
Abstract
A large number of genetic diseases have been associated with truncated or misfolded membrane proteins trapped in the endoplasmic reticulum (ER). In the ER, they activate the unfolded protein response, which can trigger cell death. Hence, a better understanding of protein misfolding features might help in developing novel therapies. Here, we have studied the molecular basis of Pelizaeus–Merzbacher disease, a leukodystrophy defined by mutations of the PLP1 gene and ER retention of two encoded tetraspan myelin proteins, PLP and DM20. In mouse oligodendroglial cells, mutant isoforms of PLP/DM20 with fewer than all four transmembrane (TM) domains are fully ER retained. Surprisingly, a truncated PLP with only two N-terminal TM domains shows normal cell-surface expression when coexpressed with a second truncated PLP harboring the two C-terminal TM domains. This striking ability to properly self-align the TM domains is disease relevant, as shown for the smaller splice isoform DM20. Here, the increased length of TM domain 3 allows for compensation of the effect of several PLP1 point mutations that impose a conformational constraint onto the adjacent extracellular loop region. We conclude that an important determinant in the quality control of polytopic membrane proteins is the free alignment of their TM domains.
Introduction
Frame-shift and missense mutations, which cause the misfolding of polytopic membrane proteins, have been associated with many neurogenetic disorders, including Pelizaeus–Merzbacher disease (PMD; OMIM #312080). PMD is an early-onset leukodystrophy associated with oligodendrocyte death, dysmyelination and demyelination, and secondary axonal loss (Seitelberger, 1995; Gow et al., 1998; Griffiths et al., 1998; Werner et al., 1998; Edgar et al., 2004; Woodward, 2008). PMD is genetically defined by perturbed expression of PLP1, an X-linked gene in mouse and man that encodes a myelin membrane protein.
PLP is a highly conserved tetraspan protein that is a major structural component of myelin in the CNS. Its two extracellular loops (EC1 and EC2) face each other at the intraperiod line in compacted myelin. Both the N and C terminus of PLP protrude into the cytosol (Fig. 1A), i.e., the major dense line of myelin. A smaller PLP splice isoform, termed DM20, differs by 35 residues in length of the cytosolic loop (Fig. 1B) (Nave et al., 1987).
In addition to PLP1 gene duplications, a major cause of PMD is point mutations leading to amino acid substitutions or truncations (Woodward, 2008). In mouse models, these mutations result in oligodendrocyte death and dysmyelination (Nave and Boespflug-Tanguy, 1996). However, because of oligodendrocyte death, PLP/DM20 retention itself cannot be adequately studied in vivo. We and others have circumvented this problem by monitoring PLP/DM20 trafficking in immortalized oligodendrocytes expressing mutant proteins (Krämer-Albers et al., 2006; Dhaunchak and Nave, 2007).
PLP1 mutations that alter the fourth transmembrane (TM) domain induce stable interactions with endoplasmic reticulum (ER)-resident chaperones such as calnexin (Swanton et al., 2003). However, what exactly constitutes PLP “misfolding” before the interaction with chaperones and ER retention is not well defined. The consequences of apparently minor structural changes in PLP are not well understood, particularly when these substitutions affect the extracellular loop or cytoplasmic domains.
We recently reported that cysteine residues located within EC2 play a critical role for PLP folding and trafficking, when harboring PMD-causing substitutions in EC2. Most likely, these mutations change the globular structure of EC2, prevent the formation of a critical intramolecular cysteine bridge, and trigger abnormal cysteine-mediated cross-links (Dhaunchak and Nave, 2007).
In these experiments, it was puzzling that some amino acid substitutions in EC2 caused the retention only of mutant PLP but not of the corresponding mutant DM20 isoform (Gow and Lazzarini, 1996). Is it possible that PLP and DM20, which differ in the predicted length of TM3 (Stecca et al., 2000), are (for the same reason) differently affected by mutations in an extracellular domain? Such a model would suggest that the alignment of TM domains organizes the overall topology of a polytopic membrane protein and that the freedom to realign TM domains determines the impact of mutations in extracellular domains. Here we show for PLP/DM20 that even protein halfmers can reconstitute a tetraspan protein that traffics normally. Moreover, experiments in which TM3 of DM20 is experimentally shortened by several residues reveal that its length in DM20 is indeed a critical feature that can even “rescue” the PMD-causing mutations affecting EC2 of PLP/DM20.
Materials and Methods
Molecular cloning.
Plasmid pPLP–EGFP was generated by fusing the enhanced green fluorescent protein to the C terminus of PLP (Dhaunchak and Nave, 2007). The product was cloned into vector pEGFP–N1 using EcoRI/NotI sites. All site-directed mutants were generated by circular amplification of this plasmid using Pfu-Turbo DNA polymerase (Stratagene), followed by DpnI digestion (New England Biolabs) and transformation into Escherichia coli. Individual clones were analyzed by analytic restriction digestion and DNA sequence analysis. Detailed cloning protocols, primer, constructs, and sequences are available on request.
DNA transfection.
One day before transfection, COS-7, oligodendroglial [oli-neu (Jung et al., 1995)], MO3.13 (McLaurin et al., 1995), and HEK293 cells were seeded on poly-l-lysine (PLL)-coated cover glasses at 50% confluency and were transfected either with Fugene 6 (Roche) or Lipofectamine 2000 (Invitrogen).
Antibodies.
Mouse monoclonal antibodies were an anti-c-myc (clone 9E10 from Sigma), anti-PLP (3F4; kindly provided by Marjorie Lees, E. K. Shriver Center, University of Massachusetts Medical School, Waltham, MA), and anti-lamp-1 (ab25630 from Abcam for COS-7 cells). Rat monoclonal antibodies were anti-lamp-1 (clone cd107a from BD Pharmingen for oli-neu cells) and anti-PLP [O10 (Jung et al., 1996)]. Polyclonal rabbit sera were raised against PLP [A431; against the C-terminal peptide GRGTKF (Jung et al., 1996)], NG2 (AB5320 from Millipore Bioscience Research Reagents), and EGFP (AB3080 from Millipore Bioscience Research Reagents). Fluorochrome-conjugated secondary antibodies were purchased from either Dianova or Jackson ImmunoResearch. Antibodies were used at the following dilutions: anti-c-myc at 1:2000, 3F4 and O10 at 1:50, anti-lamp-1 at 1:200, A431 at 1:1000, Cy2-conjugated secondary antibodies at 1:1000, and Cy3-conjugated secondary antibodies at 1:2500.
Cell culture.
COS-7, HEK293, and MO3.13 cells were maintained on untreated tissue-culture dishes (Falcon) in DMEM and 10% fetal bovine serum. Cells were grown at 37°C in a 5% CO2 atmosphere, and medium was changed every third day. For passaging cells, confluent plates were washed once with PBS, followed by a short trypsination with 0.05% trypsin–EDTA (Sigma). oli-neu cells (kindly provided by J. Trotter, University of Mainz, Mainz, Germany) were maintained in SATO medium (high glucose DMEM containing 2 mm insulin, 100 μm putrescine, 200 nm progesterone, 500 pm triiodothyronine, 220 nm sodium selenite, 500 nm l-thyroxine, 10 μg/ml transferrin, and 1–5% horse serum) on PLL-coated tissue culture dishes (Falcon). Cells were grown at 37°C and 5% CO2. For passaging, confluent plates were washed once with medium, followed by a short trypsination with 10-fold diluted trypsin–EDTA (Sigma).
Immunocytochemistry.
Immunostaining was performed 18–36 h after transfection. All steps were performed at room temperature (RT), unless stated otherwise. Cells grown on PLL-coated coverslips were washed once with TBS (in mm: 25 Tris, 136 NaCl, and 2.6 KCl, pH 7.5) and fixed for 5 min in 2% paraformaldehyde/TBS. Cell were then washed twice for 10 min in TBS, permeabilized with 0.01% saponin in TBS (for 10 min), and blocked in blocking buffer (TBS containing 2% goat serum, 2% BSA, 0.02% biotin, and 0.1% porcine skin gelatin) for at least 30 min. Primary antibodies diluted in blocking buffer were applied for at least 1 h at room temperature or overnight at 4°C. After three washes in TBS (10 min each), fluorochrome-conjugated secondary antibodies were applied for at least 45 min. After three washes with TBS (10 min each), cells were rinsed in distilled water and mounted in Aqua-Poly/Mount (Polysciences) on glass slides. For lamp-1 and calnexin immunostaining in COS-7 cells, the transfected cells were fixed and permeabilized with ice-cold acetone (for 20 min), blocked in the blocking buffer, and processed as described above.
Live staining of cells with antibody 3F4 was performed at 4°C on water/ice slurry. Cells grown on PLL-coated coverslips (24-well plates) were washed once with ice-cold DMEM, and primary antibody diluted in ice-cold DMEM was applied directly onto cells for 10 min. Cells were washed twice with Dulbecco's PBS (DPBS) and fixed with 2% PFA for 10 min. Cells were shifted to RT during fixation. After two additional washes in DPBS, Cy3-conjugated secondary antibodies diluted in DMEM were applied for at least 30 min. Cells were washed twice in DPBS, rinsed in ddH2O, and mounted. Live staining of cells with antibody O10 was performed as described previously (Jung et al., 1996).
Immunoblotting.
Before lysis in Laemmli's buffer (without reducing agent), transfected cells were rinsed once with ice-cold DPBS and incubated in ice-cold 15 mm iodoactemide in DPBS for 5 min to block free cysteines. The genomic DNA was sheared by brief sonication. Equal volumes of cell lysates were then resolved on 7.5% precast Bio-Rad XT gels under nonreducing conditions and transferred to PVDF membranes. The membranes were rinsed briefly in TBST (25 mm Tris, pH 7.4, 27 mm KCl, 137 mm NaCl, and 0.1% Tween 20) and blocked for at least 1 h at RT in blocking buffer (5% nonfat dry milk in TBST). Anti-GFP (1:2000) antibody diluted in blocking buffer was applied overnight at 4°C. After four washes in TBST, HRP-conjugated secondary antibodies were applied for 45 min, washed four times in TBST, and developed using an enhanced chemiluminescence detection kit (PerkinElmer Life and Analytical Sciences). Blots were quantified as described previously (Dhaunchak and Nave, 2007).
Image acquisition and analysis.
Fluorescent images were captured on a confocal microscope (LSM 510; Carl Zeiss) with a 63× oil plan-Apochromat objective (NA 1.4; Carl Zeiss). For final analysis, captured LSM images were exported as TIF images. Documentation and processing of TIF images were done with Photoshop CS4. For quantitative analysis of cysteine mutants, transfected cells were plated in eight-well glass chamber slides in duplicate. After live staining, the percentage of EGFP- and DAPI-positive cells labeled with 3F4 was calculated using an automated acquisition and analysis System (ImageXpressMICRO from Molecular Devices). For quantification of the various PMD mutants in Figures 8F and 9, the percentage of cells with surface expression was manually assessed by scoring cells for the presence of both microspikes and EGFP fluorescence in endosomal/lysosomal compartment.
Results
Deleting previously suggested ER retention signals from PLP/DM20
Because expression of mutant PLP and DM20 is often lethal for oligodendrocytes in the CNS, protein trafficking studies have been conducted in cultured immortalized cells. Here, we used the oligodendroglial cell line oli-neu (Jung et al., 1995) and also COS-7 cells to monitor PLP modified with a myc epitope or fused (via a C-terminal linker) to EGFP (Fig. 1A), as described previously (Trajkovic et al., 2006; Dhaunchak and Nave, 2007). Whereas this tagging of wild-type PLP (PLPwt) results in surface expression for either cell type, mutant PLP isoforms [such as PLPA242V, which causes a congenital form of PMD (Yamamoto et al., 1998)] are only stained in a reticular intracellular pattern that we refer to as “ER retained” (Fig. 2A,B).”

Structure of PLP/DM20 and mutations associated with Pelizaeus–Merzbacher disease. A, Two-dimensional model of PLP (276 residues as black beads) and its splice isoform DM20, lacking 35 residues (marked in gray) from an intracellular loop. The orientation of TM1–TM4 positions of both the N and C termini into the cytoplasm. Disulfide bonds in EC2 are joined by black lines and are critical for PLP folding (Dhaunchak and Nave, 2007). Positions of amino acids in EC2 that are substituted in patients with PMD are marked in yellow. Those that have been studied in detail here carry the single letter code of the wild-type sequence, labeled in red. Also indicated are C-terminal epitope tags used in this study (EGFP or myc) and the approximate positions of extracellular (3F4) and intracellular (A431) antibody binding sites common to PLP and DM20. B, Schematic view of the intracellular loop of PLP/DM20, flanked by TM2 and TM3, including a PLP-specific sequence (open gray circles) and 5 aa (open red circles) that extend the hydrophobicity of DM20 (see Fig. 6 for hydropathy plot). The positions of four segments (S1–S4) are indicated, each of which has been replaced individually with the myc-epitope tag, giving rise to different PLP “chimeras” (S1–S4). These serial replacement chimeras were used to test whether PLP harbors an essential ER retention signal (Fig. 5). C, A two-dimensional model of PLP, DM20, and various truncations used in the study is shown. The four TMs (block rectangles), the PLP-specific region (gray rectangle), the previously proposed retention signals (stars), and the signal peptide for proper insertion of TM4 (blue rectangle) are highlighted.
Two regions of PLP have been proposed by other groups as ER retention signals for mutant PLP: TM4 (Swanton et al., 2003) and a heptapeptide within the intracellular loop that is absent from DM20 (Southwood et al., 2007) (Fig. 1). To test the role of these motifs in trafficking, we generated truncated PLP isoforms that were misfolded and also lacking the proposed retention signals. Specifically, we expressed construct encoding only the first two (termed TM1,2), first three (TM1,2,3), second two (TM3,4), or only the last (TM4) transmembrane domain of PLP (Fig. 1D). For proper orientation of the N-terminal deletion construct (TM4), a specific signal peptide was used (Kleijnen et al., 1997). TM1,2 and TM3,4 lacked the heptapeptide sequence suggested to mediate ER retention (Southwood et al., 2007), and TM1,2 also lacked TM4, the second proposed ER retention signal (Swanton et al., 2003). The intracellular distribution of each polypeptide was examined with appropriate antibodies or by EGFP fluorescence. All four PLP truncations were retained in the ER (Fig. 2C–F), with a reticular labeling strongly concentrated in the perinuclear region. Thus, PLP harbors additional retention signals and/or the proper assembly of all four TM domains is required for surface expression of PLP.
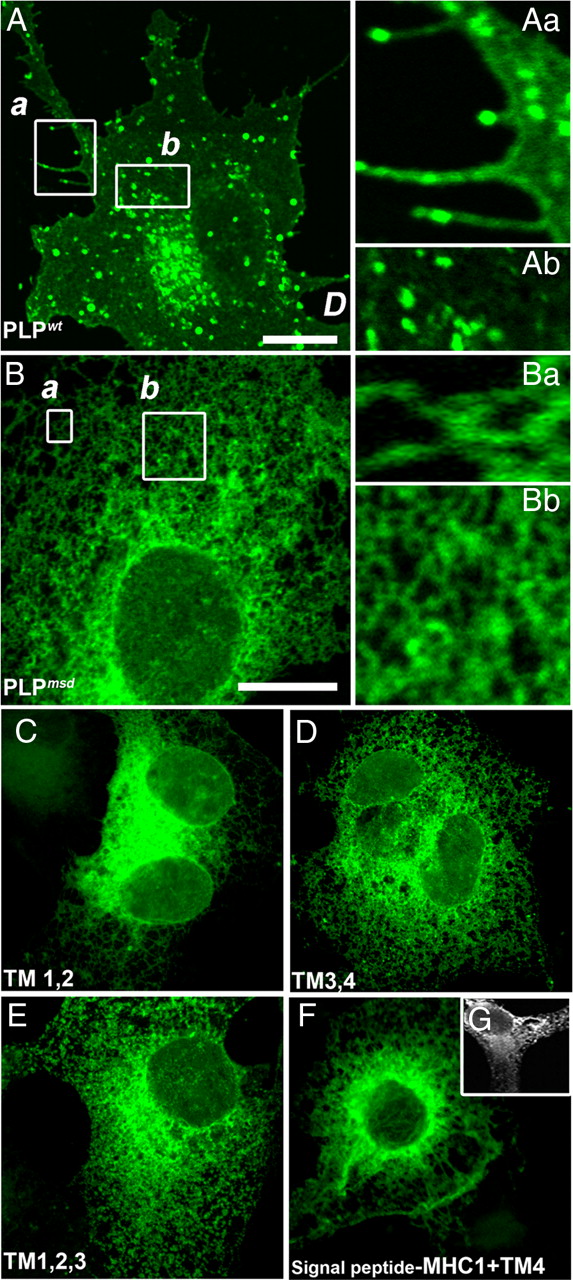
Truncated PLPs with fewer than four TMs are retained in the ER. A, When expressed, PLPwt readily traffics to the cell surface as demonstrated by green fluorescent microspikes at the tips of processes. COS-7 and glial cells (Fig. 4A) protrude numerous filopodial processes, and PLPwt accumulates in the endosomal/lysosomal compartment. Magnifications (Aa, Ab) of the boxed area in A highlight PLP-positive filopodial protrusions and endosomes/lysosomes. Scale bar, 10 μm. B, In contrast, mutant protein derived from jimpy-msd mice (PLPmsd; A242V substitution) fails to reach the cell surface. Cells lack any labeled microspikes (magnified in insets). There is a reticular distribution of EGFP fluorescence with no accumulation in the endosomal/lysosomal compartment. Magnifications (Ba, Bb) of boxed area in B are magnifications of areas in a close proximity to the cell surface. Note that there is complete absence of fine microspikes. Scale bar, 10 μm. C–F, Fixed cells permeabilized and stained for the C- or N-terminal myc epitope are shown in the left column and EGFP fluorescence is shown in the right column. Cells expressing truncated PLPs, regardless of whether they contain two or three TMs, are strongly retained in the ER (C, E). Interestingly, the second half of the protein whether consisting of TM3,4 or TM4 alone is also retained in the ER (D, F, G), exactly like PLPmsd. To facilitate proper insertion of TM4, with N terminus luminal and C terminus cytosolic, we added an MHC class I signal peptide to the N terminus (Kleijnen et al., 1997; Swanton et al., 2003) and stained methanol-fixed and digitonin-permeabilized cells (G).
Self-assembly and transport of a tetraspan protein
To test the latter hypothesis, we attempted to reconstitute the surface expression of PLP by coexpression of two truncated protein halfmers. Indeed, coexpression of TM1,2 and TM3,4 resulted in surface expression of both proteins (Fig. 3A–C). The truncated proteins were also detected in the endosomal compartment, similar to PLPwt (Figs. 2A, 3). We therefore examined the effect of missense mutations and generated TM3,4 harboring the A242V mutation. In these coexpression experiments, both halves of PLP were ER retained (Fig. 3D). Thus, the assembly of four TM domains is not only required for surface expression of PLP, but self-assembly of this four helix bundle is a very efficient process that can bind and “correctly” fold two severely truncated proteins.

Self-assembly of TM domains. TM1,2 when coexpressed in oligodendroglial (A, B) or COS-7 cells (C, D) with EGFP-tagged TM3,4 derived from PLPwt but not from PLPmsd results in surface expression of both halves of the protein (only TM3,4 is shown). In B, the endosomal/lysosomal accumulation of TM3,4 derived from PLPwt was verified by colabeling with lamp-1 (red). Also, note that, in addition to endosomal/lysosomal accumulation, glial and COS-7 cells protrude microspikes, depicted in a higher magnification of the boxed area in B (Ba) that are absent in D. Scale bar, 10 μm.
The membrane proximal disulfide bridge is critical for DM20 folding
There is an overrepresentation of mutations that map in EC2 (Nave and Boespflug-Tanguy, 1996; Woodward, 2008), suggesting that the large extracellular loop plays an important role for PLP function in myelin. We have reported previously that two pairs of cysteines within EC2 (C200–C219 and C183–C227) play a critical role in ER retention of PLP and that the membrane proximal disulfide bridge (C183–C227; see Fig. 1) is critical for PLP folding (Shaw et al., 1989; Weimbs and Stoffel, 1992; Dhaunchak and Nave, 2007). We therefore tested whether the same membrane proximal bridge is also required for normal DM20 trafficking. We analyzed the localization of cysteine-to-serine mutants in transfected oligodendroglia (Fig. 4) and in COS-7 cells (data not shown). DM20s lacking the membrane proximal bridge (C148–C192) were retained in the ER, similar to PLPmsd (Fig. 4, left). DM20 lacking the outer bridge (C165–C184), however, traffics to the cell surface (Fig. 4, right), like wild-type DM20 or PLP lacking the same bridge (Dhaunchak and Nave, 2007). Both PLP and DM20 formed cysteine-mediated cross-links in the absence of the critical (membrane proximal) cysteine bridge of EC2 (Fig. 4D). Thus, although the outer bridge is dispensable, the membrane proximal bridge in DM20 is essential for surface delivery and proper folding.

The function of extracellular disulfide bridges in DM20 folding. Of the two disulfide bridges in EC2, the “outer” one (and C165–C184) is dispensable for folding and cell-surface expression of PLP. To test the function of each cysteine bridge (see Fig. 1A) in DM20 folding and surface appearance, single and double cysteine-to-serine substitutions were engineered for each disulfide bridge. A–C, Replacing one or both cysteines of the membrane proximal bridge C148–C192 led to severe misfolding, as visualized by ER retention with no surface labeling of DM20 (in red; stained live using 3F4 antibody; left panels). Surprisingly, replacing one or both cysteines of the outer bridge and C165–C184 did not interfere with cell-surface labeling of DM20 (in red). Thus, only the membrane proximal disulfide bridge is essential for normal folding of DM20. Only merged images are shown. The percentages of cells stained with 3F4 antibody (B) or showing endosomal/lysosomal accumulation (C) are depicted in histograms on the right. As expected, DM20 lacking the membrane proximal bridge shows no accumulation in the endosomal/lysosomal compartment. D, Wild-type (W) PLP, DM20, and PLP/DM20 lacking outer (O) or inner (I) cysteines were analyzed either under nonreducing or reducing (150 mm mercaptoethanol) conditions. M and D highlight PLP/DM20 monomers and dimers, respectively. As expected, in MO3.13 cells, the wild-type, and PLP and DM20 lacking the outer cysteines do not form cysteine-mediated cross-links. The short and long exposures show that both PLP and DM20 lacking the inner bridge form cysteine-mediated cross-links (highlighted with dashed red boxes). The faint SDS and mercaptoethanol-resistant dimer seen on reducing gels constitutes between 7 and 13% (quantified from two independent transfection experiments) of the total dimer seen on nonreducing gels. Such dimers are also seen with wild-type protein but only during longer exposures and were also reported previously (Swanton et al., 2005).
Motifs regulating mutant PLP and DM20 trafficking
PLP and DM20 differ in their cytoplasmic loop, whereas the tested PMD mutations map into EC2 (Fig. 1A). It is therefore puzzling that some EC2 substitutions cause the retention of PLP but not of the corresponding DM20 mutant (Gow and Lazzarini, 1996; Gow et al., 1997; Thomson et al., 1997) in transfected fibroblasts. Also, in transfected oligodendrocytes, all EC2 mutants tested (PLPD202N, PLPR204G, PLPV208D, PLPL209H, PLPP215S) were ER retained, whereas the corresponding DM20 mutants were cell surface expressed and accumulated in late endosomes (Fig. 5A, and data not shown). We had previously discovered that the inability to close the outer disulfide bridge in EC2 leads to abnormal cysteine cross-links and is the direct cause of ER retention (Dhaunchak and Nave, 2007). Thus, intact surface expression of the corresponding DM20 mutants suggests that disulfide bridges have normally formed despite conformational constraints in EC2. We therefore asked whether DM20 would be involved in any abnormal cysteine-mediated cross-links. When transiently overexpressed in four different cell lines, we found some cysteine-mediated cross-links of PMD-causing DM20 mutants (see Fig. 8A–C) but to a much lesser extent than the cross-linking of mutant PLP.

ER retention in oligodendroglial cells distinguishes PMD-associated isoforms of PLP, DM20, and experimental chimeras. A, Wild-type PLP and DM20, here fused to a C-terminal EGFP, exit the ER and reach the cell membrane (a, d), as shown by confocal imaging revealing distal transport vesicles in glial processes and membrane-associated fluorescent microspikes (magnified in inset). Specific PMD substitutions that map into EC2 (D202N, P215S; amino acid positions refer to the sequence of PLP) cause ER retention of mutant PLP (b, c) but not of mutant DM20 (e, f). In addition to labeling the membrane of live glial cells with an antibody raised against the cell-surface protein NG2 (blue), the surface appearance of PLP was confirmed by live staining (in red) with monoclonal antibody 3F4 (b, c, e, f). The intracellular EGFP signal marks either a late endosomal compartment (a, d–f) or the reticular ER when PLP is retained (b, c). Scale bar: A–C, 10 μm. B, In searching for a PLP-specific ER retention signal, PLP–myc chimeras S1–S4 were expressed that lacked different segments (S1–S4 in Fig. 1B) of the PLP-specific intracellular loop (schematically indicated at the top). In the absence of any additional modification, chimeras S1–S3 were able to exit the ER of oli-neu cells, reaching a late endosomal compartment (a–c). Chimera S4 was ER retained, suggesting that no retention signal had been removed. C, Importantly, in combination with PMD mutations mapping into EC2 [D202N (not shown) and P215S], chimera S2 (a) and chimera S3 (c) were also ER retained, with chimera S2 showing a partial rescue of mutant PLP highlighted by lamp-1 colabeling (red). This suggests the absence of an essential retention signal in the PLP-specific cytoplasmic loop that could completely explain the differential ER retention of mutant PLP and DM20 (A).
In principle, PLP could harbor a motif within the PLP-specific loop that is missing in DM20, as proposed by Southwood et al. (2007). Such a sequence could play a role in ER retention and inhibit reorientation of EC2 in the presence of PMD-causing mutations, resulting in cross-linking of mutant PLP. To map such motifs within the PLP-specific loop, we generated chimeric constructs in which an 11 aa myc epitope replaced equally short segments in the intracellular loop of PLP (S1 to S4; Fig. 1B) and examined their subcellular distribution in transfected glial cells (Fig. 5B) and COS-7 cells (data not shown). In chimera S1, a segment common to both PLP and DM20 was replaced to control for any nonspecific effects of perturbation of the cytoplasmic loop. This construct was not ER retained (Fig. 5B). Consistent with observations in COS-7 cells (Southwood et al. 2007), the S4 replacement resulted in ER retention of chimeric PLP in oligodendrocytes (Fig. 5B). It is possible that the proximity of this segment to TM3 perturbs four TM assembly, which we have already shown to be required for correct trafficking of PLP (Fig. 3). Chimeras S2 and S3 were readily detected at the cell surface. We therefore investigated whether PMD-causing mutations in EC2 (PLPD202N and PLPP215S) would alter the trafficking of these chimeras. Mutant chimera S3 trafficked like mutant PLP and was essentially retained in the ER, whereas mutant chimera S2 showed only a partial rescue, with some colocalization with lamp-1-positive vesicles in oligodendroglia (Fig. 5C) and COS-7 cells (see Fig. 8E,F). Importantly, in both S2 and S3, the PMD-causing mutations induce cross-links to the same extent as wild-type PLP (see Fig. 8D). Hence, the intracellular loop of PLP does not harbor an essential ER retention signal that would fully explain the trafficking and cross-linking difference between EC2 mutant PLP and DM20. We therefore propose that DM20 must escape the quality control (QC) by another compensatory mechanism.
The role of TM3 in folding mutant PLP/DM20 isoforms
Because of the close proximity of the DM20-specific deletion to TM3, we hypothesized that the TM3 domain itself could be positioned differently in DM20 and that this affects trafficking of the protein. Kyte–Doolittle analysis suggested that, with fewer charged amino acids (Nave et al., 1987), TM3 is extended by five residues in DM20. This could allow TM3 of DM20 (but not of PLP) to shift its position within the lipid bilayer by more than one helical turn, thereby reducing restrictions on protein folding in EC2. Such gliding of TMs is a feature of voltage-gated ion channels and was proposed for PLP (Stecca et al., 2000). Because PLP/DM20 are proposed to have evolved from an ancestral pore-forming protein (Kitagawa et al., 1993), these proteins might indeed share such a mechanism. The conformational flexibility that is lost with the shortening of TM3 (in the evolution of PLP from the more ancestral DM20 isoform) might be responsible for the sensitivity of this protein to minor mutation-induced constraints of EC2 and the observation that mutant PLP isoforms can no longer escape the quality control of the ER.
To test this hypothesis directly, we altered mutant forms of DM20 such that they resemble PLP with respect to TM3. When Thr115 (which flanks TM3) was replaced with Lys, the protein was expressed at the cell surface (Fig. 6B), similar to wild type. When PMD mutations of EC2 were added in the construct, the DM20T115K variants showed partial ER retention (Fig. 6C,D), in support of our model. Similarly, when we replaced the last four residues that precede TM3 in DM20 (LSAT115) by the corresponding four residues of PLP (HPDK150), the DM20HPDK protein distributed like DM20wt (Fig. 6E). However, introducing additional PMD mutations (D202N and P215S) resulted in ER retention of these compound DM20 mutants (Fig. 6F,G). Thus, the motif “HPDK” in the PLP-specific loop serves as a stop-transfer signal for TM3, limiting its ability to move into the lipid bilayer and to compensate for mutation-induced misfoldings of EC2.

The length and position of TM3 determine ER retention or release of mutant DM20. A, Kyte–Doolitle hydropathy plot of PLP, DM20, and DM20LSAT–HPDK chimera. Scale bar: same for all as in PLP. Hydrophilicity plotted with a window of 11 residues, using DNA star. Negative hydrophilicity implies a highly hydrophobic stretch. Both PLP and DM20 share four highly hydrophobic TM stretches. The motif itself has been conserved among proteolipids and has been proposed to evolve from an ancestral gene encoding pore-forming proteins (Kitagawa et al., 1993). The PLP-specific region imparts a highly hydrophilic nature to intracellular loop (IC). In contrast, DM20 bears an extended hydrophobic stretch, which might allow TM3 to glide a single α helical turn up or down to reorient EC2 during local misfolding. This gliding phenomenon can be completely reversed by simply reversing the hydrophobicity of DM20-specific region to a positive hydrophilicity by replacing amino acid residues LSAT to HPDK. This engineered DM20HPDK displays hydrophilic characteristics of PLP and also traffics like PLPwt in transfected cells. The third TM domain of DM20 is potentially longer than in PLP as a result of a stretch of four uncharged residues in DM20 (positions 112–115) immediately preceding TM3. Replacing the juxtamembrane threonine by a lysine in DM20 (T115K) did not prevent cell-surface expression (B) but is likely to reduce transmembrane domain sliding of TM3. Importantly, T115K partially impaired transport of two PMD mutant isoforms (C, D). The substitution of four consecutive residues had an even greater effect on DM20 retention [HPDK150: the predicted TM3 “stop transfer” signal in PLP at the equivalent position in DM20 (LSAT115)]. Also this modification (depicted on the right) allowed DM20 to traffic normally (E) but caused complete ER retention when combined with mutations D202N (F) or P215 (G). Together, this strongly suggests that a subtle TM domain sliding of TM3 allows DM20 (but not PLP) to properly fold the globular EC2 domain in the ER lumen, despite its PMD-causing substitution. Scale bar, 10 μm.
Inhibiting TM3 reorientation in PMD bearing DM20 impair EC2 folding
Our results suggest the existence of two independent mechanisms of QC for PLP/DM20, involving luminal and TM domains of the protein. When TM3 is too short to compensate for EC2 misfolding, disulfide bridges in EC2 fail to form, causing ER retention. To confirm that EC2 misfolding is indeed the underlying mechanism of ER retention in shortened TM3 DM20s, we removed the critical cysteines in EC2 (C165–C184 corresponding to C200–C219 in PLP) in those DM20 isoforms that resembled PLP at TM3 (DM20T115K and DM20LSAT-HPDK) and that harbored additionally a PMD-causing substitution in EC2 (P215S). Importantly, all of these mutant proteins carrying multiple mutations reached the surface of transfected glial cells and accumulated in the lamp-1-positive lysosomal compartment (Figs. 7, 8E,F: COS-7 cells), exactly like wild-type PLP. In addition, removal of cysteines decreases the abnormal cross-linking of these mutants in four different cell lines (Fig. 8D). Thus, the charged residues proximal to TM3 (K or HPDK) do not comprise an essential ER retention signal, but they interfere with proper folding of EC2 in the presence of several PMD-causing mutations.

Rescue of PMD mutation bearing chimeric PLP and DM20s from ER retention suggests coordination of QC of TM domain assembly and EC2 folding. oli-neu cells transfected with EGFP fusion constructs. Surprisingly, PMD mutation bearing chimeras S2, S3, DM20T115K, and DM20LSAT–HPDK with removal of the outer cysteine pair are not retained in the ER. Quadruple mutants are also readily detectable in the lamp-1-positive endosomal/lysosomal compartment (red) as depicted in the corresponding magnified panels (right). This suggests coordination of QC of TM domain assembly and EC2 folding in PLP and DM20.

QC of EC2 folding and TM domain assembly of PLP/DM20 is not glial cell specific. A, When resolved under nonreducing conditions, the cell-surface DM20 proteins bearing PMD-causing mutations (D202N, L209H, P215S) form fewer cross-links, whereas ER-retained DM20 mutants (L223P and A242V) form cross-links to the same extent as the corresponding PLP mutants in a human oligodendroglial cell line (MO3.13). As reported previously (Dhaunchak and Nave, 2007), when the same samples were resolved under reducing conditions, no cross-links were observed at similar exposure times (data not shown). B, PMD causing PLPP215S forms twofold to threefold more cross-links when compared with the corresponding DM20P215S or wild-type PLP. C, The extent of cross-linking of PLPP215S is greater than that of the corresponding DM20P215S or wild-type PLP when analyzed in three different cell lines (COS-7, MO3.13, and HEK293). D, Removal of the outer cysteines (R) rescues the PMD-causing (P215S) mutant chimeras (M) S2, S3, DM20T115K, and DM20HPDK from cross-linking in four different cell lines. The percentage of cross-linking in MO3.13 cells quantified from two independent transfection experiments is shown on top. E, F, Removal of outer cysteines (R) releases the PMD-causing (P215S) mutant chimeras (M) S2, S3, DM20T115K, and DM20HPDK from ER retention in COS-7 cells and oli-neu cells (Fig. 7). The percentage of cell-surface PLP in COS-7 cells quantified from one of the three experiments (each performed in triplicate) is shown in F.
We next examined whether mutant PLP can also be rescued from ER retention by incorporation of the membrane proximal pentapeptide (GLSAT; highlighted in Fig. 1B) that in DM20 extends the hydrophobic stretch and allows proper EC2 folding even in presence of PMD-causing mutations (Figs. 5, 6). The chimera S5 (with GLSAT incorporated after Lys151) traffics to the cell surface and accumulates in the lamp-1 positive compartment (Fig. 9A), exactly like wild-type PLP and DM20. Although, PMD-causing mutation bearing S5 chimeras (S5D202N and S5P215S) show significant rescue over mutant PLP (Fig. 9A,B) and are detected both at the cell surface and in the lamp-1 positive compartment, a perinuclear reticular distribution is also seen. This could be attributable to a compound effect of other PLP-specific peptides that we (Fig. 5) and others (Southwood et al., 2007) have shown to play a partial role in PLP folding. The intermediate phenotype seen with S5 mutants (Fig. 9A,B) represents a shift of PLP trafficking to DM20-like when bearing PMD-causing mutations. Together, we conclude that TM3 plays a critical role in EC2 folding, which becomes relevant for the QC and trafficking of human PLP and DM20 in oligodendrocytes of patients with PMD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Experimental elongation of TM3 in PLP rescues mutant PLP from ER retention. A, Incorporating the DM20 juxtamembrane pentapeptide (GLSAT) after lysine 151 in PLP does not perturb the cell-surface expression or lysosomal accumulation of chimera S5wt but is likely to introduce transmembrane domain sliding of TM3 (Fig. 6). A representative phenotype of S5, PLP, DM20-bearing PMD mutation (P215S) is shown. Note that S5P215 accumulates in lamp-1-positive compartment similar to wild-type S5 or DM20P215S. PLPP215S, however, is present in calnexin-positive reticular compartment and is absent from lamp-1-positive compartment. The merged and individual magnification panels of boxed area are shown on right. Scale bar, 10 μm. B, When compared with corresponding PLP mutants, using unpaired t test, the S5D202N and S5P215S show significant rescue from ER retention. *p < 0.05; **p < 0.01. The quantification from two independent experiments (each done in triplicate) shows that S5D202N (54.57 ± 4.56 SEM) and S5P215S (68.52 ± 2.66 SEM) are detected at the cell surface.
Discussion
Using expression of the myelin protein PLP/DM20 as a model system, we have studied rules for membrane protein folding and trafficking that are relevant to human disease. Numerous missense mutations of the human PLP1 gene have been identified that cause severe neurological disease and premature death, but the effect of these mutations has been puzzling, in part because of different cellular consequences of the same mutation when altering the structure of PLP or its isoform DM20.
We have evidence for two distinct sites of QC, one at the luminal side of the ER and second one directly involving the TM domains. PLP/DM20 mutants that fail either one are retained in the ER. Both QC sites monitor proper folding of four TM domains, one cytoplasmic and two extracellular loops. These QCs appear to coordinate in governing proper EC2 folding and TM domain assembly in PLP and DM20.
Our data support a PMD disease model in which substitutions in the large extracellular loop domain of PLP/DM20 induce minor structural changes that prevent efficient formation of normal disulfide bridges. This exposes unpaired cysteines in the oxidative environment of the ER and competing oxidations (Dhaunchak and Nave, 2007) generate aberrant PLP dimers that fail to mature into oligomeric forms and are retained in the ER (inducing the unfolded protein response and cell death in vivo). Mutant DM20, however, is “protected” from this pathomechanism, because a longer TM3 allows for adjustment of the tetraspan to obtain sufficient folding of EC2 for the generation of intramolecular disulfide bridges.
However, if the tetraspan itself cannot form (because of truncations, frame-shift, or missense mutations affecting the TM domains), PLP/DM20 is retained in the ER. It is thought that abnormal exposure of a single hydrophobic TM can recruit chaperones or cause even a premature oligomerization in oligodendrocytes, as shown previously for PLP expression in heterologous cells (Swanton et al., 2003, 2005).
Quality control and the formation of a tetraspan membrane protein
As expected, TM domain assembly is critical for surface expression of PLP and truncated proteins with fewer than all four TM domains are retained in the ER. Quite surprisingly, however, the coexpression of two PLP halfmers (i.e., TM1,2 and TM3,4) results in the surface expression of both. This demonstrates an unexpected efficiency of TM domains in the membrane to properly self-assemble into tetraspans and is reminiscent of the role of soluble v- and t-SNARE helices in vesicle fusion reactions (Jahn and Scheller, 2006). This self-assembly of TM domains could be prevented by introducing a disease-relevant amino acid substitution into TM4, suggesting that this type of mutation indeed prevents formation of an export competent tetraspan (Ng and Deber, 2010).
We also found an explanation for the apparent differences in the ability of PLP and DM20 to cope with amino acid substitutions. Using hydropathy analysis and experimental chimeras, we found that an extended stretch of hydrophobic residues (Stecca et al., 2000) adjacent to TM3 (in DM20 but not in PLP) allows DM20 to adjust the position of TM3 in the lipid bilayer to stabilize the EC2 domain by disulfide bridges (Fig. 6). In contrast, the more recently evolved “PLP-specific” hydrophilic loop (Figs. 1B, 6) with its C-terminal basic residues inhibits such a reorientation. This model assumes, however, that there are at least two alternative thermodynamically feasible alignments of TM3 within the tetraspan (for review, see White and Wimley, 1999).
Coordination of QCs of TM domain assembly and EC2 folding
We have shown previously that various PMD-associated substitutions in PLP, all of which map into EC2, converge mechanistically by perturbing the formation of an intramolecular disulfide bridge in the lumen of the ER (Dhaunchak and Nave, 2007). Although disulfide bridges are thought to stabilize globular protein domains, only the membrane proximal disulfide bridge in PLP serves this conserved function and is critical for surface expression of PLP (Fig. 4). Importantly, the bridge itself appears dispensable for folding, as suggested by the normal trafficking of PLP when lacking both cysteine residues involved. Thus, in PMD, it is not the substituted amino acid by itself that causes PLP misfolding but an unpaired (and exposed) cysteine that causes protein cross-links and retention (Dhaunchak and Nave, 2007). Because the same mutation does not impair trafficking of DM20, it suggests that intramolecular bridges are properly formed and that the extended hydrophobicity of TM3 allows reorientation of EC2. We, however, observed cross-linking of the mutant DM20 but to a lesser extent when compared with the mutant PLP. In rescue experiments, even PLP and DM20 compound mutants (S2P215S, S3P215S, DM20T115K+P215S, and DM20LSAT-HPDK+P215S), which lack the outer cysteines, are released from ER retention (Figs. 7, 8) and are prevented from being cross-linked, demonstrating that inhibiting the sliding of TM3 impairs proper disulfide bridge formation in EC2.
Cellular mechanism of ER retention in PMD
Given that many membrane proteins harbor intramolecular disulfide bridges in their extracellular loop regions and are sensitive to point mutations in TM domains and EC loop regions (Travis et al., 1991; Umemoto et al., 1991; McDermott et al., 1999; Galon et al., 2000; Schülein et al., 2001; Yum et al., 2002; Albrecht et al., 2004; Thomas et al., 2004), we suggest that our model is likely relevant to a broad spectrum of genetic disorders. We have shown previously (Dhaunchak and Nave, 2007) that PLP dimers are generated when PMD-causing mutations affect EC2. These cysteine-dependent PLP dimers are novel oxidation products and fail to become O10 positive (Jung et al., 1996). Thus, abnormal cross-links are a plausible explanation for ER retention, the unfolded protein response, and apoptotic death of mutant oligodendrocytes in vivo.
Prolonged cycles of binding and release to specific chaperones, such as protein disulfide isomerase, may also contribute to PLP retention. Furthermore, an interaction of mutant PLP with calnexin was reported for heterologous cells (Swanton et al., 2003), and a retention signal that recruits calnexin was hypothesized for TM4.
Our rescuing experiments, in which TM1,2 and TM3,4 complement each other for surface expression, clearly demonstrate that assembly of a tetraspan is a major QC mechanism for PLP. A similar observation has been made for glycoprotein CD81, in which TM1 (a glycopolypeptide) is retained in the ER by interaction with calnexin and can be complemented with the TM2,3,4 glycopolypeptides (Cannon and Cresswell, 2001).
PLP is a non-glycosylated tetraspan, and it is possible that the interaction of calnexin with improperly aligned TM domains of PLP could involve aggregation or cross-linking into a multimeric complex (Ellgaard et al., 1999; Ellgaard and Helenius, 2003). Whether the observed binding of PLP with calnexin is the cause or consequence of ER retention remains to be determined. Because PLP and DM20 are the most abundant membrane proteins of myelinating oligodendrocytes in vivo, it is plausible that chaperones can be easily outnumbered during protein folding. This may explain in part the sensitivity of myelinating oligodendrocytes to PLP/DM20 overexpression (Kagawa et al., 1994; Readhead et al., 1994).
Because the present model was derived from observations with immortalized oligodendroglial cells (oli-neu and MO3.13) and heterologous cells (COS7 and HEK293), it is also likely to be applicable to mutant oligodendrocytes in vivo. The proposed model is likely to apply to the subset of PMD cases associated with single substitutions in EC2. The mechanism is unlikely to explain the mutations that map into EC1 or the first two TM domains of PLP. Because the tetraspan motif is evolutionarily conserved in many membrane proteins (Kitagawa et al., 1993) and EC domains often contain cysteine residues, the insight from the PLP model may be applicable to other protein families and disease entities. This could include pores and channel proteins [e.g., synaptophysin, connexins, ryanodine receptor, nAChR, GluR, GABAR, NMDAR, glycine receptors (Hille, 2001)] or proteins with less well understood functions [e.g., the retinal photoreceptor protein peripherin (Travis et al., 1991), VMA 3, and VMA 11 (Umemoto et al., 1991)], for which the consequences of single residue substitutions on protein function are not obvious. Comparable point mutations have been associated with a wide spectrum of human diseases, ranging from specific forms of sensoneurial deafness (Thomas et al., 2004) and diabetes insipidus (Schülein et al., 2001) to autoimmune disorders (McDermott et al., 1999; Galon et al., 2000), high-density lipoprotein deficiency (Albrecht et al., 2004), and Charcot-Marie-Tooth neuropathy (Yum et al., 2002).
Footnotes
- Received April 27, 2011.
- Revision received August 9, 2011.
- Accepted August 15, 2011.
A.S.D and K.A.N dedicate this paper to the late David R. Colman.
This work was supported by grants from the European Union (Neuron–Glia Nerve Development and Disease, Leukotreat), the National Multiple Sclerosis Society, the Bundesministerium für Bildung und Forschung (Leukonet), the Myelin Project, and the European Leukodystrophy Association. This study was also funded in part by Rio Tinto Alcan and The Molson Foundation. A.S.D. is supported by a Multiple Sclerosis Society of Canada Postdoctoral Fellowship. We thank members of the Nave and Colman laboratories for helpful discussions. We also thank J. Trotter for oli-neu cells, P. Talbot for MO3.3 cells, M. Lees for providing 3F4 antibody, and P. Yam and M. Pool for comments on this manuscript.
- Correspondence should be addressed to either of the following: Klaus-Armin Nave, Department of Neurogenetics, Max Planck Institute of Experimental Medicine, D-37075 Göttingen, Germany, nave{at}em.mpg.de; or Ajit Singh Dhaunchak, Department of Neurology and Neurosurgery, Montreal Neurological Institute and Hospital, McGill University, Montreal, Quebec, Canada H3A 2B4, ajit.dhaunchak{at}mcgill.ca
- Copyright © 2011 the authors 0270-6474/11/3114961-11$15.00/0