

Abstract
Human genetic studies have revealed that neurokinin B (NKB) and its receptor, neurokinin-3 receptor (NK3R), are essential elements for normal reproduction; however, the precise role of NKB–NK3R signaling in the initiation of puberty remains unknown. We investigated here the regulation of Tac2 and Tacr3 mRNAs (encoding NKB and NK3R, respectively) in female rats and demonstrated that their hypothalamic expression is increased along postnatal maturation. At puberty, both genes were widely expressed throughout the brain, including the lateral hypothalamic area and the arcuate nucleus (ARC)/medial basal hypothalamus, where the expression of Tacr3 increased across pubertal transition. We showed that central administration of senktide (NK3R agonist) induced luteinizing hormone (LH) secretion in prepubertal and peripubertal females. Conversely, chronic infusion of an NK3R antagonist during puberty moderately delayed the timing of vaginal opening (VO) and tended to decrease LH levels. The expression of NKB and its receptor was sensitive to changes in metabolic status during puberty, as reflected by a reduction in Tacr3 (and, to a lesser extent, Tac2) expression in the ARC after a 48 h fast. Yet, acute LH responses to senktide in pubertal females were preserved, if not augmented, under fasting conditions, suggesting sensitization of the NKB–NK3R–gonadotropin-releasing hormone signaling pathway under metabolic distress. Moreover, repeated administration of senktide to female rats with pubertal arrest due to chronic undernutrition rescued VO (in ∼50% of animals) and potently elicited LH release. Altogether, our observations suggest that NKB–NK3R signaling plays a role in pubertal maturation and that its alterations may contribute to pubertal disorders linked to metabolic stress and negative energy balance.
Introduction
Neurokinin B (NKB), a member of the tachykinin-peptide family, has emerged as an important modulator of reproductive function. Recent studies demonstrated hypogonadotropic hypogonadism in patients bearing inactivating mutations in TAC3 or TACR3 genes, encoding NKB and its receptor [neurokinin-3 receptor (NK3R)], respectively (Topaloglu et al., 2009). Subsequent animal studies have documented potent stimulatory effects of NK3R agonists on luteinizing hormone (LH) secretion (Amstalden et al., 2010; Billings et al., 2010; Ramaswamy et al., 2010; Wakabayashi et al., 2010; Navarro et al., 2011a). Mounting evidence suggests that this action occurs within the arcuate nucleus (ARC), through the regulation of the release of kisspeptins, encoded by Kiss1 (Ramaswamy et al., 2010; Navarro et al., 2011a). Indeed, NKB (and dynorphin) are expressed in Kiss1 neurons in the ARC, where they may act through recurrent collaterals to shape the pulsatile activity of those neurons (Navarro and Tena-Sempere, 2011).
Puberty is a key developmental period when reproductive capacity is achieved and sexual maturation is completed (Parent et al., 2003; Ojeda and Skinner, 2006; Ojeda et al., 2010). These events are centrally driven by the heightening of the neurosecretory activity of gonadotropin-releasing hormone (GnRH) neurons (Ojeda and Skinner, 2006). Compelling evidence suggests that kisspeptin signaling plays a crucial role in this process (Roa et al., 2008; Oakley et al., 2009). In support of this concept, we can observe the following: (1) absence of kisspeptin signaling prevents puberty and leads to infertility in humans and mice (Oakley et al., 2009); (2) hypothalamic Kiss1 expression and the ability of kisspeptin to induce gonadotropin secretion increases over puberty in rodents (Navarro and Tena-Sempere, 2011); and (3) transient antagonism of kisspeptin signaling delays puberty onset in rats (Pineda et al., 2010), while chronic administration of kisspeptin advances puberty onset (Navarro et al., 2004b). However, despite the proposed interplay between kisspeptins and NKB in the control of GnRH secretion in adulthood, the potential roles of NKB signaling in the timing of puberty remains virtually unexplored.
Reproduction, as an energy-demanding process, is sensitive to metabolic signals (Fernandez-Fernandez et al., 2006). Hence, sensing the metabolic status and transmitting this information to the hypothalamic reproductive centers are tightly regulated phenomena. Indeed, peripheral and central signals closely interplay to shut down the reproductive axis during energy-deficient periods. The hypothalamic Kiss1 system has been proposed to convey these inputs onto GnRH neurons (Castellano et al., 2010). Thus, a negative energy balance decreases Kiss1/kisspeptin expression at the hypothalamus, whereas exogenous administration of kisspeptin rescues puberty and reproductive function in undernourished animals (Castellano et al., 2010). However, the role of NKB signaling in the metabolic control of puberty is yet to be characterized. Of note, a recent study documented inhibition of ARC NKB expression following caloric restriction in adult rats (True et al., 2011).
In this context, we hypothesized that NKB and NK3R are involved in the timing of puberty in the female rat. We present herein the first evidence of the stimulatory role of NKB signaling in female puberty and document its modulation by conditions of metabolic stress, such as undernutrition, which are known to perturb normal pubertal maturation.
Materials and Methods
Animals and drugs
Female Wistar rats were bred in the vivarium at the University of Córdoba, in Córdoba, Spain. The animals were maintained under constant conditions of light (14 h of light, from 7:00 A.M.) and temperature (22°C). They were weaned at 21 d postpartum, when they were housed in groups of five rats per cage with free access to standard rat chow and tap water, until used in specific experiments. For hormonal tests involving intracerebroventricular cannulation, the rats were caged individually from the day before cannulae implantation until termination of experiments. Correct positioning of the cannulae was checked by visual inspection (to exclude animals showing obvious displacement or detachment) and confirmed at necropsy. Experimental procedures were approved by the University of Córdoba Ethical Committee for Animal Experimentation and were conducted in accordance with the European Union norms for the care and use of experimental animals. The NK3R agonist (senktide) and antagonist (SB222200) were purchased from Sigma Chemical and Tocris Bioscience, respectively. Doses of senktide and SB222200 were selected/adapted on the basis of previous studies (Sandoval-Guzmán and Rance, 2004; Ramaswamy et al., 2010; Navarro et al., 2011a).
Experimental Design
Developmental regulation of hypothalamic Tac2 and Tacr3 expression (Experiments 1–3).
In Experiment 1, analysis of hypothalamic expression of Tac2 (rodent ortholog of TAC3) and Tacr3 (ortholog of TACR3) mRNAs was conducted at different stages of postnatal development. Hypothalamic samples (n = 4/group) were obtained from female rats at postnatal day 1 (P1), P7, P10, P15, P20, P24, P30, P36, and P60, corresponding to the neonatal (P1), infantile (P7, P10, and P15), juvenile/prepubertal (P20 and P24), pubertal (P30 and P36), and adult (P60) stages of postnatal development; the latter (i.e., adult female rats) were checked for regular estrous cyclicity by monitoring vaginal smears and were sampled in the morning of the diestrous phase of the cycle. In Experiment 2, the neuroanatomical distribution of Tac2 and Tacr3 expression was compared between late-infantile/prepubertal and pubertal female rats. First, we mapped the distribution of Tac2 and Tacr3 mRNAs in the brain of intact female rats at the transition between the late-infantile and prepubertal stages of maturation (P20 rats) [i.e., at the middle of the transitional phase between low (P15) and high (P24) expression in both genes observed in Experiment 1]. Second, we mapped the expression of Tac2 and Tacr3 in pubertal (P36) female rats. Brains were immediately removed, gradually frozen on dry ice, and stored at −80°C until they were sectioned on a cryostat, thaw mounted, and stored at −80°C until used for in situ hybridization (ISH). Trunk blood samples for hormone measurements were also collected at the time of decapitation. In Experiment 3, the hypothalamic nuclei comprising the majority of the expression of Kiss1 [ARC and anteroventral periventricular nucleus (AVPV)], Tac2 [ARC and lateral hypothalamic area (LHA)], and Tacr3 [ARC, paraventricular nucleus (PVN), and LHA] were selectively targeted, and the expression of Tac2 and Tacr3 genes were quantitatively assayed and compared between 20- and 36-d-old female rats.
Responses to NKB agonist and antagonist in prepubertal female rats (Experiments 4 and 5).
In Experiment 4, we assessed the ability of the NK3R agonist, senktide, to influence LH secretion in prepubertal rats. To this end, central (intracerebroventricular) administration of senktide into the lateral cerebral ventricle was conducted at two developmental stages: infantile (P10) and juvenile (P25) female rats (n = 10/group). For comparative purposes, adult (P60) females in diestrus-1 (n = 10 rats/group) were also tested, as senktide has been previously described to elicit a potent secretory response of LH (Navarro et al., 2011a). A dose of 600 pmol/10 μl per rat was injected on the basis of previous studies (Navarro et al., 2011a). Briefly, animals were implanted with intracerebroventricular cannulae; to allow delivery of senktide into the lateral ventricle, the cannulae were lowered to a depth of 3 mm beneath the surface of the skull; the insert point was 1 mm posterior and 1.2 mm lateral to bregma, as described in detail previously (Navarro et al., 2004b). Animals were bled through jugular venipuncture before senktide injection (0 min), and 20 and 60 min after injection. Animals injected with vehicle (physiological saline, 0.9% NaCl) served as controls. To note, 10-d-old rats were only bled 20 min after senktide injection, due to a reduced body size that prevented us from also sampling at 60 min. Blood was collected, and serum samples were separated by centrifugation at 1600 × g for 20 min and stored at −20°C until use for hormone determinations.
As a complement to the above acute study, in Experiment 5 the effects of chronic central administration of the NK3R antagonist, SB222200, on puberty onset in immature female rats were assessed. The treatment protocol was set following our previous studies of the pubertal effects of kisspeptin antagonism (Pineda et al., 2010). The ability of SB222200 to antagonize NKB action has been recently tested in the monkey (Ramaswamy et al., 2010) and confirmed by the blockade of the NKB-dependent activation of Kiss1 neurons in the mouse (Navarro et al., 2011b). Central administration of the antagonist in the lateral cerebral ventricle was conducted as described previously (Navarro et al., 2004b), following protocols of cannulation similar to those of Experiment 4. Injection of SB222200 (3 nmol) was initiated at P28 and repeated every 12 h until P36 (n = 11). An additional group of females injected with vehicle (n = 12) was carried in parallel. In all animals, body weight and vaginal opening (VO) were monitored daily. At the end of treatment, all the animals were killed by decapitation 15 min after the last injection of SB222200. Trunk blood was collected, and the uteri were dissected out of the surrounding fat and their dry weights recorded.
Metabolic control of NKB system at puberty (Experiments 6–8).
As continuation of the previous studies, this set of experiments was conducted to evaluate whether expression and function of NKB signaling is influenced by metabolic signals at puberty. In Experiment 6, pubertal female rats (n = 10) were subjected to 48 h of fasting. A pair-aged group of animals fed ad libitum was used as control. At the end of the experiment (P36), the animals were killed by decapitation, and brains and blood were collected as described above. The brains were processed by ISH for Kiss1, Tac2, and Tacr3 genes in the same hypothalamic nuclei as in Experiment 2. In Experiment 7, the ability of senktide to acutely induce/modulate LH secretion under conditions of negative energy balance was assessed in pubertal (P36) female rats (n = 10) subjected to the same protocol of 48 h fasting used in Experiment 6. As described in Experiment 4, animals were bled before senktide injection (0 min), and 20 and 60 min after central (intracerebroventricular) injection of a 600 pmol bolus of senktide; animals injected with vehicle served as controls. For comparative purposes, similar protocols of intracerebroventricular injection and blood sampling following administration of vehicle or 600 pmol of senktide were implemented in additional groups of pubertal (P36) female rats (n = 10) fed ad libitum. Finally, in Experiment 8, we monitored the effects of repeated central administration of NKB agonist, senktide, on puberty onset in rats subjected to persistent caloric restriction. A protocol of 30% restriction in daily food intake was applied to immature female rats, staring on P23 postpartum, as this has been shown to prevent normal occurrence of VO and pubertal progression in female rats (Castellano et al., 2005). Daily repeated intracerebroventricular injection of senktide was implemented between P30 and P37 in food-restricted females (n = 11), following previously published protocols (Castellano et al., 2005). The treatment regimen was set at a dose of 600 pmol of senktide per animal in 10 μl, every 12 h. Pair-aged females (n = 12), at 30% food restriction, injected with vehicle served as controls. Body weight and VO were monitored daily as described in Experiment 5. At the end of treatment (P37), the animals were killed by decapitation, 20 min after the last injection of senktide (or vehicle), and trunk blood was collected. To determine the normal date of vaginal opening in animals fed ad libitum, an additional group of females (n = 20) without food restriction were maintained on daily inspection of canalization of the vagina up to P37.
Tissue preparation
Blood was centrifuged to isolate the serum, which was stored at −20°C until hormone measurements. Uteri were removed and weighed to provide an indirect marker of plasma E2 levels (and their biological effect). Brains were removed for ISH, frozen on dry ice, and then stored at −80°C until sectioned. Five sets of 20 μm sections in the coronal plane were cut on a cryostat (from the diagonal band of Broca to the mammillary bodies), thaw mounted onto SuperFrost Plus slides (VWR Scientific), and stored at −80°C. A single set was used for each in situ hybridization assay (adjacent sections 100 μm apart).
Radioimmunoassay for LH
Serum LH levels were measured in 50 μl samples with double-antibody RIA kits supplied by the National Institutes of Health (Dr. A. F. Parlow, National Hormone and Peptide Program, Torrance, CA). Rat LH-I-10 was labeled with 125I using IODO-GEN tubes, following the instructions of the manufacturer (Pierce). Hormone concentrations were expressed with the reference preparation LH-RP-3 as a standard. Intraassay and interassay coefficients of variation were <8 and 10%, respectively. The sensitivity of the assay was 5 pg/tube.
Semiquantitative RT-PCR of Tac2 and Tacr3 mRNAs
Real-time RT-PCR was performed in whole hypothalamic samples with the iCycler iQ Real-Time PCR detection system (Bio-Rad Laboratories). In detail, Tac2 and Tacr3 mRNA levels were assayed in samples from different stages of postnatal development in females (1-, 7-, 10-, 15-, 20-, 24-, 30-, 36-, and 60-d-old rats; the latter were monitored for regular estrous cyclicity and sampled in the diestrous phase of the ovarian cycle). For each target, RT-PCR amplification was routinely conducted with two different sets of primers, which were generated on the basis of the published sequences of the rat Tac2 gene (NM_019162.1), sequences: 153–171 (sense) and 299–319 (antisense); and the Tacr3 gene (NM_017053.1) sequences: 862–881 (sense) and 985–1008 (antisense). The synthesized cDNAs were further amplified (1:10) in triplicate by PCR with SYBR Green I as fluorescent dye and 1× iQ Supermix containing 50 mm KCl, 20 mm Tris-HCl, 0.2 mm deoxy-NTPs, 3 mm MgCl2, and 2.5 U iTaq DNA polymerase (Bio-Rad Laboratories) in a final volume of 25 μl. The PCR cycling conditions were as follows: initial denaturation and enzyme activation at 95°C for 5 min; followed by 40 cycles of denaturation at 95°C for 30 s; annealing at 57°C (Tac2), 53°C (Tacr3), or 58°C (RP-S11) for 30 s; and extension at 72°C for 10 s. Product purity was confirmed by dissociation curves and random agarose gel electrophoresis. No template controls were included in all assays, yielding no consistent amplification. Calculation of the relative expression levels of the target mRNAs was conducted based on the cycle threshold (Ct) method (Navarro et al., 2004a). The Ct for each sample was calculated using iCycler iQ real-time PCR detection system software with an automatic fluorescence threshold setting. Accordingly, the fold expression of target mRNAs over reference values was calculated by the equation 1 + e−ΔΔCt, where ΔCt is determined by subtracting the corresponding RP-S11 Ct value (internal control) from the specific Ct of the target (Tac2 or Tacr3), and ΔΔCt is obtained by subtracting the ΔCt of each experimental sample from that of the reference sample (taken as reference value 100). No significant differences in Ct values were observed for RP-S11 between the treatment groups.
Detection of Kiss1, Tac2, and Tacr3 mRNAs probe generation
The probes used for detection of Kiss1, Tac2, and Tacr3 mRNAs have been described previously (Navarro et al., 2009). Sense probes for every transcript were used as controls for the specificity of the ISH procedures, which are outlined below.
Single-label in situ hybridization of Kiss1, Tac2, and Tacr3 mRNAs
Kiss1, Tac2, and Tacr3 mRNA sense and antisense probes were transcribed with T7 or T3 polymerase (Fermentas), as described previously (Navarro et al., 2009). Briefly, radiolabeled probes were synthesized in vitro by inclusion of the following ingredients in a volume of 20 μl: 250 Ci [33P]UTP (PerkinElmer Life and Analytical Sciences); 1 μg of PCR product; 0.5 mm each ATP, CTP, and GTP; and 40 U of polymerase. Residual DNA was digested with 4 U of DNase (Ambion), and the DNase reaction was terminated by addition of 2 μl of 0.5 m EDTA, pH 8.0. The riboprobes were separated from unincorporated nucleotides with NucAway Spin Columns (Ambion). Slides with mouse hypothalamic sections from the different experimental groups were processed as reported previously (Cunningham et al., 2002; Gottsch et al., 2004).
Quantification and analysis of Kiss1, Tac2, and Tacr3 mRNAs
Brain sections were analyzed unilaterally. Slides from all of the animals were assigned a random three-letter code, alphabetized, and read under dark-field illumination with custom-designed software designed to count the total number of cells and the number of silver grains (corresponding to radio-labeled Kiss1, Tac2, and Tacr3 mRNA) over each cell (Chowen et al., 1990). Data are presented as total mRNA content, depicting the number of cells × grains per cell within the coronal sections containing the hypothalamic nucleus studied for each set, not the total mRNA in this specific nucleus. The starting and ending points of quantification were determined according to the atlas of Paxinos and Watson (2001).
Statistical analysis
All data are expressed as the mean ± SEM for each group. In addition, when appropriate, integrated LH secretory responses were expressed as the area under the curve (AUC), calculated following the trapezoidal rule (Protter and Morrey, 1964), over a 60 min period after the administration of senktide. One-way ANOVA followed by Tukey's post hoc test and Student's t test were used to assess variation among experimental groups. Significance level was set at p ≤ 0.05. All analyses were performed with GraphPad Prism.
Results
Tac2 and Tacr3 mRNA levels in rat hypothalamus during development
The profiles of hypothalamic expression of Tac2 and Tacr3 mRNAs, rat orthologs of human TAC3 and TACR3, were evaluated in hypothalamic samples from female rats at different stages of postnatal development, from the neonatal period to adulthood. Stages of postnatal maturation were defined on the basis of previous references (Ojeda and Skinner, 2006), and timing of puberty onset in the female rat was assessed following consensus criteria and recording of external signs of reproductive maturation, such as vaginal opening (Ojeda and Skinner, 2006). Persistent expression of Tac2 and Tacr3 mRNAs was detected in the hypothalamus by means of real-time RT-PCR along postnatal maturation. Expression of Tac2 and Tacr3 was virtually undetectable in neonatal females but increased progressively over the infantile (P7, P10, and P15) and juvenile/prepubertal periods (P20, P24, and P30) of postnatal maturation. Maximal expression was reached at the time of puberty onset (P36) for Tac2, whereas peak Tacr3 levels plateaued during the peripubertal transition (Fig. 1). This was followed by a decreasing trend in the expression of both genes in adult (diestrus-1) animals, which was statistically significant for Tac2. Nonetheless, hypothalamic mRNA levels of Tac2 and Tacr3 in adult females remained higher than in infantile rats.

A, B, Developmental profiles in the expression of Tac2 (A) and Tacr3 (B) in the hypothalamus of the female rat. Values represent the percentage of the lowest value (100%) measured by real-time RT-PCR and normalized to the S11 ribosomal protein mRNA. Groups with different superscript letters and asterisks are statistically different from one another (p < 0.05, by one-way ANOVA followed by Student–Newman–Keuls multiple range test and unpaired Student's t test).
Neuronal mapping of Tac2 and Tacr3 expression in prepubertal versus peripubertal rats
The neuroanatomical distribution of Tac2 and Tacr3 mRNA was assessed in medial coronal sections of the female rat brain at two representative stages of postnatal maturation by ISH. P20 was chosen on the basis of results obtained in Experiment 1 as a period of transition between low (infantile) and high (late juvenile/prepubertal) expression of both genes. Likewise, P36 was selected as the representative pubertal stage with maximal levels of Tac2 and Tacr3 mRNAs. Tac2 expression was broadly detected across brain sections with similar distribution in both ages. Tac2 transcripts were predominantly present in cortical and hippocampal areas [somatosensory area 1 (SS1), parietal region 4 (PTL4), and Ammon's horn (CA)], supraoptic area, and lateral and medial basal hypothalamic nuclei (LHA and ARC). Of note, P36 animals showed wider expression in the basal hypothalamus with detectable expression at the ventromedial nucleus, which was absent in P20 animals (Fig. 2). Tacr3 mRNA showed a scattered and differential distribution with more abundant expression at the amygdala (basolateral amygdalar nucleus and basomedial amygdalar nucleus) and LHA in prepubertal animals. Additionally, similar to Tac2 expression, Tacr3 mRNA was equally detected in both groups within cortical and hippocampal areas (SS4, PTL4, and CA) and medial basal hypothalamus (periventricular hypothalamic nucleus and ARC) (Fig. 2).

Schematic representation of the distribution of Tac2 and Tacr3 mRNAs in the brain of late-infantile (P20) and pubertal (P36) female rats. AHN, Anterior hypothalamic nucleus; BLA, basolateral amygdalar nucleus; BMA, basomedial amygdalar nucleus; LM, lateral mammilary nucleus; LV, lateral ventricle; MH, medial habenula; PH, posterior hypothalamic nucleus; PVH, periventricular hypothalamic nucleus; RSP, retroesplenial area; RT, reticular nucleus thalamus; SO, supraoptic nucleus; STN, subthalamic nucleus; ZI, zona incerta.
Quantitative changes of expression of Kiss1, Tac2, and Tacr3 over pubertal maturation
The expression levels of Kiss1, Tac2, and Tacr3 genes were quantified in representative hypothalamic nuclei, selected on the basis of the prominent presence of these targets (see neuronal mapping results), comparing late-infantile (P20) and peripubertal (P36) animals. Kiss1 expression remained unchanged in the ARC during this transition; however, it increased significantly in the AVPV (Fig. 3A, *p < 0.01). Tac2 mRNA levels did not show any significant change in any of the nuclei assessed (ARC and LHA) during the pubertal transition (Fig. 3B). In contrast, Tacr3 expression significantly augmented on P36 in the ARC, but not in the PVN or LHA (Fig. 3C, *p < 0.05).
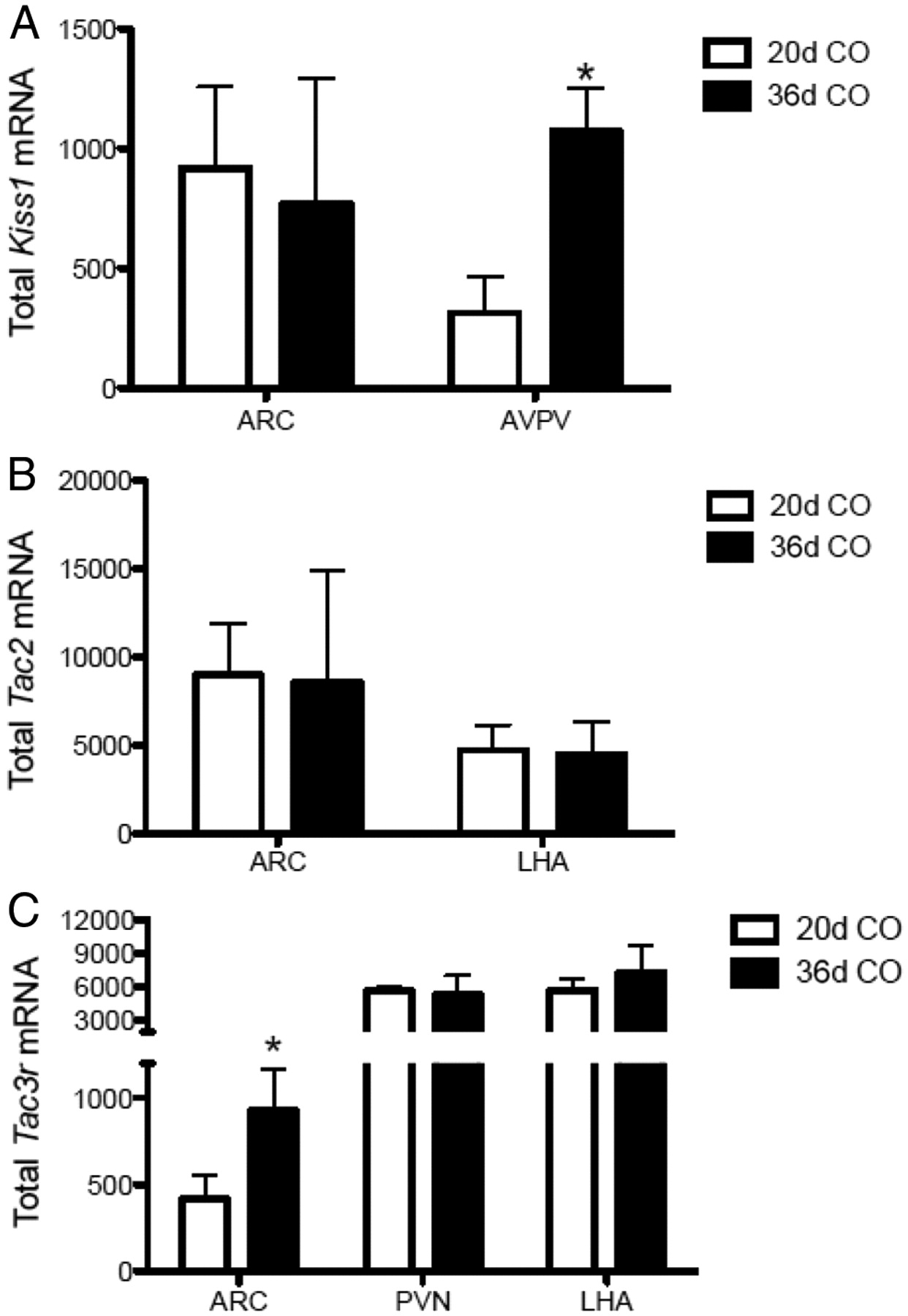
A–C, Profile of expression of Kiss1 (A), Tac2 (B), and Tacr3 (C) expression in specific hypothalamic nuclei of late-infantile (20-d-old) vs pubertal (36-d-old) female rats. Data are presented as total mRNA (number of cells × grains/cell) assessed by in situ hybridization. *p < 0.05, by unpaired Student's t test within each nucleus.
Effects of NKB agonist, senktide, on LH secretion in prepubertal and adult female rats
The NKB agonist, senktide, induced a robust increase in serum LH levels 20 min after central (intracerebroventricular) injection, in both prepubertal (P25) (p < 0.01; Fig. 4A) and adult female rats on diestrus-1 (p < 0.01; Fig. 4B); the latter is in keeping with recent reports (Navarro et al., 2011a). At both ages, LH levels remained elevated at 60 min after intracerebroventricular administration of senktide. Accordingly, the overall integrated secretory response (calculated as the AUC over a 60 min study period) showed a significant elevation in both age groups (prepubertal: vehicle = 31.0 ± 6.2 ng/ml per 60 min vs senktide = 113.7 ± 21.7, p < 0.01; adult/diestrus-1: vehicle = 52.2 ± 10.1 ng/ml per 60 min vs senktide = 145.2 ± 21.1, p < 0.01).

A, B, Serum LH levels in prepubertal 25-d-old (A) and adult female rats in diestrus-1 (B), at 0, 20, and 60 min after central intracerebroventricular administration of vehicle or 600 pmol/rat senktide. In addition to the profiles of mean LH levels in the experimental groups, the integrated secretory responses, as the AUC over the study period (60 min), are shown (in the inset as a bar graph). **p < 0.01 versus corresponding vehicle-injected controls (one-way ANOVA followed by Student–Newman–Keuls multiple range test or unpaired Student's t test; the latter for AUC data). Veh, Vehicle; Senk, senktide.
In addition, the stimulatory effects of senktide on LH secretion were also detected at an earlier age point (i.e., during the early infantile period). Thus, 10-d-old female rats displayed a robust fivefold increase of the circulating LH levels at 20 min after intracerebroventricular injection of senktide (vehicle alone: 0.32 ± 0.03 ng/ml; vs senktide: 1.69 ± 0.13, p < 0.01). Of note, because of low body size, no sampling was conducted at this age 60 min after senktide administration.
Effects of central infusion of an NK3R antagonist on puberty onset
On the basis of the stimulatory effects of senktide on LH secretion in prepubertal female rats, we next sought to determine whether chronic blockage of NK3R signaling would delay the normal onset of puberty. To this end, we tested the effect of a chronic (but intermittent) infusion of the antagonist of the NK3R, SB222200, on several markers of pubertal maturation. A model of repeated intracerebral (lateral ventricle) injection of the antagonist was selected to limit its effects to the brain. Following analogous protocols of kisspeptin antagonism (Pineda et al., 2010), pharmacological treatments were administered between P28 and P36, and the progression of puberty was monitored. As an external index of puberty onset, VO (defined as complete canalization of the vagina) was monitored daily. Such a marker was selected as it has been conventionally considered a reliable external sign of puberty (Navarro et al., 2004b). Infusion of NK3R antagonist did not alter body weight, food intake (data not shown), or uterine weight over the study period (Fig. 5). Nevertheless, repeated central administration of SB222200 to peripubertal female rats evoked a moderate delay in the overall timing of puberty onset. Thus, the percentage of VO was persistently lower in the SB222200-injected group starting at day 32, and by day 35, when nearly 80% of vehicle-treated females displayed VO, only 50% of the antagonist-injected animals did (Fig. 5). Yet, the antagonist group tended to catch up, and by day 36 no differences in terms of the percentage of VO were detected between groups. In addition, a decreasing trend in LH levels in the animals treated with the antagonist was observed at the end of the treatment (45% reduction in mean values), although it did not reach statistical significance.

Compilation of indices of pubertal maturation recorded in immature female rats chronically intracerebroventricular injected 3 nmol of SB222200 (NK3R antagonist) or vehicle. SB222200 was injected every 12 h between P28 and P36. Dates of vaginal opening (expressed as a percentage of the total number of animals per experimental group) are shown in the left panels. Data on body and uterus weights, as well as serum LH levels, in vehicle- and SB222200-injected animals are shown in the right panels. Values are the mean ± SEM. Veh, Vehicle; ANT, antagonist; BW, body weight; UW, uterus weight.
Effects of 48 h fasting on hypothalamic expression of Kiss1, Tac2, and Tacr3
To determine the effects of metabolic stress associated with negative energy balance on the hypothalamic expression of Kiss1, Tac2, and Tacr3 genes, single-label ISH was performed brain sections from pubertal (P36) female rats after 48 h of fasting. Our protocol of food deprivation induced a significant (>25%) decrease of body weight (data not shown), which was associated with a dampening of hypothalamic Kiss1 mRNA levels in both the ARC (p < 0.05) and AVPV (p < 0.05) (Fig. 6A). Likewise, a decreasing trend in Tac2 mRNA levels was observed in the ARC and LHA of fasted animals (Fig. 6B), but neither of these changes was statistically significant. Tacr3 expression was significantly inhibited in the ARC after 48 h of fasting (p < 0.05), but remained constant in the PVN and LHA (Fig. 6C).

A–C, Effect of 48 h fasting on the total mRNA levels of Kiss1 (A), Tac2 (B), and Tacr3 (C) in specific hypothalamic nuclei of pubertal (36-d-old) female rats. *p < 0.05, by unpaired Student's t test within each nucleus.
LH responses to the NKB agonist, senktide, under severe caloric restriction
The role of NKB in the regulation of gonadotropin secretion under conditions of metabolic stress associated with negative energy balance was explored by assessing the LH-releasing effects of senktide in pubertal female rats after 48 h of fasting. In age-matched (P36) rats fed ad libitum, intracerebral injection of 600 pmol of senktide induced significant LH responses (>2.2-fold increase over vehicle-injected controls), at 20 min after injection (p < 0.01) (Fig. 7A), with > 65% elevation of LH AUC in response to senktide. In fasted animals, the ability of similar doses (600 pmol/rat, i.c.v.) of senktide to elicit LH secretion in vivo was not only preserved, but notably enhanced, as evidenced by the following observations: (1) absolute LH levels at 20 min after senktide administration were twofold higher in fasted animals; (2) LH levels remained significantly elevated in fasted animals at 60 min after senktide injection in fasted, but not fed, females; and (3) integral LH response to senktide over a 60 min period (AUC) was twice as high in pubertal females submitted to prior 48 h fasting (Fig. 7B).

A, B, Serum LH levels in pubertal 36-d-old female rats fed ad libitum (A) or subjected to 48 h of fasting (B) at 0, 20, and 60 min after central intracerebroventricular administration of vehicle or 600 pmol/rat senktide in a 10 μl volume. In addition to the profiles of mean LH levels in the experimental groups, the integrated secretory responses, reported as the AUC over the study period (60 min), are shown. **p < 0.01 versus corresponding vehicle-injected controls (one-way ANOVA followed by Student–Newman–Keuls multiple range test or unpaired Student's t test; the latter for AUC data). Veh, Vehicle; Senk, senktide.
Effects of senktide on puberty onset under conditions of severe caloric restriction
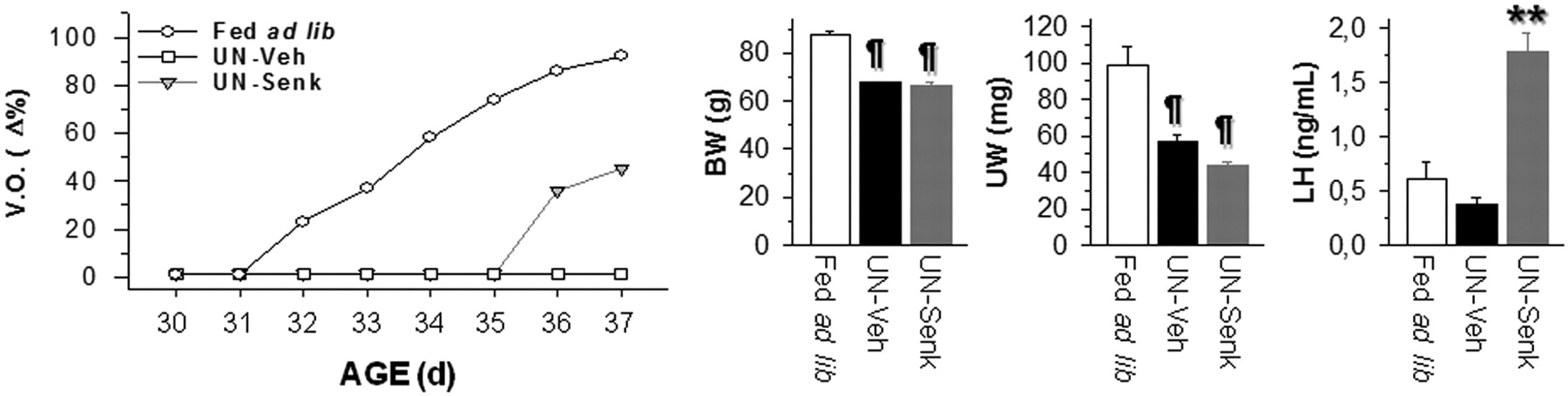
The effects of senktide upon the pubertal activation of the reproductive axis were monitored in immature female rats in a model of puberty arrest due to 30% restriction in daily food intake, initiated on P23 (Castellano et al., 2005). Repeated injections of senktide, starting at P30, were administered into the lateral ventricle. Control females fed ad libitum were also daily monitored. This group displayed normal progression of puberty, as evidenced by >85% of the animals having complete VO at P36, when mean the body weight was 87.45 ± 1.59 g. Chronic food restriction induced a significant reduction in body weight of ∼30% from controls (mean body weight on P37, 67.75 ± 0.59 g; n = 12). This regimen totally prevented normal puberty onset, as none of the vehicle-injected females submitted to undernutrition displayed VO by P37. In contrast, chronic intracerebroventricular administration of senktide (600 pmol/12 h) between P30 and P37 to food-restricted females induced complete VO in 5 of 11 treated animals (∼45%), without a significant change in terminal mean body weight compared with vehicle-injected, food-restricted females. Moreover, mean body weight in the subset of senktide-treated females displaying VO (65.4 ± 2.48 g; n = 5) was similar to that in animals from the same group without VO (67.33 ± 1.25 g; n = 6). Additionally, regardless of the presentation of VO, serum LH levels at the end of the chronic treatments were significantly elevated in senktide-injected females, which displayed a >4.5-fold increase over vehicle-injected animals at 20 min after the last intracerebroventricular injection (Fig. 8).

Compilation of indices of pubertal maturation recorded in peripubertal female rats subjected to a protocol of 30% restriction in daily food intake (UN; 70% food intake of controls), and chronically intracerebroventricularly injected with senktide (600 pmol/12 h) or vehicle between P30 and P37. For reference purposes, data from control females fed ad libitum and injected with vehicle are also shown. Dates of VO, expressed as the percentage over the total number of animals per each experimental group, are shown in the left panel. To note, although undernutrition prevented VO in all vehicle-injected animals, senktide administration was able to restore canalization of the vagina in ∼50% of cases. Body and uterus weight records in the different experimental groups at the end of the treatment are presented in the right panels. In addition, senktide treatment significantly elevated serum LH levels. ¶p < 0.01 vs controls fed ad libitum; **p < 0.01 versus UN rats injected with vehicle (one-way ANOVA followed by Student–Newman–Keuls multiple range test). Veh, Vehicle; Senk, senktide; BW, body weight; UW, uterus weight.
Discussion
Studies in humans and other mammalian species suggest that NKB–NK3R signaling plays a critical role in the control of reproductive function in adulthood, a function that appears to be mediated, at least partially, by its ability to autoregulate the activity of Kiss1 neurons in the ARC (Lehman et al., 2010; Wakabayashi et al., 2010; Navarro et al., 2011a) and is dependent on preserved kisspeptin signaling (Ramaswamy et al., 2011; García-Galiano et al., 2012). Yet, the precise role of NKB and its receptor in sexual maturation, and their possible interaction with metabolic status, remain largely unexplored. To address the putative contribution of NKB signaling to the onset of puberty, we evaluated here the developmental regulation of the hypothalamic NKB/NK3R system, with special attention to the pubertal transition. Studies in whole hypothalamic fragments, obtained serially along postnatal maturation, documented a striking profile of expression of Tac2 and Tacr3, with a progressive increase of both mRNAs from the neonatal/infantile to the peripubertal period, a profile that, compared with previously published data, appears to precede the elevation of hypothalamic Kiss1 mRNA that takes place between P20 and P30 in the female rat (Navarro et al., 2004a). Furthermore, the rise of Tac2/Tacr3 expression described herein also seems to anticipate the sharp increase of kisspeptin immunoreactivity reported in the rostral periventricular area of the female mouse hypothalamus that occurs between P25 and P30 (Clarkson et al., 2009). Altogether, these observations are compatible with an eventual stimulatory role of NKB upon Kiss1/kisspeptin expression during (early) pubertal maturation. This possibility, which is yet to be experimentally tested, is in keeping with recent clinical observations suggesting that the NKB system may play a more prominent stimulatory role on the gonadotropic axis during early stages of sexual maturation (Gianetti et al., 2010).
To gain anatomical resolution, ISH studies for mapping the expression of Tac2 and Tacr3 genes were performed in prepubertal (P20) and pubertal (P36) animals, and detected moderate regional differences in the expression patterns of both genes between these two ages and also with the previously reported distribution in adults (Navarro et al., 2011a). Nonetheless, prominent expression of Tac2 and Tacr3 was observed in the ARC and LHA nuclei (and also the PVN, selectively for Tacr3) in both prepubertal and pubertal females. At the time of puberty, a notable increase in Tacr3 expression was detected by ISH exclusively in the ARC. Given the prominent expression of Tac2 in the ARC in 20- and 36-d-old females, it was anticipated that the increase in its expression between the infantile and juvenile periods (Fig. 1) would happen (mainly) in ARC neurons. However, our ISH analyses failed to detect an overt elevation of Tac2 levels between P20 and P36 in this nucleus. As possible explanation, Tac2 expression in the ARC might have reached rather high levels already on P20, which may obscure detection of further increases in this area. In addition, the potential contribution of increased expression of Tac2 in other hypothalamic regions cannot be excluded. On the other hand, Kiss1 expression did not increase significantly in the ARC during the pubertal transition, whereas it did in the AVPV, in good agreement with previous studies (Han et al., 2005; Takase et al., 2009; Bentsen et al., 2010; Gill et al., 2010). This would suggest that in the female rat most of the pubertal increase of Kiss1 expression occurs mainly in AVPV, as recently proposed in the mouse (Gill et al., 2010). Nonetheless, it has been suggested that Kiss1 neurons in the ARC play an important role in puberty onset (Roa et al., 2011)—a contention indirectly supported also by the observations of increased Tacr3 expression in the ARC across the pubertal transition.
In agreement with a role for NKB in the timing of puberty, its agonist, senktide, robustly induced the release of LH in prepubertal females, consistent with the recently reported effects of this NKB agonist in juvenile monkeys (Ramaswamy et al., 2010) and our previous data in adult female rats (Navarro et al., 2011a). Of note, despite unambiguous hormonal responses at both ages, comparison of the magnitude of LH secretory peaks after senktide injection between P25 (juvenile) and P36 (peripubertal) females suggests a higher responsiveness to NKB stimulation during the prepubertal period, a phenomenon that may be indicative of the actual role of NKB signaling in the timing of puberty and whose physiological relevance is presently under investigation in our laboratory. Moreover, despite obvious reproductive immaturity, infantile females were also able to unambiguously respond to senktide in terms of LH secretion. This finding strongly suggests that the gonadotropic system becomes responsive to NKB stimulation well before the onset of puberty, so that low expression levels of Tac2 during the infantile period, as reported here, may play a role in preventing the precocious activation of the gonadotropic axis. Of note, a similar phenomenon (i.e., robust LH secretory responses during the infantile stage) has been described for kisspeptin stimulation in female rats (Castellano et al., 2006), an observation that is in line with the proposed mode of action of NKB as a modulator of kisspeptin secretion and, hence, actions in GnRH neurons (Navarro et al., 2009).
In turn, persistent blockade of NKB signaling during the pubertal transition did induce a modest but detectable delay in the timing of puberty. Noteworthy, the NK3R antagonist SB222200 used in our study has been shown to block direct NKB actions on mouse Kiss1 neuron (Navarro et al., 2011b) and to blunt in vivo LH responses to senktide in the monkey (Ramaswamy et al., 2010), thus confirming its efficacy and selectivity. However, its effect on puberty onset was not as effective in terms of pubertal perturbation as the chronic infusion of a kisspeptin antagonist, which resulted in a marked suppression of VO and sex organ weights at the expected age of puberty (Pineda et al., 2010). Altogether, the above observations suggest a discernible stimulatory role of NKB in the timing of puberty, which is less important than—and possibly subordinated to—that of kisspeptin signaling. Yet, it remains possible that more effective protocols of NKB antagonism (e.g., starting at earlier developmental stages) may have brought about more robust suppression of different indices of puberty.
Puberty is exquisitely sensitive to body energy status and metabolic cues that impinge on different central pathways to modulate GnRH secretory activity (Hill et al., 2008; Castellano et al., 2010). Kiss1 neurons have been proposed as nodal elements for conveying metabolic information to reproductive centers at puberty, as evidenced by the impact of metabolic stress (e.g., acute fasting) on the hypothalamic expression of Kiss1/kisspeptins in pubertal animals, and the ability of exogenous kisspeptin to rescue puberty arrest due to metabolic distress (Castellano et al., 2005). Our present findings indicate that the NKB system is also subjected to metabolic regulation during puberty. First, the expression of Tacr3, and to a lesser extent Tac2, in the ARC was markedly suppressed in pubertal female rats by metabolic stress associated with 48 h fasting, as was Kiss1 expression in the ARC and AVPV (see Fig. 6). Second, the LH responses to NKB agonist administration in pubertal (P36) rats were not only preserved but even augmented in fasting conditions, suggesting a possible sensitization of its stimulatory effects under conditions of negative energy balance; again, this is similar to previous findings on the gonadotropin-releasing actions of kisspeptins (Castellano et al., 2005; Tovar et al., 2006). Finally, as previously reported for kisspeptin (Castellano et al., 2005), repeated administration of NKB agonist was sufficient to at least partially rescue some of the indicators of puberty progression, such as VO and LH secretion, even against persistent conditions of energy deficit due to chronic subnutrition during the pubertal transition. Together, our studies suggest a role of metabolic cues in the modulation of NKB signaling at puberty, which is likely involved in transmitting information pertaining to body energy status to the Kiss1/GnRH pathway governing puberty onset. Given our current data and the similarities with Kiss1/kisspeptin responses, it is highly plausible that such an effect is mediated, at least partially, via modulation of kisspeptin output in the ARC. However, we cannot rule out the contribution of additional, kisspeptin-independent pathways for the stimulatory effects of NKB on the gonadotropic axis at puberty.
Puberty is a key developmental event that is under the control of an intricate network of interacting peripheral signals and central transmitters that cooperate to ensure proper pubertal maturation. Our current data are the first to thoroughly document the roles of NKB signaling in this phenomenon, a physiological function that is seemingly linked (and possibly subordinated) to that of kisspeptins and sensitive to metabolic cues, which are likely to conduct (part of) their modulatory effects on puberty onset via regulation of the hypothalamic NKB system.
Footnotes
This research was supported by NIH Grant R01 HD 049651 and the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement U54 HD 12629; the Marie Curie Outgoing International Fellowship within the 7th Framework Programme of the European Union; Mary Gates Endowment for undergraduate students (to S.J.H.; University of Washington); and Ministerio de Ciencia e Innovación, Spain, Research Grants BFU 2008-00984 and BFU 2011-25021, Junta de Andalucía, Spain, Grant P08-CVI-00603, and EU Research Contract DEER FP7-ENV-2007-1. CIBER Fisiopatología de la Obesidad y Nutrición is an initiative of the Instituto de Salud Carlos III.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Víctor M. Navarro or Manuel Tena-Sempere, Department of Cell Biology, Physiology and Immunology, University of Córdoba, 14004 Córdoba, Spain, nalovic{at}gmail.com or fi1tesem{at}uco.es





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}