
Abstract
In developing peripheral nerves, differentiating Schwann cells sort individual axons from bundles and ensheath them to generate multiple layers of myelin. In recent years, there has been an increased understanding of the extracellular and intracellular factors that initiate and stimulate Schwann cell myelination, together with a growing appreciation of some of the signaling pathways involved. However, our knowledge of how Schwann cell growth is regulated during myelination is still incomplete. The mammalian target of rapamycin (mTOR) is a core kinase in two major complexes, mTORC1 and mTORC2, that regulate cell growth and differentiation in a variety of mammalian cells. Here we show that elimination of mTOR from murine Schwann cells prevented neither radial sorting nor the initiation of myelination. However, normal postnatal growth of myelinating Schwann cells, both radially and longitudinally, was highly retarded. The myelin sheath in the mutant was much thinner than normal; nevertheless, sheath thickness relative to axon diameter (g-ratio) remained constant in both wild-type and mutant nerves from P14 to P90. Although axon diameters were normal in the mutant at the initiation of myelination, further growth as myelination proceeded was retarded, and this was associated with reduced phosphorylation of neurofilaments. Consistent with thinner axonal diameters and internodal lengths, conduction velocities in mutant quadriceps nerves were also reduced. These data establish a critical role for mTOR signaling in both the longitudinal and radial growth of the myelinating Schwann cell.
Introduction
Myelinating Schwann cells in the mammalian peripheral nervous system (PNS) reach the promyelinating stage when they have adopted a one-to-one relationship with axons after sorting them to the periphery of axon bundles (Peters and Muir, 1959; Jessen and Mirsky, 2005; Sherman and Brophy, 2005). Schwann cell proliferation during development is required for individual axon ensheathment and is stimulated by axonal neuregulin (Nrg1), acting via ErbB2/3 receptors (Martin and Webster, 1973; Webster et al., 1973; Wood and Bunge, 1975; Salzer et al., 1980; Stewart et al., 1993; Morrissey et al., 1995; Riethmacher et al., 1997). The Nrg1/ErbB system is also a key regulator of myelination and remyelination after injury (Michailov et al., 2004; Nave and Salzer, 2006; Newbern and Birchmeier, 2010; Fricker et al., 2011), although it is not essential for maintenance of the mature sheath (Fricker et al., 2011).
ErbB receptors can act through a variety of cytoplasmic signaling pathways. In Schwann cells, they activate the phosphatidylinositol-3-kinase (PI3K)/Akt pathway (Maurel and Salzer, 2000; Li et al., 2001). Phosphatidylinositol 3,4,5-trisphosphate, the product of PI3K, is hydrolyzed by the lipid phosphatase and tensin homolog (PTEN), and ablation of the gene causes hypermyelination in both the CNS and PNS (Goebbels et al., 2010). In the PNS, Dlg1 is believed to stabilize PTEN, thus potentiating the ability of PTEN to modulate myelin growth (Cotter et al., 2010). Nevertheless, overexpression of a constitutively active form of Akt enhances myelination in the CNS but has no effect on PNS myelination (Flores et al., 2008).
The mammalian target of rapamycin (mTOR) kinase is a downstream effector of PI3K signaling and functions in two major complexes, mTORC1 and mTORC2, to regulate cell growth and differentiation in a variety of mammalian cells (Zoncu et al., 2011). There is considerable evidence that mTOR has a role in regulating myelination in the CNS (Bibollet-Bahena and Almazan, 2009; Narayanan et al., 2009; Tyler et al., 2009, 2011; Goebbels et al., 2010). The mTORC1 and mTORC2 complexes include the specific regulatory proteins raptor and rictor, respectively, and act through distinct if overlapping signaling pathways (Zoncu et al., 2011). Although once considered to be rapamycin-insensitive, mTORC2 can be inhibited by long-term treatment with rapamycin, and this occurs in oligodendrocytes in culture (Sarbassov et al., 2006; Tyler et al., 2011). Nevertheless, it is not yet known whether rapamycin treatment of intact animals inhibits both complexes in oligodendrocytes, or whether the effects observed in the CNS are the consequence of influencing mTOR function in both neurons and glia.
Here, we have used a conditional gene inactivation approach to ask whether ablating mTOR function specifically in Schwann cells influences their ability to myelinate. Thus, we eliminate uncertainty over the target cell and can be sure that both mTORC1 and mTORC2 are disrupted. Further, rictor can still function outside the TORC2 complex, where it can stimulate phosphorylation at S473 of Akt by interacting with integrin-linked kinase (ILK) (McDonald et al., 2008). We show that in mutants, the myelin sheath is thin, Schwann cells do not elongate normally, and the radial growth of axons is compromised with collateral effects on axon diameter and nerve function. We propose that signaling through the mTOR complexes has a critical role in regulating both myelin thickness and overall Schwann cell growth during myelination.
Materials and Methods
Mice and genotyping.
All animal work conformed to UK legislation (Scientific Procedures) Act 1986, and to the University of Edinburgh Ethical Review Committee policy. Cnp1-Cre mice have been described previously and have been shown to be effective in promoting Cre-mediated recombination in mouse embryonic peripheral nerves (Lappe-Siefke et al., 2003; Grove et al., 2007). Mice carrying an mTOR floxed allele where one LoxP site was upstream of the promoter and the second site was in the intron preceding exon 6 and their use to inactivate mTOR in skeletal muscle have been described previously (Gangloff et al., 2004; Risson et al., 2009). Both Cre and floxed mouse lines were back-crossed to a minimum of F8 with C57BL/6 mice. Genotyping of the recombined allele by PCR used the primers 5′-TTCATTCCCTTGAAAGCCAGTCTCACC-3′ and 5′-TCATTACCTTCTCATCAGCCAGCAGTT-3′, and was predicted to yield a product of 552 bp from the recombined allele (Gangloff et al., 2004).
Histology, immunostaining, and Western blotting.
The perineurium was removed from all peripheral nerves before immunostaining of teased fibers or Western blotting. Further preparation and the method for immunostaining of cryosections or teased fiber preparations was as described previously (Sherman et al., 2005; Grove et al., 2007). Primary antibodies were rabbit unless specified otherwise and were used at the following dilutions for immunostaining or Western blotting, as indicated; all were from Cell Signaling Technology unless otherwise noted: anti-mTOR (catalog no. 2983, 1:200 for immunostaining, 1:1000 for Western blotting), anti-phosphoS235/236-S6 (catalog no. 4858, 1:100 for immunostaining, 1:1000 for Western blotting), anti-S6 (catalog no. 2217, 1:1000), anti-S473 phospho-Akt (catalog no. 4058, 1:1000), anti-myelin-associated glycoprotein (anti-MAG; 1:3000; a gift from Dr. David Colman, McGill University, Montreal, Quebec, Canada), mouse monoclonal anti-S473 phospho-Akt (catalog no. 4051, 1:1000), anti-T308 phospho-Akt (catalog no. 2965, 1:1000), anti-Akt (catalog no. 9272, 1:1000), anti-phospho T37/46–4E-BP1 (catalog no. 2855 1:1000), anti-4E-BP1 (catalog no. 9644, 1:1000), anti-Krox20 (1:100 for immunostaining, 1:500 for Western blotting; Covance), anti-Nrg1 (catalog no. SC-348, 1:300; Santa Cruz Biotechnology); anti-Sox10 (1:1000; a gift from Dr. Michael Wegner, University of Erlangen, Bavaria, Germany), chicken anti-Po (1:200 for immunostaining, 1:2000 for Western blotting; Aves Labs), mouse monoclonal anti-DLG1 (1:1000; Enzo Life Sciences), anti-radixin (raised against the peptide DKYKTLRQIRQGNTK, 1:400), mouse monoclonal anti-neurofilament H, SMI-34 (1: 1000; Covance) and SMI-32 (1:400; Covance). All other primary and secondary antibodies and nuclear stains were described previously (Sherman et al., 2005; Grove et al., 2007). Conventional fluorescence microscopy was performed using an Olympus BX60 microscope and images were captured using a Hamamatsu Orca-ER camera and Improvision Openlab software. For confocal microscopy, we used a Leica TCL-SL confocal microscope with either a 40× or a 63× objective, 1.4 NA, and Leica proprietary software. The acquired stacks were assembled using the maximum projection tool and all figures were prepared using Adobe Photoshop CS4 extended version 11. Western blotting was performed as described previously (Sherman et al., 2005) on P21 sciatic nerve lysates containing phosphatase inhibitors. For electron microscopy, mice were perfused intravascularly with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4. Nerves were removed, fixed for 2 h at room temperature then for 18 h at 4°C in the same fixative, postfixed in OsO4, and embedded in Araldite. Ultrathin sections were stained with uranyl acetate and lead citrate and examined on a Phillips BioTwin CM120 electron microscope equipped with either a Gatan Orius or an Olympus Morada digital camera.
Electrophysiology and rotarod.
Acutely prepared quadriceps nerves, taken from 42-day-old control and mutant mice, were placed in oxygenated mammalian HEPES physiological solution (137 mm NaCl, 5 mm KCl, 2 mm CaCl, 1 mm MgCl, 5.5 mm d-glucose, and 5 mm HEPES, pH 7.2–7.4) and nerve conduction velocities measured as described previously (Court et al., 2004). Rotarod analysis of mice at P42 (minimum of three per group) was performed at two different speeds, 24 and 32 rpm, as previously described (Court et al., 2004), except mice were conditioned for 2 d.
Morphometry.
Quantitation of the relative numbers of Sox10- and Krox20-positive Schwann cells at P21 was performed on longitudinal cryosections of quadriceps nerves and a minimum of 300 Sox10-positive Schwann cells was counted in each of two animals per condition. Quantitation of the absolute numbers of Sox10-positive Schwann cells in the quadriceps nerves from control and mutant animals was performed on cross-sections and a minimum of five sections per animal (five animals per genotype) were counted. Cross-sectional areas of 0.5 μm toluidine blue-stained sections of nerve, excluding the perineurium, were measured using ImageJ after construction of montages of images in Photoshop captured at 100×. The numbers of sorted, promyelinated, and myelinated axons were quantitated in cross-sections using montages of electron micrographs assembled in Photoshop. A minimum of 200 axons were counted per animal, with a minimum of three animals per condition. The diameters of axons and axons plus myelin were measured in electron micrographs by first measuring the circumference of each by hand-tracing using ImageJ, then by calculating areas from which diameters were derived. Their g-ratios were calculated from these diameters. A minimum of 100 axons was counted per animal and a minimum of three animals was used per condition. Internodal lengths were measured as described using Openlab (PerkinElmer) (Court et al., 2004).
Statistical analysis.
Statistical analysis was by unpaired t tests with Graphpad Prism 5.0c software.
Results
Conditional inactivation of mTOR in Schwann cells
We generated mice without functional mTOR in Schwann cells as described in Materials and Methods, above, by crossing mice homozygous for a floxed allele of mTOR (Risson et al., 2009) with a Cnp1-Cre driver line specific for myelin-forming glia (Lappe-Siefke et al., 2003). Cre-mediated inactivation of this mTORflox/flox mouse line (control mice or floxed mTOR mice) has previously been shown to abrogate mTOR expression efficiently in skeletal muscle (Risson et al., 2009), and the Cnp1-Cre driver line is effective in Schwann cells during embryonic development before the radial sorting of axons (Grove et al., 2007). Throughout this paper, we refer to mice with the genotype mTORflox/flox/Cnp1-Cre+/− as mTOR mutants. The DNA fragment generated by PCR of peripheral nerve genomic DNA after Cre-mediated recombination showed effective inactivation of the mTOR gene (Fig. 1A). Loss of mTOR in Schwann cells was confirmed by immunofluorescence and the substantial decrease in mTOR protein observed by Western blotting of peripheral nerve lysates (Fig. 1B,C). Persistence of residual mTOR protein in nerve lysates is best explained by the fact that sciatic nerves contain, in addition to Schwann cells, capillary endothelial cells and fibroblasts, and axons (Fig. 1B).

Conditional inactivation of mTOR in Schwann cells. A, PCR of genomic DNA isolated from mTORflox/flox and mutant quadriceps nerves shows efficient recombination in mutant nerves. B, Immunofluorescence analysis of P21 teased individual quadriceps nerve fibers confirms the loss of mTOR from the cytoplasm of Schwann cells in the mutant. The Schwann cell nucleus stained with TOTO-3 is in blue and mTOR is in green. Scale bar, 10 μm. C, Western blot analysis of lysates from P21 sciatic nerve shows that mTOR protein is reduced in the mutant and that the mTOR downstream targets S6 and 4E-BP1 are less phosphorylated. In contrast, there is a substantial increase in phosphorylation at T308 of Akt, and no decrease in phosphorylation at S473 of Akt. Gamma-actin was the loading control. D, Immunofluorescence staining of P21 teased quadriceps nerve fibers confirmed the loss of phospho (P)-S6 in Schwann cells lacking mTOR. Myelin was visualized using antibodies against Po in red, phospho-S6 in green, and Schwann cells nuclei were stained with TOTO-3. Scale bar, 10 μm. E, Nerve conduction velocities in the quadriceps nerves of control and mutant mice are represented in a vertical scatter plot. Each point is the mean of measurements from both nerves from one mouse; eight mice were analyzed. Horizontal lines are the SEMs and p < 0.005. F, Rotarod testing was at two speeds of rotation; mutant mice were significantly worse at both speeds. Values are expressed as mean time to fall ± SEM (n = 6, *p < 0.05, **p < 0.005).
Ribosomal S6 kinase (S6K) is a downstream target of mTORC1 and the reduced phosphorylation of one of its substrates, the ribosomal S6 protein, indicated that the mTORC1 pathway was no longer active (Fig. 1C,D). The percentage of Schwann cells immunopositive for phospho-S6 at P21 was reduced from 100% in the control to 1% in the mutant nerves, which attests to the efficiency of Cnp1-Cre-mediated recombination and ablation of mTOR expression. Phosphorylation of a second well characterized target of mTORC1, the eukaryotic initiation factor 4E-BP1, was also substantially reduced (Fig. 1C). Phosphorylation of Akt at S473 is a downstream target of mTORC2 signaling (Sarbassov et al., 2005), but can also be mediated in an mTOR-independent fashion by the ILK/rictor complex (McDonald et al., 2008). Indeed, we did not observe a decrease in phospho-S473Akt in mutant sciatic nerve lysates (Fig. 1C) and we did observe enhanced phosphorylation at the T308 site (Fig. 1C). Blotting for the latter epitope suggested that there were two species of Akt phosphorylated at T308 that migrated close together (Fig. 1C), and successive incubation with antibodies that recognized S473 then T308 indicated that the major upper band was doubly phosphorylated at both sites, whereas the lower band was singly phosphorylated at T308 (data not shown).
Mutant mice were born in predicted numbers and were no different in weight from controls up to P90. At birth they were indistinguishable from littermates and displayed no overt clinical phenotype. Nevertheless, the nerve conduction velocities of mutant nerves were severely reduced from 38.7 ± 0.7 m/s to 11.7 ± 1.5 m/s (n = 8, ± SEM, p < 0.005; Fig. 1E). Analysis of motor behavior revealed that these reduced conduction velocities were associated with poorer performance in the Rotarod test (n ≥ 3, p < 0.05 at 24 rpm, p < 0.005 at 32 rpm; Fig. 1F).
Loss of mTOR retards myelination
We have previously shown in wild-type nerves that by P3 there is extensive radial sorting with the establishment of a one-to-one relationship of Schwann cells with axons at this so-called promyelination stage (Grove et al., 2007). Radial sorting of axons in mutant nerves was normal at P2 (Fig. 2A); Schwann cell differentiation was also unimpaired, as shown by the normal percentage of Schwann cells that were both Sox10+ and Krox20+ in the mutant at P21 (Fig. 2B,C). The amount of Krox20 protein in P21 sciatic nerve lysates, as assessed by Western blot, was not decreased either; if anything, it was slightly increased (Fig. 2D). In contrast, there was a decrease in the amount of the major peripheral myelin protein P0, suggesting a deficit in myelin production (Fig. 2D). The increase in Krox20 levels was likely due to the fact that there was a 1.9-fold increase in the number of Sox10+ Schwann cells from 38.9 ± 1.1 to 73.1 ± 2.5 in cross-sections of the mutant nerve at P21 (values are means ± SEM, n = 5, p < 0.0001). This increase in Schwann cell number probably also accounts for the fact that there was a slight increase in the amount of the mammalian discs large homolog 1 protein (Dlg1), a proposed negative regulator of myelination (Cotter et al., 2010). Appropriate signaling from the axon was indicated by the fact that Nrg1 levels were not reduced (Fig. 2D). Nevertheless, there was a dramatic reduction in the cross-sectional area of mutant nerves (Fig. 2E), which was first significant at P7 (values are ± SEM, n ≥ 3, p < 0.01 at P7, P14, and P21; p < 0.001 at P42 and P90; Fig. 2F). To account for the reduced size of peripheral nerves in the mutant, we first asked whether myelination occurred normally. Unmyelinated promyelinating figures were more numerous in mutant nerves compared with floxed mTOR mice (Fig. 3A,B), indicating a delay in the progression of myelination. Nevertheless, promyelinating Schwann cells were associated with a wide range of low to large caliber axons (Fig. 3C) and 95% of all sorted axons were ultimately myelinated by P90 (Fig. 3B). Hence, the delay in initiating myelin deposition in a fraction of the sorted axons was not because they were of an unusually small diameter.

Mutant Schwann cells sort axons and differentiate, but their nerves are abnormally small. A, Electron micrographs of cross-sections of quadriceps nerves from floxed mTOR and mutant mice at P2, a time when axon sorting is normally well advanced, showed that formation of 1:1 promyelinating figures was normal in mutant mice. Scale bar, 5 μm. B, C, Immunofluorescence staining of longitudinal sections of quadriceps nerves with antibodies against Sox10 (for all Schwann cells) and Krox20 (transcription factor required for progression beyond the promyelinating stage) and quantitation revealed no difference in double-labeled Schwann cells between floxed mTOR and mutant mice at P21, showing that loss of mTOR did not prevent Schwann cell differentiation. Scale bar, 20 μm. D, Western blot of sciatic nerve lysates showing no decrease in Krox20, Dlg1, or the active cleaved fragment of axonal Nrg1 in the mutant but reduced amounts of the myelin protein P0. Gamma-actin was the loading control. E, F, Electron micrographs of quadriceps nerves at P21 show that the cross-sectional area of mutant nerves is much smaller than normal, which may explain why the density of Schwann cells revealed by Sox10 staining in the mutant shown in C appears to be increased. The decreased cross-sectional area was first significant at P7 and became more pronounced with age (means ± SEM, n ≥ 3, **p < 0.01, ***p < 0.001). Scale bar, 20 μm.
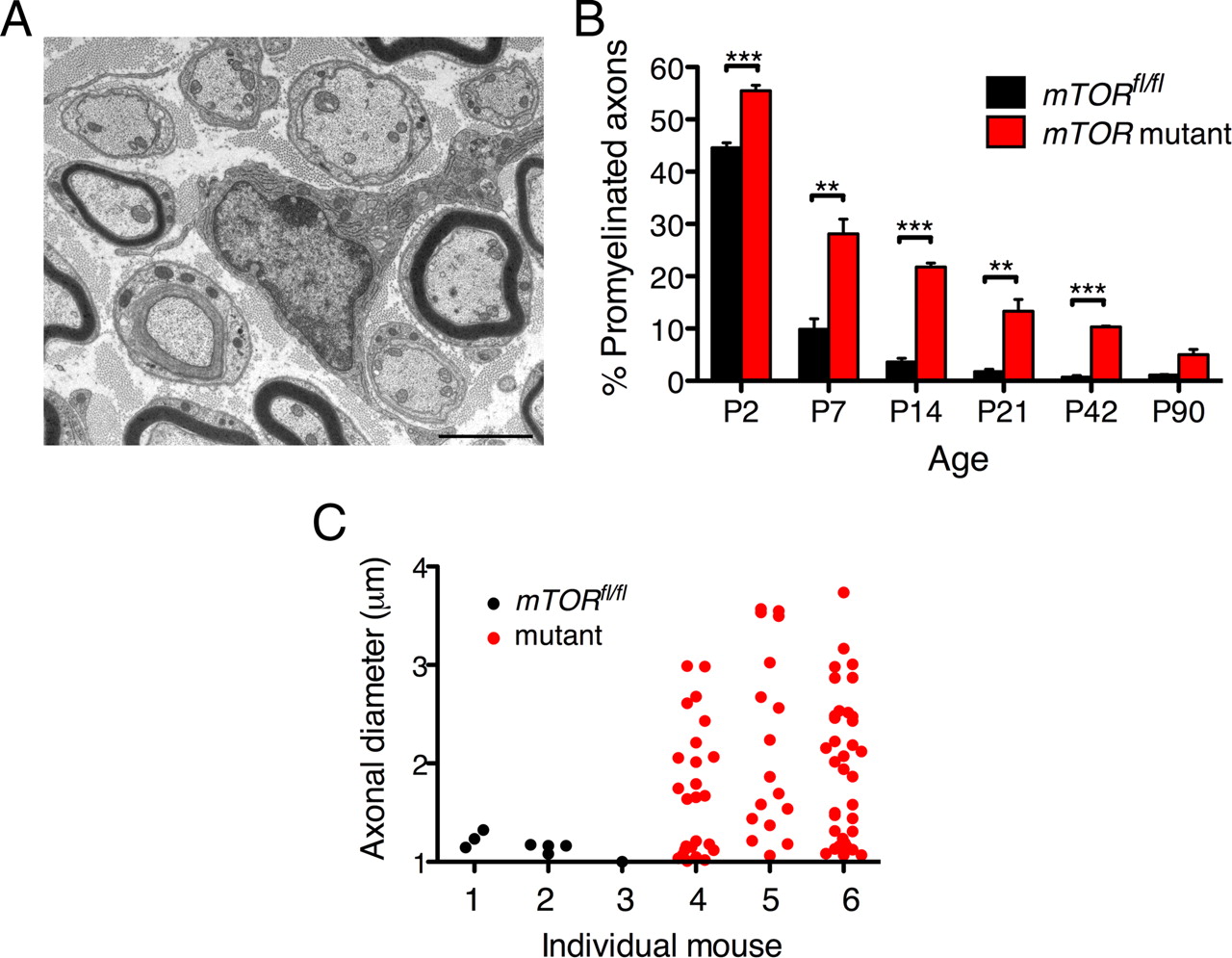
Myelination is delayed in the absence of mTOR in Schwann cells. A, B, Electron microscopy revealed a large number of sorted but unmyelinated, promyelinating profiles at P21. When quantitated, the difference in the number of promyelinated profiles as a percentage of all sorted axons was significantly higher in mutant quadriceps nerves from P2 to P42 (means ± SEM, n ≥ 3, **p < 0.01, ***p < 0.001). However, by P90, the percentage of unmyelinated axons in the mutant had declined to 5%. Scale bar, 2 μm. C, Vertical scatter plots of quadriceps axonal diameters for promyelination-stage axons in three independent control mice (1–3) and three mutant mice (4–6) at P42. All the control values were <1.3 μm. However, in the mutant, many large axons were sorted into a 1:1 relationship with Schwann cells that had still not myelinated (4–6). The median values for animals 4, 5, and 6 were 1.7, 1.9, and 1.9 μm respectively.
Mutant nerves have thinner myelin and reduced axon diameter
The fact that the reduced nerve growth in the mutant only became apparent with age and after the bulk of axon sorting had occurred suggested that either myelination did not proceed normally or radial growth in axon diameter was impaired, or both. Electron microscopy revealed that, consistent with the reduced amount of P0 protein (Fig. 2D), myelin in the mutant was consistently thinner than normal; this aberrant ensheathment persisted from P7 until P90 (Fig. 4). Quantitation of the g-ratio at different ages revealed that the growth of the myelin sheath, relative to axon diameter, was not only retarded (Fig. 5A) but was also arrested (Fig. 5B). Although the mean g-ratio of the mutant nerves was always significantly different to control at each age (p < 0.005; Fig. 5B), there was no significant change in the g-ratio of mutant nerves from P14 to P90, as was the case for controls (Fig. 5B). Hence, similar to the g-ratio during normal myelination, the abnormal ratio in mutant nerves was set at an early age and did not change as the mice matured (Fig. 5B).

Myelin produced by mTOR mutant Schwann cells is thinner than normal and stays thin during development. Electron micrographs of cross-sections of quadriceps nerves shows sorted axons in mutant nerves but thin myelin from P7 to P90. Scale bar, 5 μm.

Mutant nerves have thin myelin and reduced axon diameters. A, B, Quantitation of g-ratios as a function of axon diameter from P14 to P90 indicated strong diversion of the regression lines through the data for control compared with mutant quadriceps nerves, which was confirmed by analysis of mean values ± SEM (n ≥ 3). At each age, there was a significant difference between control and mutant (**p < 0.05, ***p < 0.005). However, there was no significant difference between the g-ratios measured at P14 and P90 for control nerves or between g-ratios measured at P14 and P90 for mutant nerves. C, Panel A indicated that there were fewer large caliber axons in the mutant at older ages. This was confirmed by measuring mean axonal diameters ± SEM (n ≥ 3) for control and mutant. A significant reduction in axonal diameter in the mutant was found from P21 onwards (**p < 0.01).
Plots of g-ratio versus axon diameter (Fig. 5A) indicated that mutant axons were of reduced caliber, although these were not significantly different from controls until P21 (Fig. 5C), a time when >75% of sorted axons in the mutant were already myelinated (Fig. 3B). Reduced axon caliber in dysmyelinating mouse mutants has previously been shown to be attributable to abnormal axonal interactions with ensheathing Schwann cells in the PNS and oligodendrocytes in the CNS and to be associated with reduced neurofilament phosphorylation (de Waegh et al., 1992; Kirkpatrick and Brady, 1994; Brady et al., 1999; Martini, 2001). In agreement with this, we found substantial deficits in the phosphorylation of neurofilament H in the axons of mutants (Fig. 6A). Neurofilament H is known to be crucial in the development of large axons (Elder et al., 1998) and phosphorylation is believed to regulate axonal caliber through its effect on neurofilament packing (Cole et al., 1994). Consistent with this view, large unmyelinated mutant fibers displayed a high density of axonal cytoskeletal elements, including neurofilaments (Fig. 6B). It has been proposed that the MAG might act as a key molecule in signaling between Schwann cells and their ensheathed axons because the peripheral nerves of MAG-deficient mice show reduced axonal calibers, decreased neurofilament spacing, and reduced neurofilament phosphorylation, even though they myelinate normally (Yin et al., 1998). It seems unlikely that deficiencies in MAG-mediated signaling caused reduced axonal caliber, since mTOR mutant peripheral nerves were actually enriched in MAG compared with controls (Fig. 6A). This was likely due to the fact that MAG in peripheral nerves is predominantly localized to noncompact myelin, including the periaxonal membrane apposed to the axon (Trapp and Quarles, 1982).

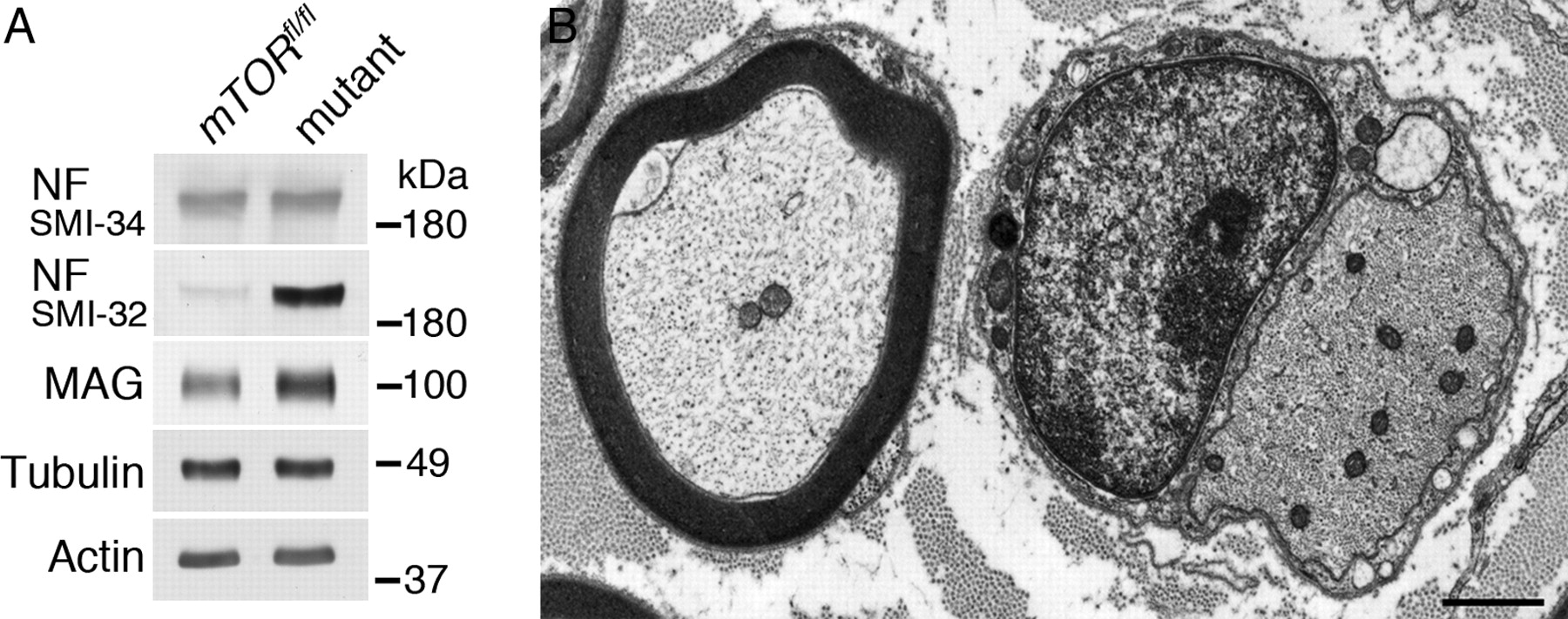
Mutant nerves have reduced neurofilament phosphorylation and high neurofilament packing density. A, At P21, there is a significant difference in axonal caliber between control and mutant quadriceps axons (Fig. 5C). Western blotting of sciatic nerve lysates at the same age showed a major decrease in the extent of neurofilament H phosphorylation, as determined by the antibody SMI-32, which recognizes an epitope normally masked by phosphorylation and which is uncovered on dephosphorylation. Other phosphorylated epitopes recognized by the antibody SMI-34 were unaffected, and MAG was not reduced. Tubulin and actin were loading controls. B, Electron micrograph of mutant quadriceps at P90 shows the increased density within a promyelinating axon (diameter, 3.1 μm beside a myelinated axon of comparable diameter, 3.3 μm). Scale bar, 1 μm.
Aberrant growth including short internodes in the absence of mTOR
In addition to arrested radial growth, mutant Schwann cells also displayed reduced longitudinal growth (Fig. 7A,B). Internodes in mutant quadriceps nerves at P21 showed a wider range of lengths compared with control nerves and mean values were reduced from 488 ± 12.9 μm to 241 ± 17.8 μm (±SEM, n ≥ 3). Consistent with this 50% reduction in intermodal length, there was a 1.9-fold increase in the number of Schwann cells in mutant nerves compared with controls (see Loss of mTOR retards myelination, above). Other derangements, such as myelin outfoldings, were also observed at low frequency in the mutant from P7 onwards but not in controls (Fig. 7C). However, these were not associated with a decrease in Dlg1 expression (Fig. 2D), as has been proposed in other mouse models (Cotter et al., 2010). In unmyelinated, large diameter axons, in addition to some vacuolation and redundant basal lamina, we commonly observed that the axonal membrane was highly irregular in outline (Figs. 6B, 7D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mutant nerves have shorter internodes and aberrant myelin. A, B, Immunofluorescence imaging (A) of a teased mutant fiber using an antibody against radixin (green), which is concentrated at microvilli, and TOTO-3 to visualize Schwann cell nuclei (blue). Arrows indicate the position of nodes of Ranvier. This reveals some variability in internodal length, which is quantitated in the histogram (B) showing the frequency distribution of internodal length in control and mutant quadriceps nerve. Scale bar, 50 μm. C, D, Electron micrographs of aberrant profiles from mutant nerves observed at all ages from P90. Outfoldings of thin myelin are shown in C. Arrows point to redundant basal lamina with vacuolation in the Schwann cell cytoplasm surrounding an unmyelinated axon. Scale bars: C, 2 μm; D, 1 μm.
Discussion
We have shown by inactivating the mTOR gene that mTOR signaling plays a central role in regulating the growth of the myelin sheath in the PNS. Mice with Schwann cells lacking mTOR displayed reduced rates of nerve conduction associated with impaired performance in the rotarod test, which were consistent with a combination of smaller axonal calibers and shorter internodal length (Court et al., 2004). The Cnp1-Cre driver line would also be expected to ablate mTOR expression in oligodendrocytes and the deficits in motor function might also have resulted in derangements in the CNS. However, although beyond the scope of this work, preliminary analysis of the CNS did not reveal the dramatic hypomyelination that we observed in the PNS.
Mutant Schwann cells were able to sort axons from bundles to adopt the 1:1 relationship characteristic of the promyelinating stage and most sorted axons were eventually myelinated. Expression of Krox20, a transcription factor known to be required for progression beyond the promyelinating stage (Topilko et al., 1994), was consistent with cells' ability to proliferate, differentiate, and myelinate. A key feature of the cell biology of mTOR mutant Schwann cells is that they produce thin myelin and short internodes. Further, the fact that the extent of myelin ensheathment relative to axon caliber as measured by the g-ratio does not change in the mutant from P14 to P90 suggests that, as in control animals, the g-ratio is still set at an early stage of myelination. Nevertheless, normal levels of Nrg1 in the mutant suggest that signaling from the axon is not deficient.
Work on the Trembler dysmyelinating mutant has strongly implicated neurofilament phosphorylation and its predicted influence on filament packing density as determining reduced axon caliber, and evidence for a relationship between neurofilament phosphorylation and axon size has also been found in a human peripheral neuropathy, CMT1A (de Waegh et al., 1992; Watson et al., 1994). Schwann cell MAG has been proposed as a key aspect of the signal regulating neurofilament phosphorylation (Yin et al., 1998); however, we did not observe reduced levels of MAG in mTOR mutant nerves.
How might the absence of mTOR influence the growth of Schwann cells during myelination? Well characterized down-stream targets of mTORC1 are the eukaryotic initiation factor 4E-BP1 and the ribosomal S6K, both of which regulate mRNA translation (Russell et al., 2011). Stimulation of TORC1 causes phosphorylation of 4E-BP and the release of a second initiation factor, eIF4E, which promotes translation (Fingar et al., 2002). Activation of TORC1 also activates S6K, which phosphorylates several substrates known to be involved in translation initiation, including a regulatory subunit (eIF4B) of RNA helicase (Holz et al., 2005). Interestingly, rapamycin does not always inhibit 4E-BP1 phophorylation, despite efficiently preventing S6K activity (Choo et al., 2008; Feldman et al., 2009). This suggests that rapamycin-mediated inhibition of mTORC1 may not always be a reliable way to investigate downstream pathways.
We have shown that phosphorylation of both 4E-BP1 and S6 is highly reduced in the mutant; hence, it seems possible that compromised rates of protein synthesis might contribute to impaired myelin growth and Schwann cell elongation. This might also explain why myelinating Schwann cells, which actively extend both longitudinally and radially, could be particularly vulnerable to mTOR ablation compared with other cell types in the nerve that also express mTOR. However, the possibility that other functions known to involve mTOR might also be affected in Schwann cells cannot be excluded. Mitochondrial energy metabolism and mitochondrial biogenesis are downstream targets of mTORC1 (Chen et al., 2009), and disruption of mitochondrial function through inactivation of the mitochondrial transcription factor Tfam has recently been shown to cause demyelination and axonal deficits (Viader et al., 2011). Further, since lipids comprise 76% by weight of the peripheral myelin sheath (O'Brien et al., 1967), dysregulation of lipid biosynthesis would disrupt myelin accumulation and the mTORC1 complex regulates SREBP, a key protein in the control of lipogenesis (Peterson et al., 2011). Indeed, Krox20 and SREBP synergistically regulate lipogenesis in Schwann cells (Leblanc et al., 2005). A possible role for dysregulation of autophagy is also possible, based on recent studies on myelination in vitro using dorsal root ganglion explant (DRG) cultures from mutant mice (Rangaraju et al., 2010). The unc-51-like kinase, a component of the autophagy complex, is phosphorylated by mTORC1 which inhibits autophagy (Wullschleger et al., 2006). Rapamycin has been shown to rescue myelination in DRG cultures from mice expressing a mutant form of the myelin protein PMP22, and this has been attributed to inhibition of mTOR function and concomitant activation of autophagy (Rangaraju et al., 2010): nevertheless, no enhancement of myelin synthesis was observed in rapamycin-treated explants from wild-type animals. Finally, the actin-based cytoskeleton is a target of mTORC2 and Schwann cells lacking N-WASP, a regulator of actin dynamics, produce thin myelin (Jacinto et al., 2004; Jin et al., 2011; Novak et al., 2011).
In the CNS, rapamycin treatment of mice reverses hypermyelination caused by expression of a constitutively active form of Akt in oligodendrocytes (Flores et al., 2008; Narayanan et al., 2009). Although rapamycin did not affect the extent of myelination in the CNS of adult wild-type mice, it did cause modest reductions in the levels of CNS myelin proteins in younger animals (Narayanan et al., 2009). Also, it has been shown that mice with mutations in either neuronal TSC1 or TSC2 (inhibitors of mTORC1), which are associated with hypomyelination, are partially rescued by rapamycin treatment (Meikle et al., 2008), and rapamycin treatment of purified oligodendrocytes or their precursors has revealed a role for mTOR in the differentiation of oligodendrocyte precursors (Tyler et al., 2009). Translation of the transcription factor Olig2 appears to be a likely target responsible for this effect on differentiation (Narayanan et al., 2009). Recently, it has been shown that eliminating the activator of mTORC1, Rheb1, in both neurons and glia leads to a substantial reduction in the amounts of CNS myelin proteins and myelin sheath, whereas overexpression of Rheb1 led to precocious myelination (Zou et al., 2011).
Akt is the best characterized target of the mTORC2 complex (Russell et al., 2011). This complex phosphorylates S473 of Akt, but persistent phosphorylation at this site in the mutant suggests that either this arises from other cell types in peripheral nerve, including axons, or the possible involvement of another kinase in Schwann cells, as has been proposed previously for skeletal and cardiac muscle (Bentzinger et al., 2008; Risson et al., 2009; Zhang et al., 2010). Interestingly, it is known that, in a variety of cell lines, there is enhanced ILK/rictor complex-dependent phosphorylation at S473 in the absence of mTOR (McDonald et al., 2008). Since there is a population of Akt in mutant (but not control) lysates that is phosphorylated on T308 but not S473, it seems most likely that this signal arises from Schwann cells and that normally mTORC2 is primarily responsible for Akt S473 phosphorylation. Enhanced phosphorylation on T308 is likely to be a consequence of the suppression of the feedback loop, S6K to AKT 308 via insulin receptor substrate and PI3K (Harrington et al., 2004; Shah et al., 2004). If elevated Akt S473 phosphorylation occurs in axons, it will be of interest to explore any possible relationship to the regulation of neurofilament phosphorylation.
Mutant Schwann cells, with their thin myelin, had a striking effect on axonal diameter. This was apparent by P21, by which time >75% of the sorted axons in mutant nerves were myelinated. Further, the diameters of unmyelinated fibers in 1:1 relationship with axons at this age exhibited a wide range from 1 to 4 μm, suggesting that it was not simply the absence of myelin that caused the overall reduction in axon diameter. Other hypomyelinating mutants in mice also display reductions in axon caliber (Robertson et al., 1997; Frei et al., 1999). Hence, it seems likely that thin myelin sheaths themselves and the disturbances in axon–glia interactions that they reflect influence axon growth. Conversely, overproduction of myelin in the PTEN knock-out results in increased axon diameter (Goebbels et al., 2010). Further, it has been proposed that Dlg1, by influencing PTEN activity, might act as a brake on the production of myelin outfoldings and subsequent demyelination (Cotter et al., 2010). However, mutants were not deficient in Dlg1 and the outfoldings that we observed at low frequency did not lead to demyelination, at least up to 10 months of age.
Although highly retarded in mutants, myelin continues to grow both radially and longitudinally and the relationship between the thickness of the myelin sheath and the diameter of the axon remains constant from an early stage. Clearly, it will be of great interest in the future to determine whether aberrant transcriptional regulation, for example by SREBP, plays a part in the postnatal arrest of myelination we observed in the PNS. In view of the many regulators and signaling pathways that converge on the mTORC complexes, it will also be important to determine whether as yet incompletely characterized variants of the inherited peripheral neuropathy, Charcot-Marie-Tooth disease, present with the arrested myelination phenotype we describe here.
Footnotes
- Received September 21, 2011.
- Revision received November 3, 2011.
- Accepted November 24, 2011.
This study was supported by the Wellcome Trust and the Novartis Foundation. We thank Heather Anderson and Stuart Fleming of Edinburgh University for excellent technical assistance and Dr. Dominique Langui of the ICM Centre de Recherche, CHU Pitié-Salpêtrière, Paris for EM access and assistance.
- Correspondence should be addressed to Peter J. Brophy, Centre for Neuroregeneration, University of Edinburgh, Chancellor's Building, 49 Little France Crescent, Edinburgh EH16 4SB, UK. Peter.Brophy{at}ed.ac.uk
- Copyright © 2012 the authors 0270-6474/12/321817-09$15.00/0