phosphatidyl inositol-linked prion protein (Spielhaupter and Schätzl, 2001) also suggest that synapsin may act as an extracellular protein and interact with cell surface receptors. Here, we provide evidence that recognition of oligomannose on synapsin by NCAM triggers synapsin-dependent neurite outgrowth. In addition, since we found that synapsin also binds to oligomannoses on AMOG, which is exclusively present on astrocytes, it is possible that this interaction triggers biological responses in astrocytes.
Possible role of synapsin in neural cell interactions
Glia–neuron interactions, which can be modulated by carbohydrates, play a critical role in brain development and function. Here, we showed that oligomannose-mimicking peptides or oligomannose-carrying RNase B inhibit adhesion between cerebellar neurons and glial cells. These results indicate that glia–neuron interactions depend, at least partially, on oligomannoses. In the present study, we demonstrate that cultured glial cells express synapsin and release it via exosomes into the extracellular space. Cell surface biotinylation indicated that a significant portion of this extracellular synapsin is present as a cell surface-associated molecule, supporting its possible role in mediating and/or modulating glia–neuron interactions. We failed to demonstrate synapsin at the cell surface of astrocytes by immunostaining, possibly because synapsin on live glial cells is not accessible to the antibody or the number or density of synapsin molecules at the cell surface is too low to be detectable by immunostaining. So far, synapsin has been detected by immunohistological methods only in neuronal cells, while non-neuronal cells did not show detectable staining of synapsin (De Camilli et al., 1983). However, one cannot exclude the possibility that synapsin is also expressed in vivo in non-neuronal cells, because detection by immunostaining depends on specificity and sensitivity of antibodies, quantity and accessibility of antigens, and other experimental constraints. In our in vitro experiments, we also showed that GFAP-positive astrocytes in culture were scarcely positive for synapsin staining. However, we observed strong synapsin immunoreactivity on NG2-expressing cells, which represent a distinct subpopulation of glial cells that form contacts with astrocytes and neurons [for review, see Wigley and Butt (2009), Bergles et al. (2010), and Trotter et al. (2010)]. The functions of NG2-expressing cells, which are also called synantocytes or polydendrocytes, are unknown. Since we observed an intense staining of synapsin in NG2-positive glial cells, it is possible that only NG2-expressing glial cells express and release synapsin. Another explanation is that these glial cells do not release, but accumulate, synapsin and, thus, are strongly synapsin immunopositive. On the other hand, we cannot exclude the possibility that the glial cells that are only weakly synapsin immunopositive release synapsin via exosomes and thus, showed a weak immunostaining. Since we obtained no indications that cultured neurons release synapsin, we exclude the possibility that the release of synapsin via exosomes in glia-enriched cell cultures is due to the presence of only a few neurons in glial cell cultures.
Extracellular synapsin is released by glia-derived exosomes
For glia–neuron communication, not only direct cell-to-cell contact but also the exchange of soluble factors appears to be important. Exosomes have been suggested to be involved in these intercellular communications and intercellular protein transport [for review, see Simons and Raposo (2009)]. In the present study, we found that exosomes released from glial cells carry synapsin, and we observed in in vitro experiments that synapsin was released from isolated exosomes in the presence of 75–80 mm KCl, while other cargo proteins, such as actin or hsc70, are not released. The selective release of synapsin from glial-derived exosomes under distinct conditions suggests a regulated release mechanism for synapsin at high KCl concentrations. In vivo, it may be possible that synapsin is released from exosomes by a more moderate increase in KCl concentrations and/or by other factors, such as the extracellular calcium and magnesium concentrations. Indeed, the release of synapsin from exosomes was inhibited in vitro in the presence of 1.5 mm MgCl2/CaCl2, and thus, low extracellular magnesium and calcium concentrations may be a prerequisite for the release of synapsin from exosomes under in vivo conditions. In the intact nervous tissue, transient elevations of up to 80 mm KCl are observed during propagation of SD waves (for review, see Irwin and Walz, 1999). SD waves have been observed in the normal and diseased or injured brain under pathological conditions, such as focal ischemia/stroke or migraine [for review, see Irwin and Walz (1999) and Lauritzen et al. (2011)]. Interestingly, the extracellular levels of magnesium and calcium change in association with pathological stress conditions such as ischemia or hypoglycemia and low extracellular calcium and magnesium concentrations have been implicated in seizures and migraine and have been shown to evoke calcium waves that resemble those observed during SD (Stout and Charles, 2003, and references therein). SD waves in healthy brains lead to activation of microglia and astrocytes, which can secrete neurotrophic factors, exert neuroprotective functions, and play beneficial roles in preischemic conditioning, making the tissue more resistant to subsequent insults. Interestingly, hypoxia or ischemia has been shown to result in an increase of synapsin levels in various brain regions (Ploughman et al., 2005), suggesting that synapsin is involved in the brain's response to stress conditions and that it is associated with neuroprotective mechanisms. One may speculate that SD waves or oxidative stress lead to increases in extracellular KCl concentrations, which trigger the release of synapsin from glia-derived exosomes.
Extracellular synapsin stimulates neurite outgrowth and is neuroprotective
Several reports indirectly indicate that synapsins modulate neuronal development through the promotion of neurite outgrowth and synaptogenesis. The expression pattern of synapsin closely parallels the dynamics of neuropil development and synapse formation, thus suggesting a functional link between the onset of synapsin expression and the maturation of the neuronal network (Fornasiero et al., 2010). Lack of synapsin was reported to cause a general growth impairment of hippocampal neurons in in vitro culture during the first days, but no longer at later stages (Chin et al., 1995). Since synapsins are actin-binding proteins and interact with multiple cytoskeletal proteins (Cesca et al., 2010), it is currently believed that the synapsin effects on neuronal development are mainly attributable to a regulation of actin cytoskeleton dynamics. Our study indicates that glial-derived synapsin when released into the extracellular space may contribute to the synapsin's effects on neuronal development.
Moreover, our results show that, when applied extracellularly, synapsin is neuroprotective under stress conditions. Interestingly, it has been shown that aged synapsin I-deficient mice display an increased loss of neurons in the neocortex and hippocampus (Corradi et al., 2008) and that stress-related memory induced by a glucocorticoid-triggered signaling pathway is synapsin dependent (Revest et al., 2010), indicating an important function of synapsin for neuronal survival following exposure to stress.
In summary, results from our study provide evidence that, in the nervous system, glia-derived exosomes and glia-derived synapsin released into the extracellular space represent a means to provide particular cargo proteins, such as synapsin. In turn, these cargo proteins may modulate glia–neuron interactions and communication as well as serve neuroprotective functions.
Footnotes
M.S. is supported by the New Jersey Commission for Spinal Cord Research and is a consultant at the Center for Neuroscience at Shantou University Medical College, China. We are grateful to Ute Bork, Sönke Hader, and Chudamani Raithore for excellent technical assistance, Drs. Paul Greengard (The Rockefeller University, New York, NY) and Hung-Teh Kao (Brown University, Providence, RI) for providing the synapsin TKO mouse strain, Marina Nanni for cell culture, Eva Kronberg for excellent animal care, Claus Schafer-Nielsen (Copenhagen, Denmark) for synthetic peptides, and the Deutsche Forschungsgemeinschaft for support (KL-1378/1-1 and /1-2).
- Correspondence should be addressed to Melitta Schachner, Zentrum für Molekulare Neurobiologie, Universität sklinikum Hamburg, Martinistrasse 52, D-20246 Hamburg, Germany. melitta.schachner{at}zmnh.uni-hamburg.de





{kind=link}
{kind=link}
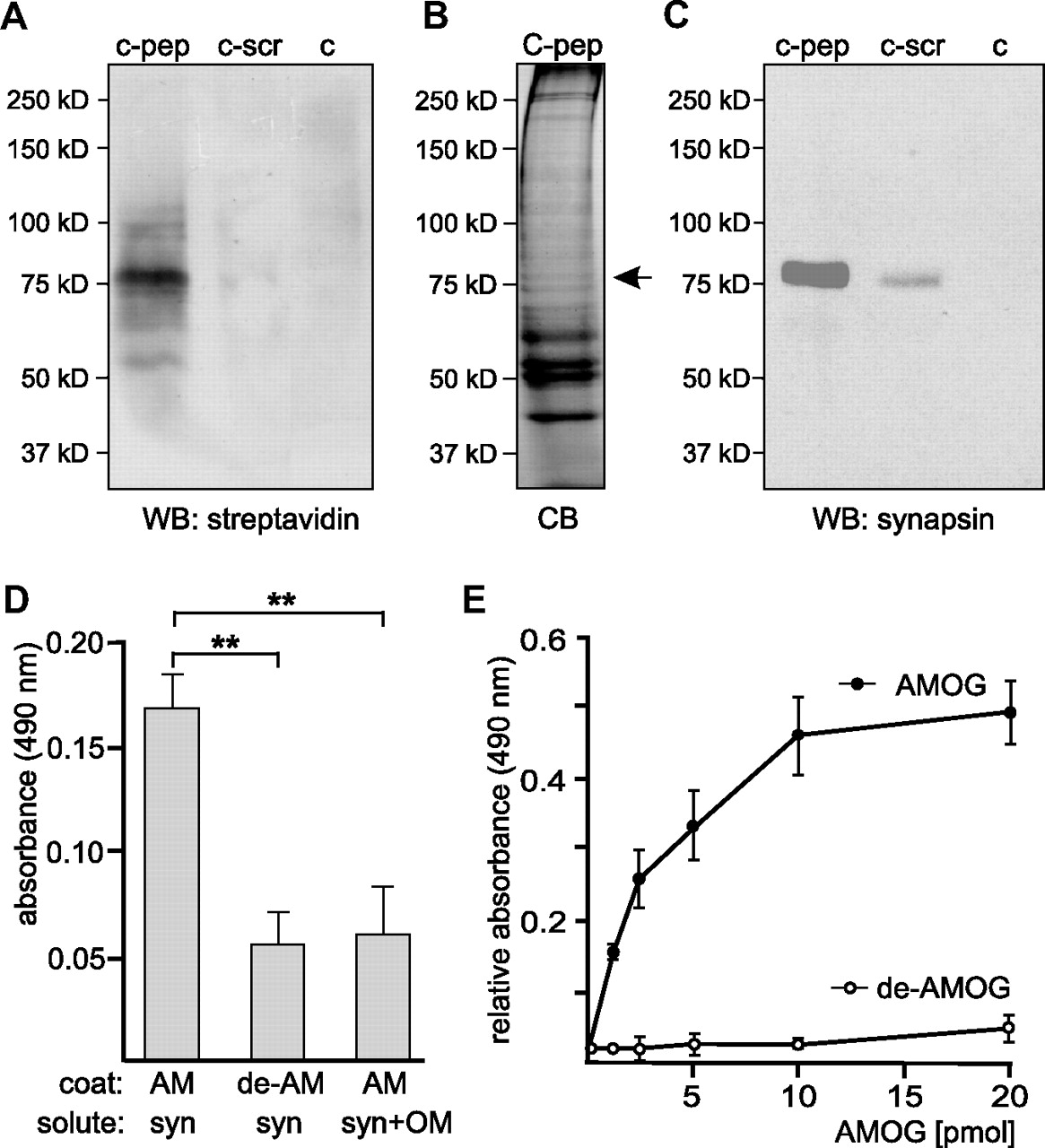{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}