

Abstract
The A/VN/1203/04 strain of the H5N1 influenza virus is capable of infecting the CNS of mice and inducing a number of neurodegenerative pathologies. Here, we examined the effects of H5N1 on several pathological aspects affected in parkinsonism, including loss of the phenotype of dopaminergic neurons located in the substantia nigra pars compacta (SNpc), expression of monoamines and indolamines in brain, alterations in SNpc microglia number and morphology, and expression of cytokines, chemokines, and growth factors. We find that H5N1 induces a transient loss of the dopaminergic phenotype in SNpc and now report that this loss recovers by 90 d after infection. A similar pattern of loss and recovery was seen in monoamine levels of the basal ganglia. The inflammatory response in lung and different regions of the brain known to be targets of the H5N1 virus (brainstem, substantia nigra, striatum, and cortex) were examined at 3, 10, 21, 60, and 90 d after infection. In each of these brain regions, we found a significant increase in the number of activated microglia that lasted at least 90 d. We also quantified expression of IL-1α, IL-1β, IL-2, IL-6, IL-9, IL-10, IL-12(p70), IL-13, TNF-α, IFN-γ, granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, macrophage colony-stimulating factor, eotaxin, interferon-inducible protein 10, cytokine-induced neutrophil chemoattractant, monocyte chemotactic protein-1, macrophage inflammatory protein (MIP) 1α, MIP-1β, and VEGF, and found that the pattern and levels of expression are dependent on both brain region and time after infection. We conclude that H5N1 infection in mice induces a long-lasting inflammatory response in brain and may play a contributing factor in the development of pathologies in neurodegenerative disorders.
Introduction
A number of neurodegenerative disorders, including Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis, have been shown to have a significant inflammatory component to their pathologies (Appel et al., 2010; Glass et al., 2010; Tansey and Goldberg, 2010). While the proximate cause of inflammation can be varied, it has clearly been demonstrated that viruses, including influenza, can be encephalitic (Hayase and Tobita, 1997; Jang et al., 2009a; Sejvar and Uyeki, 2010). In fact, involvement of the CNS during influenza infections can be fatal, particularly in young patients (Smith et al., 1997). Neurological symptoms associated with influenza infection have been reported as far back as 1385, and intermittent outbreaks with similar symptoms have occurred at other times during influenza outbreaks (Menninger, 1926). The most deadly influenza pandemic of the 20th century, the Spanish Flu, occurred in 1918. It is estimated that ∼25–30% of the world population was infected and upward of 50 million people died (Taubenberger and Morens, 2006). About the same time, the world was hit by an unusual epidemic of neurological disease. Post-influenza psychosis and post-encephalitic parkinsonism were reported in Europe and in the United States. More than 500,000 patients who survived this influenza infection were thought to have developed some type of nervous system disorder (Ravenholt and Foege, 1982) including encephalitis lethargica, also known as von Economo's encephalitis (von Economo, 1917). Additionally, a significant number of persons who recovered from this “sleeping sickness” later developed a form of encephalitic parkinsonism (Dickman, 2001).
Although rare, experimental evidence has shown that some type A influenza viruses are neurotropic, i.e., they can travel into the nervous system following systemic infection (Tanaka et al., 2003; Klopfleisch et al., 2006; Rigoni et al., 2007), a finding confirmed with certain A/VN/1203/04 (H5N1) influenza viruses (Rimmelzwaan et al., 2006; Jang et al., 2009b). H5N1 infection in mice can initiate a parkinsonian pathology that includes bradykinesia, loss of the substantia nigra pars compacta (SNpc) dopaminergic neuron phenotype, increased levels of α-synuclein phosphorylation and aggregation, and activation of microglia. Each of these symptoms persists at least 60 d after resolution of the infection (Jang et al., 2009b). To determine whether the pathology worsens with age, we examined the SNpc tyrosine hydroxylase-positive (TH+) dopaminergic neuron numbers, striatal dopamine, and its metabolites through 90 d after infection. We also studied the inflammatory effect of infection by quantitatively measuring the total number of resting and activated microglia in the SNpc and the production of cytokines in regions of the brain infected by H5N1. We found that infection with H5N1 induces a significant, but transient, loss of both dopaminergic neurons in the SNpc and dopamine (and its metabolites) in the striatum. Examination of other indolamines demonstrated a significant and sustained loss of serotonin in regions of the brain infected with H5N1. We also observed that areas of the brain infected with H5N1 expressed increased levels of proinflammatory cytokines, chemokines, and growth factors that appeared both dependent and independent of induction of a peripheral cytokine storm (Yuen and Wong, 2005).
Materials and Methods
All experiments using the highly pathogenic influenza virus A/VN/1203/04 were conducted in a Biosafety Level 3+ laboratory approved for use by the U.S. Department of Agriculture and the Centers for Disease Control and Prevention. This facility is authorized for the exclusive use of the Division of Virology and other approved scientists at St. Jude Children's Research Hospital.
Virus stock preparation and inoculation of mice with H5N1.
Stock viruses were prepared by propagating neurotropic A/VN/1203/04 H5N1 in the allantoic cavity of 10-d-old embryonated chicken eggs for 40–48 h at 37°C. Virus stock was aliquoted and then stored at −70°C until use. Viral infectious titers were determined using the method of Reed and Muench (1938) and are expressed in log10 of the 50% egg infectious dose per 1.0 ml of fluid. C57BL/6J female mice (Jackson Laboratory), 6–8 weeks old, were anesthetized by isofluorane inhalation and infected intranasally with 30 μl of allantoic fluid diluted in PBS to the target virus infectious titer (102 50% egg infectious dose). One group of animals received 0.9% saline and was used as an age-matched negative control. Infected mice showed a mortality/morbidity rate similar to that described in Jang et al. (2009b), so that all measurements taken in animals before day 7 were from the complete pool of animals, while only animals that were visibly sick were used 10 d after infection.
Immunocytochemistry.
Mice were deeply anesthetized with Avertin and transcardially perfused with 0.9% saline followed by 10% neutral buffered formalin at 0, 10, 60, and 90 d after infection. Brains were removed and postfixed for 3 weeks in 10% neutral buffered formalin to ensure that the virus particles present in the tissue had been killed. Brains were cryoprotected with 30% sucrose in PBS, serially sectioned in the coronal plane at 40 μm, and placed in PBS-filled 24-well plates. Free-floating sections were immunolabeled overnight with antibodies directed against TH (rabbit, P40101–0, 1:500, Pel-Freez) to identify dopaminergic neurons or ionized calcium-binding adapter protein-1 (Iba-1) (rabbit, 019–19741, 1:500, Wako Chemicals) to identify microglia. Primary antibodies were visualized using the ABC method (PK-6101, Vector Laboratories) and visualized using either 3,3-diaminobenzidine (SK-4100, Vector Laboratories) for dopaminergic neurons or the Vector VIP substrate (SK-4600 Vector Laboratories) for microglia as chromogens, respectively.
For identification of neurons undergoing cell death, we used antibodies directed against activated caspase-3 (1:500; catalog #559565; BD Biosciences) or TUNEL (DeadEnd Colorimetric Apoptosis Detection System; Promega) to mark apoptotic neurons, while FluoroJadeB was used to identify necrotic neurons (Histo-Chem). FluoroJadeB staining was performed as previously described (Faherty et al., 1999; Jensen et al., 2004; Boyd et al., 2007). To examine cell division, we used an antibody directed against Ki-67 (1:2000, NovoCastra). Each protocol was performed as previously described (Faherty et al., 1999; McKeller et al., 2002; Jang et al., 2009b).
After staining, sections were mounted on Superfrost-Plus (12–550, Fisher) slides, counter-stained with neutral red to visualize Nissl substance, and then dehydrated, cleared, and mounted with Permount (SP15–500, Sigma).
Stereological neuron and microglial cell counts.
The total number of TH+ dopaminergic neurons (days 0, 10, 60, and 90 after infection) and resting and activated microglia (days 0, 60, and 90 after infection) in both hemispheres of the SNpc was estimated using the optical fractionator method (West et al., 1991) in the StereoInvestigator software (version 7.0, MicroBrightField) (Baquet et al., 2009). The outline of the SNpc in both hemispheres was delineated at low power (4× magnification) and an unbiased counting frame (60 × 60 μm) was placed at the intersections of a grid (frame size, 200 × 200 μm) randomly superimposed on a video image of these contours. Sections were examined under a high-power 100× objective lens (numerical aperture, 1.3) on a BX51 microscope (Olympus) with a MAC5000 motorized XYZ axis computer-controlled stage and a CX9000 CCD video camera (MicroBrightField). In each counting area, TH+ dopaminergic neurons and microglial cells were counted at the depth at which each nucleus came into focus. The reliability of the estimates was measured by calculation of the coefficient of error (West et al., 1990). Gundersen coefficients of error for m = 1 were all ≤0.10. Statistical significance was calculated using a one-way ANOVA followed by Student–Newman–Keuls post hoc test (Baquet et al., 2009).
Neuron size, determined by the longest length of neuronal cell body, was measured using the Neurolucida program (version 7.0, MicroBrightField). Raw data were converted to percentage of control.
Biochemical measurement of monoamine neurotransmitters.
At 10, 60, and 90 d after infection (n = 5 at each time point), C57BL/6J mice were deeply anesthetized with Avertin and transcardially perfused with ice-cold 0.9% saline to remove the majority of the blood from the brain vasculature. Brains were rapidly removed and placed in a brain matrix (Model BS-AL-5000C, Braintree Scientific) and sliced into 2-mm-thick sections and placed on an ice-cooled plate. Tissues were dissected using the following coordinates: substantia nigra (bregma: −2.00 to −4.00 mm), striatum (bregma: 0.00 to 2.00 mm), brainstem (bregma: −5.00 to −7.00 mm), cortex (bregma: −1.00 to −3.00 mm), and the hippocampus (bregma: −1.00 to −3.00 mm) (Paxinos and Franklin, 2001). Individual dissected tissues were then homogenized in chilled 0.3 m perchloric acid and centrifuged at 10,000 × g for 15 min at 4°C. A variety of monoamine transmitters, such as dopamine and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA); norepinephrine (NE); 5-hydroxytryptamine (5-HT) and its metabolite 5-hydroxyindoleacetic acid (5-HIAA), were analyzed using reverse-phase ion-pairing HPLC combined with electrochemical detection under isocratic elution conditions. The amount of monoamine neurotransmitters in the tissues was determined by injecting a known concentration of monoamine neurotransmitters and extrapolating from a standard curve. Statistical difference was determined using a one-way ANOVA followed by Student–Newman–Keuls post hoc test (Smeyne et al., 2007).
Generation of postnatal substantia nigra cultures and dopamine treatment.
Brains from postnatal days 0–5 (P0–P5) C57BL/6J mice were removed from the calvaria and placed into dissociation media (DM). Substantiae nigrae were dissected from the brains as previously described (Smeyne and Smeyne, 2002), placed in fresh DM, minced into small pieces, and incubated at 37°C in papain following manufacturer's protocol (Worthington Biochemical). After digestion in papain, the tissue was rinsed and triturated through a 5 ml serological glass pipette. The cell suspension was layered over plating media (PM) containing BSA (100 mg/ml) and ovomucoid albumen (100 mg/ml). The cell suspension was centrifuged, resuspended in PM containing 2% rat serum and 8% FBS, and plated at 400,000 cells per well in Lab-Tek four-well Permanox chamber slides previously coated with laminin (10 μg/ml, Collaborative Biomedical Products) and poly-d-lysine (200 μg/ml, Collaborative Biomedical Products) using 0.5 ml per well. Cells were placed in a Tri-Gas incubator and maintained at 37°C, 5% CO2, and 5% O2. Twenty-four hours after plating, one-third of the media was changed with feeding media plus 2% rat serum. Cytosine arabinofuranoside (3 μm final concentration) was added to all wells to prevent excessive glial proliferation.
To determine cell number in the cultures, wells were fixed in 3% paraformaldehyde in 1× PBS, followed by three rinses with PBS. The individual wells were then processed for TH and Iba-1 staining with the protocol listed above. At the conclusion of antibody staining, wells were removed and coverslips were applied to the Permanox slides with Vector aqueous mount containing DAPI. Cells in each well were counted using stereological methods (fractionator) applied to cell culture protocols.
Quantification of cytokines, chemokines, and growth factors.
Using the Luminex 200 system (Luminex) and the Milliplex mouse cytokine kit (MPXMCYTO-70K-20, Millipore), dissected brain regions and lung, as well as sera and cells from substantia nigra cell cultures, were analyzed for concentrations of interleukin (IL)-1α, IL-1β, IL-2, IL-6, IL-9, IL-10, IL-12(p70), IL-13, tumor necrosis factor α (TNF-α), interferon γ (IFN-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating factor (M-CSF), eotaxin, interferon-inducible protein 10 (IP-10), cytokine-induced neutrophil chemoattractant (KC), monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein (MIP)-1α, MIP-1β, and vascular endothelial growth factor (VEGF) proteins.
For tissue samples, at 3, 10, 21, 60, and 90 d after infection, dissected tissues were homogenized in a buffer containing 50 mm Tris-HCl, pH 7.4, 2.5 mm EDTA, 0.1% Triton X-100, and 150 mm NaCl with a protease and phosphatase inhibitor mixture (Complete mini, PhosphoStop, Roche). The tissue lysates were then incubated for 30 min on ice and centrifuged at 12,000 × g for 15 min. Supernatants were aliquoted and stored at −70°C until used.
For measurement of cytokine/chemokines in primary substantia nigra cultures 1 week after plating, dopamine hydrochloride (Sigma) was prepared as a 1 m stock in HPLC grade water and diluted to a 500 μm solution in feeding media. The final concentration of dopamine was either 500 nM or 5 micromilar after addition to each culture well. Aliquots of media removed at feeding (50–75 μl) were collected in vials on dry ice and stored at −80°C after initial media changes and 8, 24, and 48 h following dopamine addition. At 24 and 48 h, cells in each of the individual wells corresponding to media aliquots saved at 24 or 48 h, respectively, were rinsed once in 1× PBS after media removal, and fixed with 2.5% formalin prepared in PBS for 10 min at room temperature.
To quantify cytokine levels, the supernatants were incubated with the microspheres coated with capture antibodies for each analyte for 2 h. After rinsing, biotinylated detection antibodies were added into each well and incubated for 1 h. Streptavidin-phycoerythrin conjugates were added, and fluorescence intensity was measured by the Luminex 200 reader (Luminex). The concentration of each cytokine was calculated by generating a standard curve using known concentrations of each cytokine. All results were normalized with total protein concentration measured from each tissue lysate (picograms per 25 μg of total protein). For cell culture, cytokine levels were measured as described above and corrected based on the total cell number in each well.
Results
H5N1 infection transiently reduces the number of TH+ dopaminergic neurons in the SNpc
Infection with H5N1 leads to a wide array of pathologies in humans and mice, including diffuse damage to the lung; hemophagocytic damage to spleen, lymph nodes, and blood vessels; and alterations in bone marrow (Dybing et al., 2000; Nishimura et al., 2000; Yuen and Wong, 2005; Korteweg and Gu, 2008). Additionally, several identified strains of H5N1 are neurotrophic in mice, inducing inflammation, neuronal death, and induction of pathologies seen in Parkinson's disease, including formation of aggregates of phosphorylated α-synuclein (Wang et al., 2008; Jang et al., 2009b).
To determine whether H5N1 infection can directly induce the parkinsonian pathologies by damaging dopaminergic neurons, we stereologically assessed the number of TH+ dopaminergic neurons in the SNpc (Fig. 1A) and empirically determined the total amount of dopamine, HVA, and DOPAC in the substantia nigra, striatum, brainstem (Fig. 2), cortex, and hippocampus (data not shown).

H5N1 infection alters TH+ neurons in SNpc. A, Following H5N1 infection, the number of TH+ dopaminergic neurons in the SNpc was reduced ∼60% at 10 d after infection and 20% at 60 d after infection from the noninfected control mice. At 90 d after infection, the number of TH+ dopaminergic neurons in the SNpc was similar to that of control mice. B, At day 10–60 after infection, dopaminergic neurons in the SNpc of H5N1-infected mice are shrunken compared with those of saline-administered mice. The longest length of TH+ neuronal cell bodies was reduced ∼20% at day 10 after infection. However, the size of these cells recovered and appeared similar to that seen in the control mice at 90 d after infection. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls post hoc tests (n = 5 for each time point, *p ≤ 0.05, **p ≤ 0.001 vs control mice). Error bars indicate SEM.

Percentage change in dopamine, DOPAC, and HVA in striatum and brainstem following systemic H5N1 infection. In striatum, the amount of dopamine was significantly decreased at day 10 after infection compared with control noninfected mice. By day 60 after infection, dopamine levels had recovered to baseline levels. This pattern of a transient decrease 10 d after infection followed by recovery by day 60 after infection was also seen in levels of HVA and DOPAC. In the brainstem, dopamine, DOPAC, and HVA levels sharply increased at 10 d after infection, and then returned to their basal levels by day 60 after infection. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls post hoc tests (n = 5 for each condition, *p ≤ 0.05, **p ≤ 0.001 vs control mice). Error bars indicate SEM.
At day 10 after infection, we find ∼60% loss of TH+ neurons in the SNpc, compared with non-infected control mice. By 60 d after infection, we saw a recovery in the TH+ dopaminergic neuron number, and by 90 d after infection, we saw no difference in the number of SNpc TH+ dopaminergic neurons (Fig. 1A). From 10 through 60 d after infection, the TH+ dopaminergic neurons appeared shrunken and atrophic. The length of the longest TH+ neuronal cell bodies was reduced by 20%. However, the size of these cells recovered and appeared similar to that seen in the noninfected control mice at day 90 after infection (Fig. 1B).
To determine whether the loss of TH+ neurons was a result of cell death and the subsequent recovery in number was due to repopulation via neurogenesis, we examined expression of activated caspase-3, TUNEL, and FluoroJadeB staining as well as expression of Ki-67. In regions of the brain where the A/VN/1203/04 H5N1 virus had been detected, we found a few activated caspase-3-positive apoptotic but not FluoroJadeB necrotic cells (data not shown). The apoptotic cells were only visible through 10 d after infection and, although not systematically quantitated, did not appear to be numerous enough to account for a 20% reduction in substantia nigra TH+ neuron number. Examination of cells in the SNpc showed no evidence of cell division using immunohistochemical (Ki-67) methods through 90 d after infection. Therefore, there is no compelling evidence that SNpc dopaminergic neurons underwent any form of neurogenesis. Thus, it is most likely that active H5N1 infection induces SNpc dopaminergic neurons to transiently reduce their metabolic capacity, which both compromises TH activity and reduces cell size (Fig. 1A,B), each leading to a loss of the dopaminergic phenotype in neurons.
Effect of H5N1 infection on dopamine, DOPAC, and HVA levels in the CNS
We used reverse-phase HPLC with electrochemical detection to determine whether infection with H5N1 affected the levels of dopamine and its metabolites in the striatum, the major target of SNpc dopaminergic neurons, as well as in the brainstem, substantia nigra, and cortex. The amount of striatal dopamine and its metabolites, DOPAC and HVA, were each significantly decreased by ∼40% compared with intranasal saline-treated control mice at 10 d after infection. By 60 d after infection, dopamine levels returned to baseline levels, and this change was stable through 90 d after infection. This pattern of a transient decrease at 10 d after infection followed by recovery at 90 d after infection was also seen when examining levels of HVA and DOPAC (Fig. 2).
We also compared the turnover ratio of dopamine in the striatum [(DOPAC + HVA)/dopamine] to see whether infection with H5N1 altered dopamine metabolism. Despite alterations in levels of dopamine, its turnover was unchanged due to concurrent fold changes in DOPAC and HVA.
In brainstem, the pattern of dopamine levels was reversed. At 10 d after infection, we found a transient 300% increase in dopamine and DOPAC and a 500% increase in HVA (Fig. 2), which resolved by 60 d after infection. As with the striatum, no changes in dopamine turnover were detected since the relative ratios of dopamine to DOPAC and HVA did not change at each time point.
No changes in dopamine, DOPAC, or HVA levels were detected in substantia nigra or cortex at any time after influenza infection (data not shown).
Effect of H5N1 infection on NE and 5-HT levels in the CNS
We used reverse-phase HPLC with electrochemical detection to determine whether infection with H5N1 affected levels of NE, 5-HT, and its metabolite 5-HIAA in the substantia nigra, striatum, brainstem, cortex, and hippocampus.
In striatum, we observed a doubling of striatal NE at 10 d after infection, which resolved to baseline levels by 60 d after infection (Fig. 3). Unlike NE, 5-HT levels dropped by ∼60% and remained low through 90 d after infection. A similar reduction in 5-HIAA was detected (Fig. 4).

Percentage change in the amount of NE in brain following H5N1 infection. No significant changes were seen in NE levels in striatum, substantia nigra, or brainstem. In hippocampus, NE levels significantly increased at day 60 after infection, and returned to basal levels at 90 d after infection. The cortex showed a significant loss of NE at 10 d after infection, which returned to baseline by 60 d after infection. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls post hoc tests (n = 5 for each condition, *p ≤ 0.05, **p ≤ 0.001 compared with control mice). Error bars indicate SEM.

Percentage change in 5-HT and 5-HIAA in brain following H5N1 infection. The level of 5-HT was significantly reduced in the striatum, substantia nigra, and cortex starting at day 10 after infection and remained reduced through day 90 after infection. A similar reduction pattern was seen in the level of 5-HIAA, although there was a return to baseline levels in the substantia nigra. Although the mean levels of 5-HT and 5-HIAA in hippocampus and brainstem trended lower, none of these changes achieved statistical significance. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls post hoc tests (*p ≤ 0.05, **p ≤ 0.001 vs control mice). Error bars indicate SEM.
In substantia nigra, we measured a slight increase in NE at 10 d after infection, although this change did not reach statistical significance (Fig. 3). In regard to 5-HT, we observed a 50% decrease that stayed significantly below control levels through 90 d after infection (Fig. 4). A similar reduction was observed in 5-HIAA levels, although by 90 d after infection, these levels returned values similar to those of control animals (Fig. 4).
In hippocampus, no changes in NE levels were seen until 60 d after infection, where we observed a 75% increase in NE (Fig. 3), which returned to baseline levels at 90 d after infection. No statistically significant changes were seen in 5-HT or 5-HIAA levels following H5N1 infection compared with control mice (Fig. 4).
In cortex, we measured a significant 70% reduction in NE at 10 d after infection, which recovered to control levels by 60 d after infection (Fig. 3). In regard to 5-HT, we observed a 90% decrease, which stayed significantly below control levels through 90 d after infection. A similar reduction was observed in 5-HIAA levels (Fig. 4).
No changes in NE, 5-HT, or 5-HIAA were detected in brainstem.
H5N1 infection increases the number of microglia in the SNpc
Microglia, the resident immune cells of the CNS, are derived from cells in the monocyte lineage (Hickey and Kimura, 1988; Simard and Rivest, 2004). In surveillance mode, they are said to be resting (Gehrmann et al., 1993; Raivich, 2005) and have a characteristic histological appearance with long slender tendrils emanating from their bodies (Fig. 5A). Once exposed to infection, injury, or trauma (Harry and Kraft, 2008), they undergo a transformation in which they retract and thicken their processes and assume a more amoeboid morphology (Fig. 5B) (Graeber and Streit, 2010). To determine whether exposure to H5N1 alters the morphology and number of microglia in the SNpc, we used the optical fractionator to assess the number of resting and activated microglia. Control C57BL/6 mice administered saline intranasally were found to have ∼7500 total Iba-1-positive microglia in the SNpc. Of these, ∼10% were structurally classified as activated, while 90% were classified as resting. Sixty days after intranasal inoculation of H5N1, we found a 67% increase in total microglial number. Examination of microglial subtype revealed a 300% increase in activated microglia and a 33% increase in resting microglia. From day 60 to 90, this number of microglia, as well as the percentage of activated and resting microglia remained constant (Fig. 5C). This suggests that neurotropic influenza exposure in the brain induces a long-term, if not permanent, increase in activated microglia.

H5N1 infection increases the number of activated microglia in the SNpc. A, B, Photomicrographs showing the typical appearance of resting (A) and activated microglia (B). Both resting and activated microglia were observed in the substantia nigra of noninfected control and H5N1-infected mice. C, The number of activated microglia increased approximately threefold in the H5N1-infected group, compared with the control group. The number of microglia also increased ∼67% in the H5N1-infected group, compared with the control group. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls post hoc tests (n = 4 for each group, **p < 0.001 vs control mice). Error bars indicate SEM.
Effect of H5N1 infection on levels of cytokines and chemokines in the lung and CNS
Activated microglia have been shown to produce a variety of cytokines, chemokines, and growth factors following exposure to infection as well as other insults to the CNS (Hirsch et al., 2003; Kim and Joh, 2006; Tansey and Goldberg, 2010). This “inflammatory response” has been shown to be varied and specific to the type of insult (Perry et al., 2003). Some cytokines (IL-1α, IL-1β, IL-2, IL-9, IL-12, IFN-γ, and TNF-α) function primarily to induce inflammation (proinflammatory), while others (IL-6, IL-10, and IL-13) suppress inflammation (Opal and DePalo, 2000). A class of cytokines function as chemokines, which act as chemoattractants and include eotaxin, KC, IP-10, MCP-1, MIP-1α, and MIP-1β (Fernandez and Lolis, 2002), while others can act as growth and differentiation factors (GM-CSF, M-CSF, and VEGF) (Metcalf, 1985). In this study, we examined cytokine, chemokine, and growth factor profiles in regions of the CNS infected by the H5N1 virus, both during (day 0–21) and after (day 60–90) the acute infectious stage (Jang et al., 2009b). To determine whether any alterations were specific to the CNS or were in response to, or coincident with, humoral activation of cytokines, we also measured these proteins in lung, which is the primary site of H5N1 influenza infection in mice (Jang et al., 2009b) and, traditionally, in humans (Yuen and Wong, 2005).
Examination of cytokines expression profiles in tissues generally demonstrated the following four distinct profiles of induction: in Pattern 1, proteins were transiently increased during the initial phase of infection through day 10 after infection and then returned to baseline levels; in Pattern 2 an initial decrease in expression was followed by a continued loss or a return to baseline levels; in Pattern 3, cytokine/chemokine expression demonstrated an initial transient increase followed by a return to baseline levels and then a reinduction at times after the influenza virus was no longer detectable by immunohistochemical methods (visualization of H5N1 influenza nucleoprotein); and Pattern 4 of cytokine/chemokine expression involved no changes during the active phase of infection (through day 10 after infection), but later, induction was detected. Examples of these patterns of expression are seen in Figure 6A–D.

Patterns of cytokine, chemokine, and growth factor expression observed following H5N1 infection. The following four distinct temporal patterns were observed in the expression of cytokines, chemokines, and growth factors along the time course after H5N1 infection: Pattern 1, a transient increase at initial phase of infection followed by a return to basal levels; Pattern 2, an initial transient decrease in expression followed by a return to baseline levels; Pattern 3, an initial transient increase in expression followed by a return to baseline levels and then a reinduction at day 60 after infection (dpi); and Pattern 4, no changes during the active phase of infection, but induction after day 60 after infection.
In the lung, most of the proinflammatory cytokines and chemokines displayed one of these four patterns. One (IL-9) sustained a decrease in expression, a unique pattern not seen in any of the brain regions. We saw that some of these proteins followed Pattern 1, transiently increasing expression during the initial phase of infection (through day 21 after infection) and then returning or decreasing to baseline levels. The proinflammatory cytokines/chemokines that expressed this profile were IL-6, IL-12, GM-CSF, G-CSF, IFN-γ, IP-10, KC, MCP-1, MIP-1β, MIP-1α, and TNF-α. IL-10, an anti-inflammatory cytokine/chemokine, also expressed this profile. Following Pattern 2, an initial transient decrease in expression followed by a return to baseline levels, were the proinflammatory cytokines/chemokines IL-1β, IL-2, eotaxin, and VEGF. Pattern 3 of cytokine/chemokine expression was also demonstrated in lung. This pattern involves an initial transient increase in expression followed by a return to baseline levels, and then a reinduction after the influenza virus was no longer detectable by immunohistochemical methods (visualization of H5N1 influenza nucleoprotein). The proinflammatory cytokines/chemokines that expressed this profile were IL-1α and M-CSF. Pattern 4 was not seen in the cytokine/chemokine profile in the lung. IL-9 had a unique response: downregulation without return to baseline. IL-13 was not detected in lung (Fig. 7).

Expression of cytokines, chemokines, and growth factors in the lung following intranasal H5N1 infection. Two distinct patterns were observed in the lung. The proinflammatory cytokines, chemokines, and growth factors IL-6, IL-12, G-CSF, GM-CSF, IFN-γ, KC, MIP-1α, MIP-1β, and TNF-α, and anti-inflammatory IL-10 increased during the initial phase of infection (through day 10 after infection) and then returned to baseline levels (Pattern 1). The proinflammatory cytokine IL-2 showed an initial decrease in expression at 10 d after infection followed by a return to baseline levels (Pattern 2). The growth factor VEGF showed an initial transient increase followed by a decrease below the baseline levels and then reinduction at 60 d after infection (Pattern 3). Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls test. Cytokine levels (picograms per milliliter) are presented as mean ± SEM (n = 4 for each group and time point, *p ≤ 0.05, **p ≤ 0.001 vs control baseline).
In the CNS, we examined the expression of cytokines and chemokines in four separate regions: brainstem (Fig. 8), substantia nigra (Fig. 9), striatum (Fig. 10), and cortex (Fig. 11).

Expression of cytokines, chemokines, and growth factors in the brainstem following intranasal H5N1 infection. Three distinct patterns were observed in the brainstem. The proinflammatory cytokines, chemokines, and growth factors IL-1α, IL-12(p70), IL-13, eotaxin, G-CSF, GM-CSF, IP-10, KC, M-CSF, MCP-1, MIP-1α, MIP-1β, and TNF-α, and anti-inflammatory IL-10 increased during the initial phase of infection (through day 10 after infection) and then returned or decreased to baseline levels (Pattern 1). The proinflammatory cytokines IL-2 and IL-9 showed an increase, return to baseline, and re-expression (Pattern 3), while the proinflammatory cytokines, chemokines, and growth factors, including IL-1β, IL-2, and VEGF, did not show changes during the active phase of infection, but later displayed induction (Pattern 4). Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls test, and cytokine levels (picograms per milliliter) are presented as mean ± SEM (n = 4 for each group and time point, *p ≤ 0.05, **p ≤ 0.001 vs baseline control).

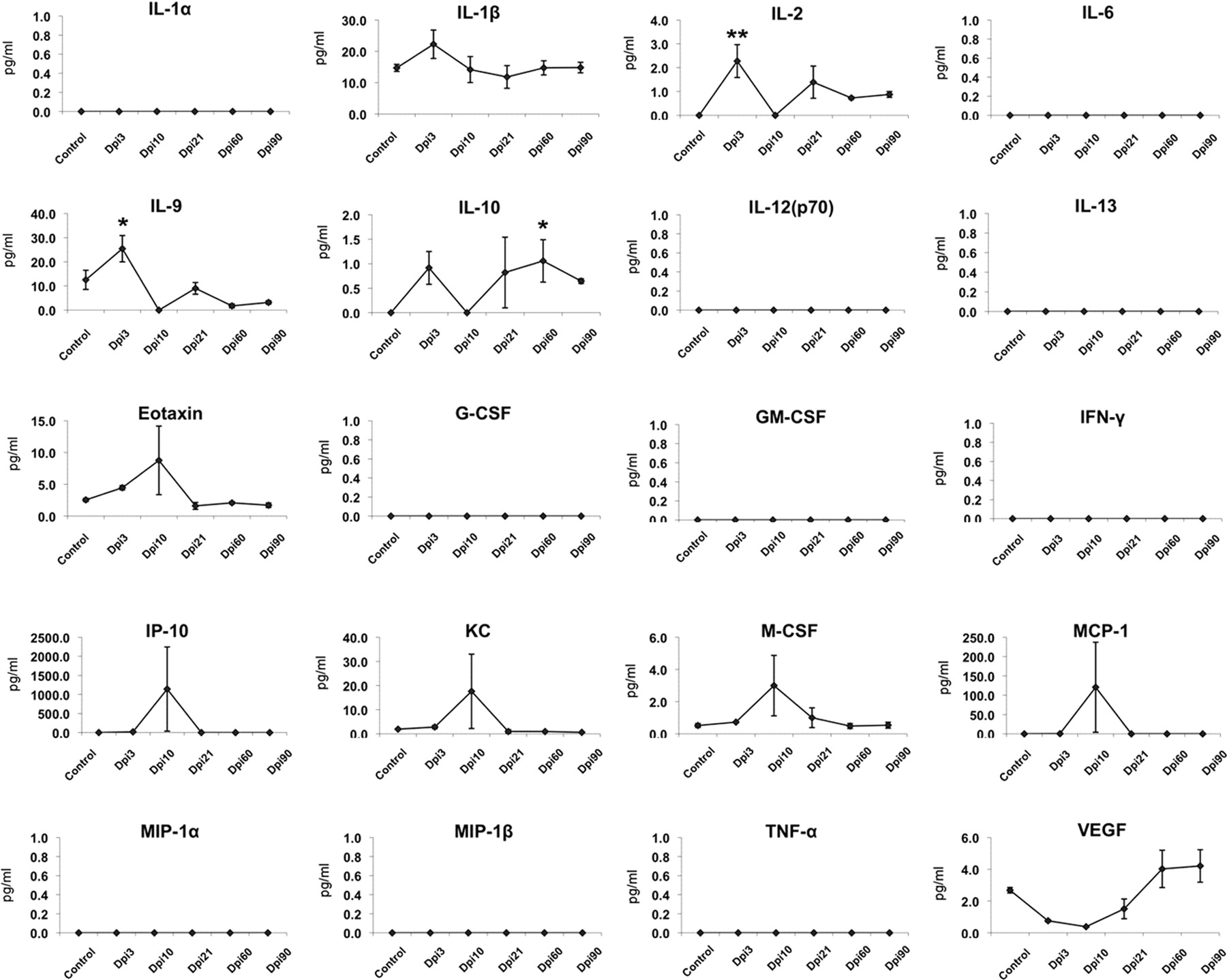
Expression of cytokines, chemokines, and growth factors in the substantia nigra following intranasal H5N1 infection. In substantia nigra, the proinflammatory cytokines/chemokines IL-1β, IL-2, IL-6, G-CSF, M-CSF, and MCP-1 increased during the initial phase of infection (through day 10 after infection) and then returned or decreased to baseline levels (Pattern 1). The proinflammatory cytokine IL-13 initially increased and then returned to baseline levels before reinduction at 60 d after infection (Pattern 3). The proinflammatory chemokine MIP-1β and growth factor GM-CSF did not show changes during the active phase of infection (through day 10 after infection), but later displayed induction (Pattern 4). Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls test, and cytokine levels (picograms per milliliter) are presented as mean ± SEM (n = 4 for each group and time point, *p ≤ 0.05,**p ≤ 0.001 vs baseline control).

Expression of cytokines, chemokines, and growth factors in the striatum following intranasal H5N1 infection. In striatum, the proinflammatory chemokines and growth factors eotaxin and M-CSF increased during the initial phase of infection (through day 10 after infection) and then returned to baseline levels (Pattern 1). The proinflammatory cytokine IL-2 initially increased and then returned to baseline levels, after which it reinduced at 60 d after infection (Pattern 3). The anti-inflammatory cytokine IL-10 did not show changes during the active phase of infection, but later displayed induction (Pattern 4). Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls test, and cytokine levels (picograms per milliliter) are presented as mean ± SEM (n = 4 for each group and time point, *p ≤ 0.05,**p ≤ 0.001 vs baseline control).

Expression of cytokines, chemokines, and growth factors in the cerebral cortex following intranasal H5N1 infection. In cortex, the proinflammatory cytokines, chemokines, and growth factors IL-β, IL-9, eotaxin, IP-10, KC, M-CSF, and MCP-1 increased during the initial phase of infection (through day 10 after infection) and then returned or decreased to baseline levels (Pattern 1). The proinflammatory cytokine IL-2 and anti-inflammatory IL-10 initially increased, returned to baseline levels, and then reinduced at 60 d after infection (Pattern 3). The proinflammatory growth factor VEGF did not show changes during the active phase of infection, but later displayed induction (Pattern 4). IL-1α, IL-6, IL-12, IL-13, G-CSF, GM-CSF, IFN-γ, MIP-1α, MIP-1β, and TNF-α were not detected in cortex following exposure to influenza. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls test, and cytokine levels (picograms per milliliter) are presented as mean ± SEM (n = 4 for each group and time point, *p ≤ 0.05, **p ≤ 0.001 vs baseline control).
In the brainstem, the expression of cytokines, chemokines, and growth factors displayed three of the different patterns described above. The proinflammatory cytokines, chemokines, and growth factors that expressed Profile 1 were IL-1α, IL-12(p70), IL-13, eotaxin, G-CSF, GM-CSF, IP-10, KC, M-CSF, MCP-1, MIP-1α, MIP-1β, and TNF-α. The anti-inflammatory cytokines IL-10 also followed this profile. The level of proinflammatory cytokines and growth factors IL-1β, IL-2, and VEGF followed Pattern 4. They were not changed immediately upon detection of the virus, but increased later when the H5N1 influenza nucleoprotein was no longer evident in the region (Fig. 8).
In the substantia nigra, the expression of cytokines, chemokines, and growth factors displayed Profiles 1, 3, and 4. The cytokines, chemokines, and growth factors that expressed Profile 1 were IL-1β, IL-2, IL-6, G-CSF, M-CSF, IP-10, and MCP-1. Their expression increased at 3 or 10 d after infection and then returned to baseline levels. IL-13 exhibited Pattern 3, with its levels increasing before day 21 followed by a return to baseline and another rise in level later after the active infection (60 d after infection) was over. Levels of the chemokines and growth factors GM-CSF and MIP-1β in the substantia nigra exhibited Pattern 4, where expression did not change immediately upon detection of the virus, but increased later when the H5N1 influenza nucleoprotein was no longer evident in the region. Neither MIP-1α nor TNF-α was detected in the substantia nigra following exposure to influenza (Fig. 9).
In striatum, the expression of cytokines, chemokines, and growth factors displayed Profiles 1, 3, and 4. The chemokines and growth factors that expressed Profile 1 were eotaxin and M-CSF. The cytokine that expressed Profile 3 was IL-2, while the anti-inflammatory IL-10 displayed Profile 4. IL-1α, IL-6, IL-12(p70), IL-13, G-CSF, GM-CSF, IFN-γ, MIP-1α, MIP-1β, and TNF-α were not detected in striatum following exposure to H5N1 influenza (Fig. 10).
In cortex, the expression of cytokines, chemokines, and growth factors displayed Profiles 1, 2, and 4. The proinflammatory cytokines that expressed Profile 1 were IL-2 and IL-9. The growth factor VEGF expressed Profile 2, while an anti-inflammatory cytokine, IL-10, displayed Profile 4. IL-1α, IL-6, IL-12(p70), IL-13, G-CSF, GM-CSF, IFN-γ, MIP-1α, MIP-1β, and TNF-α were not detected in cortex following exposure to influenza (Fig. 11).
Effect of dopamine on cytokine expression in substantia nigra cell culture
The reinduction of cytokine/chemokine expression between 60 and 90 d, long after the H5N1 virus was absent from the CNS, appeared to coincide temporally with the re-expression of TH in the substantia nigra and the appearance of dopamine in the striatum. To determine whether some of the inflammatory response was induced by this reintroduction of dopamine to the basal ganglia, we examined the pattern of cytokine expression using an in vitro postnatal substantia nigra culture system (Smeyne and Smeyne, 2002). We allowed substantia nigra cultures to stabilize in a 5% O2/5% CO2 environment for 1 week and then added either 500 nm or 5 μm dopamine to each culture. Supernatants were taken at 8, 24, and 48 h after the addition of dopamine and 22 cytokines/chemokines were measured. Of the 22 cytokines/chemokines measured, 12 were detected in the supernatants (Fig. 12). The addition of dopamine increased expression of 4 of the 12 detected cytokine/chemokines, including IL-1β (24 h after addition of 5 μm dopamine), TNF-α (8 h after 500 nm dopamine), IL-13 (8 h after 500 nm dopamine), and GM-CSF (24 h after at 5 μm dopamine).

Expression of cytokines, chemokines, and growth factors at 8, 24, and 48 h following addition of dopamine to primary substantia nigra cultures. In primary substantia nigra cultures, addition of 5 μm dopamine results in a significant increase in IL-1β; addition of 500 nm dopamine results in a significant increase in TNF-α and IL-13. Statistical significance was determined by one-way ANOVA followed by Student–Newman–Keuls test, and are presented as mean ± SEM (n = 4 for each group and time point, *p < 0.05, vs baseline control).
Discussion
The A/VN/1203/04 strain of the H5N1 influenza virus induces both short-term and long-term effects in the CNS of mice. During the acute phase of the infection, which lasts through day 10 after infection (Jang et al., 2009b), the virus induces in the basal ganglia a transient loss of the dopaminergic neurotransmitter phenotype. However, over a period of 80 d from P10 to P90, there is a steady recovery of both TH+ substantia nigra neurons and striatal dopamine. Because we saw no evidence of any significant cell death in the substantia nigra, as judged by immunostaining with activated caspase-3 or TUNEL, nor any expression of the mitotic marker Ki-67, we feel that the loss and subsequent recovery was due to injury to the biosynthetic machinery involved in dopamine production rather than a direct influenza-induced cell death and subsequent neuron regeneration. The recovery of dopamine following transient depletion has been well documented in other models of parkinsonism, including 6-OHDA (Robinson et al., 1994) and MPTP (Bazzu et al., 2010).
In addition to the transient loss of dopamine in substantia nigra and striatum, we also see an acute but long-lasting increase in inflammation within the brain, characterized by an increase in the number of resting and activated microglia and differential expression of a number of cytokines and chemokines. Inflammation in the nervous system has been associated with both the initiation and progression of a number of neurological disorders, including Parkinson's disease (Tansey and Goldberg, 2010). Some of the cardinal pathologies seen in Parkinson's disease include dopaminergic neuronal death in the SNpc (and other regions of the brain), aggregation of ubiquitinated proteins, including α-synuclein (Lewy bodies) throughout the brain, and generalized increases in cerebral oxidative stress (Dauer and Przedborski, 2003). Coincident with these processes, there is an activation of the microglia within the brain. Thus, what remains to be determined is whether the inflammatory reaction induced by H5N1 is a primary defect, occurs in response to initiation of these underlying pathologies, or both. There is significant support of the first possibility. For example, increases in microglial number, morphology, and production of cytokines have been shown to occur in response to overexpression of α-synuclein (Zhang et al., 2005; Su et al., 2008; Reynolds et al., 2009; Sanchez-Guajardo et al., 2010), death of neurons (Minghetti et al., 2005), and increased oxidative stress (Kuhn et al., 2006; Hu et al., 2010; Rojo et al., 2010). Others have shown that activation of the immune system is likely a predisposing factor that contributes to the initiation and progression of these pathologies (Gao et al., 2008; Hu et al., 2008; Liu et al., 2009; Peng et al., 2009).
In this study, we find that microglia become activated coincident with the presence of the H5N1 virus, and it is only after the microglia become activated that we observe loss of the dopaminergic cellular phenotype (lack of TH production), aggregation of phosphorylated α-synuclein (Jang et al., 2009b), and induction of cell death. The rapid microglial activation following peripheral inoculation of the influenza virus raises an important question: is the initial microglial activation due to the direct sensing of the virus within the brain parenchyma or does it result from signals initiated outside of the brain, i.e., response to induction of a “cytokine storm” in lung? Support for a direct innate effect comes from studies demonstrating that both microglia (Wang et al., 2008) and neurons (Yao et al., 2008) are capable of directly interacting with the hemagglutinin protein on the surface of the influenza virus by binding to sialic acid-α2,3-galactose receptors, where they can induce inflammatory cytokine production (Yokota et al., 2000; Wang et al., 2008). Other studies, including studies of the pandemic 1918 H1N1 influenza, lend support for an indirect adaptive mechanism involving a humoral–microglial interaction (Tumpey et al., 2005; de Jong, 2008). The increased presence of activated T-cells in the brains of Parkinson's disease patients also supports this possible mode of humoral–innate cross talk (Appel et al., 2010). In addition to interactions with the physical influenza viral protein, it is also possible that circulating activated T-cells previously exposed to the H5N1 influenza virus may secrete a soluble factor (chemokines/cytokines) that induces microglial activation through signaling of the B7, CD-40, and CD-23 pathways (Yong and Marks, 2010) without the presence of viral particles.
Based on our findings of virus in the brain and probable interactions with circulating T-cells, we suggest that a combination of mechanisms play a role. We find that cytokine expression, a measure of immune activation/response, in most cases closely follows a pattern initiated in lung, including expression of IL-1α, IL-1β, IL-6, eotaxin, G-CSF, IFN-γ, IP-10, KC, MCP-1, MIP-1α, MIP-1β, TNF-α, and VEGF, all acutely, and GM-CSF, with a delayed response, and thus are likely to be, at least in part, a response in brain from a lung-initiated cytokine storm. However, there are clear examples, including those involving IL-9, IL-12, and IL-13, where cytokine expression in the brain has no apparent relationship to the response observed in lung. In substantia nigra, which contains cells especially susceptible to oxidative stress (Jenner, 1998; Yokoyama et al., 2008) and is the brain region most affected by Parkinson's disease in humans (Parent and Parent, 2010), the pattern of activation of IL-13 and GM-CSF were particularly interesting. IL-13 showed a rapid induction followed by return to baseline levels and then a reinduction long after H5N1 virus, marked by the absence of H5N1 nucleoprotein expression, is absent from the brain. GM-CSF was never induced until 60 d after H5N1 infection. The stimulus for this cytokine reinduction is unknown. However, we find it interesting that the timing of cytokine reinduction closely approximates the timing of TH re-expression in both the substantia nigra and striatum. A comparison of our in vivo and in vitro data shows that IL-13 and GM-CSF are induced by addition of dopamine in substantia nigra culture supernatants. We also found a rapid increase in TNF-α in our cultures, although we were not able to detect this in the tissue samples. Several studies have examined the role of dopamine in microglial activation and found that dopamine can act as an inducer of oxidative stress via mechanisms that include dopamine quinone formation (Smith et al., 1994; Kuhn et al., 2006; Mastroeni et al., 2009; Gonçalves et al., 2010) and microglia cell activation (Gonçalves et al., 2010). Thus, at least for a subpopulation of the measured cytokines, our results support the hypothesis that dopamine itself may induce an inflammatory reaction and perhaps even be a factor in Parkinson's disease progression.
Based on our previous study (Jang et al., 2009b), we found that the regions of the CNS affected the earliest and most severely by H5N1 influenza infection were the brainstem and midbrain. Each of these subdivisions contains structures that degenerate in Parkinson's disease, including locus ceruleus and substantia nigra, respectively. Based on our cytokine analysis, these regions also show the largest cytokine inductions. In these regions, 16 of the 22 cytokine/chemokines analyzed show at least a twofold increase in protein levels at the peak of expression, while 9 of these had a change of at least 500%. The largest increases were in IL-6 (500% brainstem, 400% substantia nigra), IL-13 (500% brainstem, 800% substantia nigra), eotaxin (500% brainstem, 200% substantia nigra), G-CSF (1000% brainstem, 8000% substantia nigra), IFN-γ (6000% brainstem and substantia nigra), IP-10 (100,000% brainstem, 500,000% substantia nigra), KC (5000% brainstem, 10,000% substantia nigra), MCP-1 (60,000% brainstem and substantia nigra), and MIP-1α (6000% brainstem and substantia nigra). Additionally, the brainstem was the only place in the CNS that we were able to still see an increase in TNF-α (100% brainstem) when examined 10 d after the infection, even though this inflammatory protein is usually downregulated soon after the initial insult (Ghoshal et al., 2007).
The source of the secreted cytokines/chemokines/growth factors in brain is critical to understanding the process of inflammation following influenza infection. We clearly see an increase in the number of activated microglia, and these have been shown to secrete a number of cytokines when challenged by bacterial or viral infection (Hayashi et al., 1995; Persidsky et al., 1999; Wang et al., 2008). Additionally, these cells secrete other factors (MCP-1, MIP-1α) involved in macrophage homing (Carr et al., 1994; Menten et al., 2002) and IFN-γ (Dufour et al., 2002), which has been shown to inhibit viral replication, possibly part of the brain's innate neuroprotective mechanism.
Our findings therefore suggest that any neurotropic influenza virus that activates the immune system in the brain, whether directly or indirectly, could contribute to CNS disorders of protein aggregation; and more generally that viruses may be an important etiological agent in the developmental sequelae of neurodegenerative diseases, including Parkinson's disease. Our studies and the aforementioned articles from other laboratories suggest that activation of an immune response may be a precipitating factor for developing later parkinsonism. Our work using the A/VN/1203/04 H5N1 influenza virus shows that this strain can get into the CNS and activate the innate immune system. This is sufficient to induce transient damage to neurons as well as mediate long-term changes (deposition of phosphorylated α-synuclein and permanent activation of microglia), but is insufficient alone to cause Parkinson's disease. Further work on the role of influenza as well as immune system modulation by viruses will be necessary to determine whether modulation of these changes can alter the pathogenesis of this disease.
Footnotes
This project was funded in part by the Michael J. Fox Foundation for Parkinson's Research (R.J.S.); the National Institute of Neurological Disease and Stroke, National Institutes of Health (NIH) Grant NS058310 to R.J.S.; the National Institute of Allergy and Infectious Diseases, NIH, Department of Health and Human Services, under contract No. HHSN266200700005C to R.W.; and by the American Lebanese Syrian Associated Charities.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Richard Jay Smeyne, Department of Developmental Neurobiology, St. Jude Children's Research Hospital, 262 Danny Thomas Place, Memphis, TN 38015. Richard.smeyne{at}stjude.org





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}