

Abstract
Adolescence is a period in which the developing prefrontal cortex (PFC) is sensitive to maladaptive changes when exposed to nicotine. Nicotine affects PFC function and repeated exposure to nicotine during adolescence impairs attention performance and impulse control during adulthood. Nicotine concentrations experienced by smokers are known to desensitize nicotinic acetylcholine receptors (nAChRs), but the impact thereof on PFC circuits is poorly understood. Here, we investigated how smoking concentrations of nicotine (100–300 nm) interfere with cholinergic signaling in the mouse PFC. nAChR desensitization depends on subunit composition. Since nAChR subunits are differentially expressed across layers of the PFC neuronal network, we hypothesized that cholinergic signaling through nAChRs across layers would suffer differentially from exposure to nicotine. Throughout the PFC, nicotine strongly desensitized responses to ACh in neurons expressing β2* nAChRs, whereas ACh responses mediated by α7 nAChRs were not hampered. The amount of desensitization of β2* nAChR currents depended on neuron type and cortical layer. β2*-mediated responses of interneurons in LII–III and LVI completely desensitized, while cholinergic responses in LV interneurons and LVI pyramidal cells showed less desensitization. This discrepancy depended on α5 subunit expression. Two-photon imaging of neuronal population activity showed that prolonged exposure to nicotine limited cholinergic signaling through β2* nAChRs to deep PFC layers where α5 subunits were expressed. Together, our results demonstrate a layer-dependent decrease in cholinergic activation of the PFC through nAChRs by nicotine. These mechanisms may be one of the first steps leading up to the pathophysiological changes associated with nicotine exposure during adolescence.
Introduction
Despite negative health consequences, tobacco smoking remains a persistent drug addiction worldwide (World Health Organization, 2012). First experiences with cigarette smoking often take place during adolescence (Escobedo et al., 1993; Currie et al., 2008). The prefrontal cortex (PFC), which is involved in higher order processes such as attention, impulse control, and working memory (Groenewegen and Uylings, 2000; Miller, 2000), continues to develop during this period (Gogtay et al., 2004). As a consequence, exposure to nicotine during adolescence compromises normal PFC development (Counotte et al., 2011b; Goriounova and Mansvelder, 2012a). Repeated exposure to nicotine transiently increases nicotinic acetylcholine receptor subunit (nAChR) expression and GABAergic synaptic transmission in the PFC (Counotte et al., 2012). Secondary to this, a decrease of mGluR protein persists into adulthood and causes altered synaptic learning rules and attention behavior (Counotte et al., 2011a; Goriounova and Mansvelder, 2012b). Despite these insights into long-term changes of PFC function after nicotine exposure, it is still unclear what the initial mechanisms are by which nicotine alters cortical processing at the neuronal network level.
Rapid, phasic cholinergic signaling within the PFC is crucial for attention behavior (Parikh et al., 2007; Sarter et al., 2009) and disturbances in cholinergic signaling impair attention (Turchi and Sarter, 1997; Newman and McGaughy, 2008). nAChRs are fast ionotropic receptors and their activation kinetics suggests that they are efficiently activated by rapid increases in acetylcholine. Attention performance depends on functional nAChRs in the medial PFC (Guillem et al., 2011). Nicotinic receptors activate the PFC in a layer-specific manner (Poorthuis et al., 2013). In superficial layers only interneurons are activated, whereas in deeper layers pyramidal neurons and interneurons are modulated by nAChRs. Short exposure to nicotine alters synaptic transmission and rules for plasticity induction (Couey et al., 2007). However, during smoking, blood levels of nicotine in smokers remain elevated and reach peak levels of 300–600 nm (Matta et al., 2007). These concentrations desensitize neuronal nAChRs (Mansvelder et al., 2002; Wooltorton et al., 2003; Grady et al., 2012). It is not known whether desensitization plays an important role in the PFC. The presence of α5 subunits protects β2-containing receptors in layer VI pyramidal neurons from desensitization (Bailey et al., 2010). In the PFC, α5 nAChR subunits are highly expressed (Counotte et al., 2012), but α5 subunit expression has been reported to be much lower in superficial cortical layers (Wada et al., 1990; Winzer-Serhan and Leslie, 2005). It is unknown how nicotine affects cholinergic transmission in these layers and whether α7 nAChR activation is affected by nicotine.
We tested the hypothesis that nicotine interferes with cholinergic activation of the PFC network through nAChRs and that this effect is more prominent in superficial layers. Using electrophysiological recordings and two-photon network imaging, we find that desensitization in response to nicotine is cell type and layer specific and that this can be explained by the presence of the nAChR α5 subunit. As a consequence, in the presence of nicotine, cholinergic signaling through β2* nAChRs is restricted to layer VI.
Materials and Methods
Prefrontal cortical slice preparation.
Prefrontal coronal cortical slices (300 μm) were prepared from P14–P21 and P34–P43 C57BL/6 mice or α5 wild-type and α5-null littermates P34–P43 of either sex, in accordance with institutional and Dutch license procedures. Following rapid decapitation, the brain was removed from the skull in ice-cold artificial CSF containing 125 mm NaCl, 3 mm KCl, 1.25 mm NaH2PO4, 3 mm MgSO4 1 mm CaCl2, 26 mm NaHCO3, and 10 mm glucose (∼300 mOsm). After removal of the cerebellum the brain was glued on this plane to create a coronal orientation for cutting slices. Slices were then transferred into holding chambers containing aCSF 125 mm NaCl, 3 mm KCl, 1.25 mm NaH2PO4, 1 mm MgSO4 2 mm CaCl2, 26 mm NaHCO3, and 10 mm glucose (∼300 mOsm) and bubbled with carbogen gas (95% O2/5% CO2) to recover for at least an hour.
Electrophysiology.
Slices were transferred to the recording chamber and perfused with standard aCSF (2–3 ml/min). All experiments were performed at 31−34°C. Cells were visualized using differential interference contrast microscopy. Recordings were made using Multiclamp 700B amplifiers (Molecular Devices), sampled at a frequency of 20 kHz, digitized by the pClamp software (Axon), and later analyzed off-line. Patch pipettes (3–5 MOhms) were pulled from standard-wall borosilicate capillaries and were filled with intracellular solution: 140 mm K-gluconate, 1 mm KCl, 10 mm HEPES, 4 mm K-phosphocreatine, 4 mm ATP-Mg, and 0.4 mm GTP (pH 7.2–7.3, pH adjusted to 7.3 with KOH; 290–300 mOsm) and biocytin (4 mg/ml; used for EPSC and puff application experiments, reversal potential chloride ∼-127 mV, hence IPSCs in this case are detected as outward currents). Action potential profiles of cells were made using hyperpolarizing and depolarizing current steps. For IPSC experiments a modified intracellular solution was used with a high chloride concentration (70 mm K-gluconate and 70 mm KCl) to augment GABAergic currents (reversal potential for chloride is ∼-16 mV, hence GABA currents are detected as inward currents). All IPSC experiments were done in the presence of DNQX (10 μm). All experiments recording IPSCs or EPSCs were done in the presence of atropine (200 nm) to prevent muscarinic receptor stimulation. For network experiments, acetylcholine (1 mm) was bath applied. Nicotine (Sigma, 300 or 3000 nm) was bath applied in all experiments.
Nicotinic receptor currents on interneurons and pyramidal neurons were tested by pressure ejection of acetylcholine (Sigma, 1 mm) for 100 ms using a Picospritzer III (General Valve Corporation) from a glass electrode with a tip opening of ∼1 μm. The puffer pipette was located ∼20 μm from the soma and placed in perpendicular direction with respect to the pial surface. The presence of atropine (200 nm) prevented stimulation of muscarinic receptors, and during all experiments DNQX (10 μm) and bicuculline (1 μm) were used to block synaptic transmission. Nicotine (Sigma, 100 and 300 nm) was bath applied in all experiments.
Analysis and statistics for electrophysiological experiments.
Frequency of EPSCs or IPSCs was analyzed using MiniAnalysis (Synaptosoft). Local pressure application experiments were analyzed using custom made software for Matlab (MathWorks). The effect of nicotine on cholinergic signaling was determined by calculating the charge of ACh-induced currents before, during, and after exposure to nicotine. In case cells showed a mixed α7/β2-mediated nAChR current, the charge of the β2 current was calculated after the α7 current ended (∼300 ms). The different receptor currents were well distinguishable by the different rise times of the two components and the full α7 component remained after desensitization. In addition, the β2 currents are >10 times longer than α7 currents (∼3–10 s), hence taken out the α7 had little influence on determining the charge of the β2 receptor. In Figure 1, A3 and B3, only the charge of the β2 component was plotted, while the α7 component was not plotted. To test for frequency differences in PSCs we used a Student's t test. To test for effects of pharmacology or genotype effects on nAChR charge induced by puff application of Ach, a Student's t test was used. Statistical tests for stable baseline currents were done on the raw data. Statistical tests for effects of desensitization were done on normalized data and by comparing the last data point before nicotine application with the first data point after 10 min of nicotine. In all desensitization experiments, analysis was done on the charge of the nAChR currents. Significant results were obtained with p <0.05. p values between 0.05 and 0.01 are shown as <0.05. p values between 0.01 and 0.001 are shown as p < 0.01 and p values lower than 0.001 are shown as p < 0.001.
Two-photon calcium imaging: loading.
Slices were made as described before, but in an alternative slicing solution (27 mm NaHCO3, 1.5 mm NaH2PO4, 222 mm sucrose, 2.6 mm KCl, 0.5 mm CaCl2, 3 mm MgSO4. Hereafter, slices were incubated in regular aCSF at 35°C for 20 min and in room temperature for another 40 min. For bulk loading, a modified protocol based on the study by Trevelyan et al. (2006) was used. Briefly, slices were first preincubated at 37°C for 5 min in 3 ml of aCSF containing 8 μl of Cremophor EL solution (0.5% Cremophor EL in DMSO). After this, 1 μl of Fura-2AM solution (25 μg of Fura-2AM in 4.5 μl of DMSO and 0.5 μl of pluronic acid) was pipetted on top of each slice. Then, the slices were left for incubation for 35–40 min after which they were put back in the slice chamber with aCSF at room temperature for at least 45 min. Imaging experiments were performed in aCSF (perfusion speed 2.5 ml/min), continuously bubbled with 95% O2/5% CO2, at 32°C. Imaging was performed using a multibeam two-photon laser-scanning microscope system (Trimscope, Lavision BioTec) coupled to a Ti:Sapphire laser (Chameleon, Coherent, excitation at 820 nm) and a CCD camera (C9100 Hamamatsu). The objective used had a 20× magnification and a 0.95 numerical aperture. The imaged plane was always in the same orientation with respect to the pia and the distance between them was determined for later analysis. The imaged area was 400 × 400 μm (pixel size of 0.8 μm, binning 2 × 2) and the imaging frequency was 9 Hz.
Experimental protocol.
Baseline activity was imaged during a 4 min period. After this, nicotine (300 nm) was applied for 10 min. During the first 4 min of nicotine perfusion, the activity in the slice was imaged. Then, ACh (1 mm) and nicotine (300 nm) were applied for 2 min after which the drugs were washed out (8 min). During these periods imaging took place.
Analysis.
Analysis was done using custom-made software for Matlab (MathWorks). This program detected cell contours and extracted the fluorescence within these contours as a function of time. After this, cell activity was determined per minute in a blind fashion. Cells were divided in three depth groups, corresponding to the measured thicknesses of the three layers in the PFC. Neurons that were between 100 and 300 μm, between 300 and 550 μm, and between 550 and 800 μm were considered to be part of layer II/III, V, and VI, respectively. For determining the activity in the different drug conditions, the percentage of neurons showing at least one calcium event was calculated per slice per minute. If slices included multiple layers, then the slice was split up into two new slices containing just one layer. Effects of drugs, layer, and condition were tested using repeated-measures ANOVA, followed by Fisher's LSD post hoc tests. After this, for direct comparison of the activations in the different cell types in the different conditions, it was determined per neuron whether the activity after ACh application was higher, lower, or equal to the amount of calcium events in the minute before ACh application. χ2 tests were performed to test whether this statistic was different for the multiple layers, condition, and neuron types. In addition, binomial tests were used to determine the significance of the activation for every combination.
Determination of cell identity.
High resolution z-stacks were made to optimize the possibilities for identification (voxel size: 0.4 × 0.4 × 0.5 μm). For the majority of neurons, proximal dendrites showed strong fluorescence. Cells were only taken into account if dendritic fluorescence was sufficient and cells could be identified as interneurons or pyramidal neurons according to the following criteria: (1) the presence of a clear apical dendrite, (2) a pyramidal-shaped cell body for pyramidal neurons; (3) a clear nonpyramidal cell body morphology; and (4) bipolar or multipolar dendrite morphologies for the interneurons. Criteria 1 and 2 classified the neuron as pyramidal. Criteria 3 and 4 classified a neuron as interneuron. If the dendrites were not visible in the z-stack, the neurons were not categorized. Identification of cells was done in a blind manner, i.e., the experimenter was unaware of whether neurons were activated by nicotine receptor stimulation or not, excluding the possibility of a bias. After morphological identification, data were compared with electrophysiological experiments. If neurons could not be unequivocally identified, they were excluded from statistics on cell type-specific activation.
Results
Desensitization of LII–III β2*-nAChR current responses by smoking concentrations of nicotine
To test the hypothesis that nAChR currents desensitize more strongly in PFC LII–III than in LVI, we first targeted layer II–III nonfast-spiking (NFS) interneurons (Fig. 1A2), the only cell type in this PFC layer that expresses β2-containing nAChRs (Fig. 1A1) (Poorthuis et al., 2013). ACh-induced β2* nAChR-mediated currents had slow rise and decay times, were blocked by dihydro-β-erythriodine (DHβE), and were absent in β2-null mice (Fig. 1A3) (Poorthuis et al., 2013). nAChR currents were induced by pressure application of ACh (1 mm, 100 ms) at 2 min intervals (Fig. 1A4). These applications induced repeatable postsynaptic currents that were stable over time (Fig. 1B1, the third vs the first response, 100% vs 98.4 ± 14%, Student's t test, p = 0.49). We then tested the effect of a 10 min nicotine application of 300 nm, which resembles arterial blood concentration profiles during cigarette smoking (Matta et al., 2007), on these ACh-induced currents. After 10 min of nicotine application, responses to ACh were strongly reduced on LII–III NFS interneurons (Fig. 1B1,3; n = 5, 17.4 ± 0.06% remaining response, p < 0.01). The reduction of ACh-induced currents remained after nicotine was washed-out from the bath for up to 45 min (Fig. 1B1; n = 4; at 15 min, 45.2 ± 10% remaining response, p < 0.01; at 30 min, 64.7 ± 9.0% remaining response, p < 0.05; at 45 min, 83.4 ± 11% remaining response, p = 0.11; at 60 min, 78 ± 6% remaining response, p = 0.40). This suggests that β2* nAChRs expressed by PFC LII–III NFS cells were desensitized by exposure to smoking concentrations of nicotine.

Desensitization of LII–III β2*-nAChR responses by smoking concentrations of nicotine. A1, Schematic showing nAChR receptor distribution in PFC LII–III microcircuitry. FS, Fast-spiking interneuron; NFS, nonfast-spiking interneuron; P, pyramidal neuron. Gray synapse, Glutamatergic input; black synapse, inhibitory input. β2* nAChRs and α7 nAChRs are indicated with turquoise and purple colored ovals. Right panel shows the recording configuration used to test for desensitizing effects of nicotine on LII–III β2*-nAChR responses. A2, Morphological staining of a LII–III NFS interneuron in the adolescent PFC. Scale bar, 100 μm. A3, β2* nAChRs on NFS interneurons are characterized by slow rise and decay kinetics and are blocked by DHβE (wild type example traces). In β2-null mice these current are absent and only short-lasting currents with a fast rise-time characteristic of α7 nAChRs remain [right example traces, see the study by Poorthuis et al. (2013)]. A4, Example trace showing β2* nAChR currents in LII–III of the adolescent PFC evoked by puff application of ACh (1 mm) every 2 min. Low concentrations of nicotine (300 nm, 10 min, pink shading) completely abolish β2* nAChR currents in LII–III. B1, Average surface area of current responses of juvenile LII–III NFS interneurons to local ACh (1 mm) application during bath exposure to nicotine (300 nm, 10 min). Current charge remains reduced when nicotine is washed out of the bath for up to 45 min. B2, Same as in B1, but now for adolescent NFS interneurons. In gray the effect of exposure to 100 nm nicotine is shown. Note that the desensitization rate is slower and recovery from desensitization quicker. B3, Summary histogram quantifying the desensitizing effect of a 10 min nicotine (300 nm) application on the current charge of β2* nAChRs in juvenile (n = 6, Student's t test, p < 0.01) and adolescent LII–III NFS interneurons (n = 7, p < 0.01). The degree of desensitization was not different between the age groups (p = 0.15). The right bar shows that 100 nm nicotine also strongly interfered with ACh-induced β2*-mediated currents in adolescent LII–III NFS neurons (n = 6, p < 0.01), but less compared with 300 nm nicotine ($p = 0.03). C1, Histogram showing that nicotine abolished the effect of ACh on inhibitory transmission to layer II–III pyramidal neurons. Response without nicotine is shown in gray. C2, Same experiment as in A1, but now for 3000 nm nicotine. C3, Summary showing the effect of nicotine on ACh induced increase of inhibitory transmission to layer II–III pyramidal neurons. Nicotine completely abolished cholinergic control over inhibitory transmission (300 nm, n = 7, p = 0.03; 3000 nm, n = 6, p = 0.02). All statistical tests for Figures 1⇓⇓⇓⇓–6 used Student's t test. * Denotes significance within test group, $ denotes significance between test groups.
Adolescence (P34–P43) is a period in which rodents are in particular vulnerable to the effects of nicotine on PFC-dependent cognitive functioning (Counotte et al., 2011a). Nicotinic AChR subunit expression changes during development and may therefore alter the sensitivity of receptors for nicotine and desensitization. To test whether nAChR-mediated currents in the adolescent PFC similarly desensitize, we performed the same experiment in mice at this developmental period. Acetylcholine application induced stable currents (Fig. 1B2, first vs third response, 100% vs 87.9 ± 12.5, p = 0.96). Nicotine application abolished ACh-induced β2*-mediated currents in adolescent LII–III NFS neurons (Fig. 1B2,3; n = 7, 6.7 ± 2.5% remaining after 10 min of nicotine, p < 0.01). Similar to the ACh responses in juvenile neurons, β2*-nAChR-mediated responses were reduced for a prolonged period of time in adolescent neurons (Fig. 1B2; n = 3, at 15 min, 10.9 ± 2.6% remaining response, p = 0.02; at 30 min, 37.1 ± 2.9% remaining response, p = 0.06; at 45 min, 61.8 ± 4.2% remaining response, p = 0.28; 60 min 73.7 ± 6.0% remaining response, p = 0.99), suggesting that also in adolescent PFC neurons β2*-nAChRs strongly desensitize. Two of seven recorded cells contained a mixed β2*- and α7-nAChR-mediated response. In these cells, the α7 component was not desensitized by nicotine (data not shown). We also tested whether a lower nicotine concentration, as observed in smokers between cigarettes in the afternoon (Matta et al., 2007), would have a desensitizing effect on nicotinic receptor currents. Application of 100 nm nicotine strongly reduced ACh-induced β2*-mediated currents in adolescent LII–III NFS neurons (Fig. 1B2,3; n = 5, 21.4 ± 6.3% remaining after 10 min of nicotine, p < 0.01), but the reduction was less compared with 300 nm nicotine (Fig. 1B3, p = 0.03).
Activation of β2* nAChRs enhances GABAergic signaling onto pyramidal neurons in the PFC (Couey et al., 2007; Poorthuis et al., 2009, 2013). We tested whether nicotine (300 nm) interferes with cholinergic modulation of IPSCs received by LII-III pyramidal neurons by applying nicotine for 10 min followed by coapplication of nicotine and ACh (1 mm). In the absence of nicotine, ACh dramatically increases the frequency of IPSCs in layer II–III pyramidal neurons (Fig. 1C3; n = 10, 505.3 ± 148.2%, p < 0.01). After exposure to nicotine, ACh hardly increased IPSC frequency anymore (Fig. 1C1–3; 300 nm nicotine, n = 7, 122.7 ± 11.3%, p = 0.06; 3000 nm nicotine, n = 6, 104.8 ± 6%, p = 0.09; ACh-control vs ACh-nicotine (300 nm), p = 0.03). Together, these data suggest that smoking concentrations of nicotine desensitize β2* nAChRs in LII/III. Thereby, nicotine interferes with cholinergic control through nAChRs over inhibitory circuits in superficial layers of the PFC.
Smoking concentrations of nicotine do not affect cholinergic signaling through α7 nAChRs
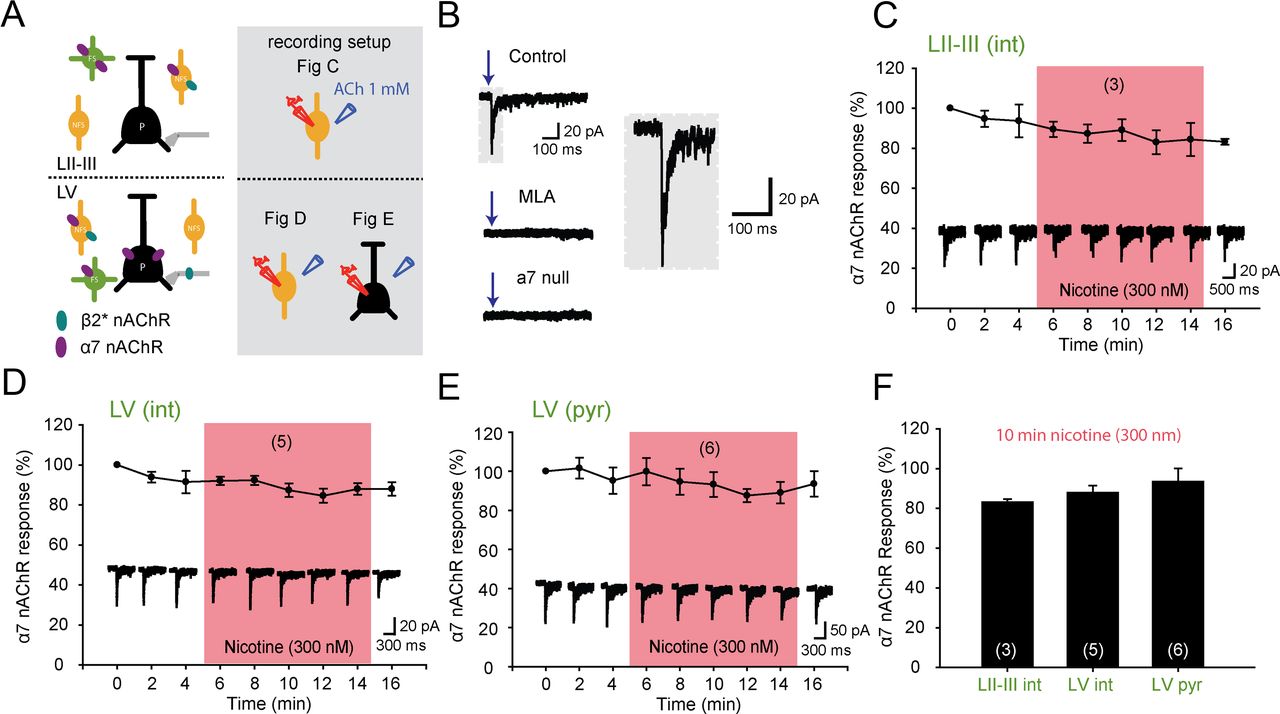
In other brain areas, nAChRs containing α7 subunits suffer less from desensitization by low concentrations of nicotine than β2-containing nAChRs (Mansvelder et al., 2002; Wooltorton et al., 2003). In the PFC, α7 nAChRs are expressed by LII–III and LV fast-spiking and nonfast-spiking interneurons, as well as by LV pyramidal neurons (Fig. 2A) (Poorthuis et al., 2013). We hypothesized that in the PFC cholinergic signaling through α7 nAChRs is not influenced by concentrations of nicotine experienced by smokers. We targeted interneurons in LII–III positive for α7 nAChRs. α7 nAChR-mediated currents had a fast rise and decay time, were blocked by methyllycaconetine (MLA), and were absent in α7-null mice (Fig. 2B) (Poorthuis et al., 2013). Similar to β2* nAChRs, repeated ACh-induced currents mediated by α7 nAChRs were stable and showed a constant amount of charge (Fig. 2C, third vs first response, 100% vs 94 ± 8%, p = 0.3). Subsequent exposure of the receptors to 300 nm nicotine for 10 min did not significantly alter ACh-induced currents (Fig. 2C,F; 83.0 ± 1.4% remaining after 10 min of nicotine, p = 0.31). A similar result was obtained for layer V interneurons (Fig. 2D,F; 88.0 ± 3.4% remaining after 10 min of nicotine, p = 0.69) as well as layer V pyramidal neurons (Fig. 2E,F; 93.5 ± 6.6% remaining after 10 min of nicotine, p = 0.25). Hence, these data show that nicotine concentrations seen in smokers during cigarette smoking do not hamper cholinergic stimulation of α7 nAChRs in the PFC.

Smoking concentrations of nicotine do not affect α7 nAChR currents. A, Left, nAChR modulation of PFC microcircuitry in LII–III and LV. β2* nAChRs and α7 nAChRs are indicated with turquoise and purple colored ovals. Right panel in gray shading shows the recording setup for the different experiments. B, Characteristics of α7 nAChRs. Currents show rapid activation and desensitization kinetics and are blocked by MLA and absent in α7-null mice (Poorthuis et al., 2013). In gray shading a magnification of an α7 current is shown. C, Effect of nicotine (pink shading, 300 nm, 10 min) on α7 nAChR current responses induced by ACh (1 mm) application on juvenile LII–III interneurons. Nicotine does not interfere with a7 nAChR activation in the PFC. D, Same as in C but now for interneurons in layer V. E, Same as in C but now for layer V pyramidal neurons. F, Summary bar graph showing the effect of smoking concentrations of nicotine on α7 (300 nm, 10 min) nAChR currents. Nicotine exposure did not desensitize α7 nAChR currents throughout the PFC (Student's t test, p = 0.31, p = 0.69, and p = 0.25 for LII–III interneurons, LV interneurons, and LV pyramidal neurons, respectively).
Partial interference of nicotine with β2* nAChR-mediated cholinergic responses in LV
In layer V of the PFC, β2* nAChRs are found on glutamatergic inputs and nonfast-spiking interneurons. Stimulating the latter increases inhibitory inputs to pyramidal neurons (Fig. 3A1) (Poorthuis et al., 2013). Nonfast spiking interneurons in juvenile mice were targeted and tested for the effect of nicotine on β2* nAChR-mediated cholinergic responses. A 10 min application of nicotine (300 nm) strongly reduced β2* nAChR-mediated responses (Fig. 3A2,A3; 30.6 ± 4.0% remaining charge, n = 9, p < 0.01). However, compared with the reduction in ACh-induced current by nicotine in LII–III NFS neurons, the reduction in NFS neurons in LV was less complete and a substantial ACh-induced current remained (Fig. 3A2,A3). Thus, β2-containing nAChRs expressed by LV NFS neurons desensitized to a lesser extent than β2-containing nAChRs expressed by LII–III NFS neurons (Fig. 3A3, p < 0.05).

Partial interference of nicotine with β2* nAChR-mediated currents in LV. A1, Microcircuitry showing nAChR distribution in layer V of the PFC. On the right, in gray shading, the recording setup for Figure 3A–C is shown. A2, Average current responses of juvenile LV NFS interneurons to local ACh (1 mm) application during bath exposure to nicotine (300 nm, 10 min, pink shading). ACh-induced currents are not completely abolished after 10 min. Currents remain smaller for up to 45 min when nicotine is washed out of the bath. A3, Summary histogram showing the desensitizing effect of nicotine on ACh-induced β2* nAChR responses. Nicotine significantly interferes with β2* nAChR currents in LV NFS-interneurons (*p < 0.01), but the desensitization is less compared with LII–III (Student's t test, $p < 0.05). B1, Histogram showing nicotine only partially interferes with the effect of ACh on inhibitory transmission to layer V pyramidal neurons. Response without nicotine is shown in gray. B2, Same experiment as in A1, but for 3000 nm nicotine. B3, Summary showing the effect of nicotine on the ACh-induced increase of inhibitory transmission to layer V pyramidal neurons (300 nm, p < 0.01; 3000 nm, p < 0.01). C1, Histogram showing that nicotine strongly interfered with the effect of ACh on glutamatergic transmission to layer V pyramidal neurons. Response without nicotine is shown in gray. C2, Same experiment as in A1, but for 3000 nm nicotine. C3, Summary bar graph showing the desensitizing effect of nicotine on the ACh-induced increase of excitatory transmission to layer V pyramidal neurons (300 and 3000 nm, p < 0.01).
Spontaneous IPSCs received by LV pyramidal neurons were strongly enhanced by ACh application (Fig. 3B1,3; n = 16, 351 ± 41%, p < 0.01). After nicotine application, ACh still increased IPSC frequency (Fig. 3B1,3; n = 10, 171 ± 22%, p = 0.03), but less than in control conditions (p < 0.01). A high dose of nicotine (3000 nm) abolished ACh modulation through nAChRs of IPSCs (Fig. 3B2,3; n = 8, 112 ± 5%, p = 0.08). Thus, in line with the results on LV NFS neurons, nicotine only partially interfered with cholinergic modulation through β2* nAChRs of IPSCs received by LV pyramidal neurons.
Activation of β2* nAChRs strongly enhances glutamate release from thalamic projections to PFC LV pyramidal neurons (Lambe et al., 2003). Nicotine (300 nm) partially reduced the ACh-induced increase in frequency of spontaneous EPSCs (Fig. 3C1,3; control n = 21, 992 ± 172%, p < 0.01, nicotine n = 11, 340 ± 34%, p < 0.05, nicotine vs control, p < 0.05). This reduction was more prominent with a higher dose of nicotine (Fig. 3C2,3; n = 6, 118 ± 12%, p = 0.96, control vs nicotine, p < 0.05). Together, these data show that in PFC LV, nicotine partially interferes with β2* nAChR signaling on NFS interneurons and glutamatergic inputs received by LV pyramidal neurons.
Differential desensitization of β2*-mediated nAChR currents in layer VI
Layer VI pyramidal neurons are relatively spared from desensitization because of the presence of α5 subunits (Bailey et al., 2010). Whether this holds true for LVI interneurons, which are also modulated by β2* nAChRs (Fig. 4A) (Poorthuis et al., 2013), is not known. To investigate possible differences we targeted these two cell types. A 10 min application of nicotine completely abolished β2* nAChR-mediated responses to ACh application on nonfast-spiking interneurons (Fig. 4B,C,F; 13.1 ± 3.4% remaining charge, n = 7, p < 0.01). In contrast, β2* nAChR-mediated responses to ACh application of pyramidal neurons (Fig. 4B) did not desensitize completely (Fig. 4B,D,F; 29.1 ± 3.1% remaining charge, n = 5, p < 0.01). The degree of desensitization was significantly less for LVI pyramidal neurons compared with interneurons in LVI (p < 0.01). During development, expression of nAChR subunits in LVI pyramidal neurons changes (Kassam et al., 2008). In the adolescent PFC, ACh-induced currents in LVI pyramidal neurons showed a similar degree of desensitization when exposed to nicotine as in the juvenile PFC (Fig. 4E,F; 33.6 ± 8.5% remaining charge, n = 5, p < 0.01; juvenile vs adolescence, p = 0.64). LVI interneurons showed significantly stronger desensitization of ACh-induced β2* responses than LVI pyramidal neurons (Fig. 4F, p < 0.01) and LV interneurons (Fig. 4F, p < 0.01). Desensitization of β2* nAChR-mediated ACh-induced currents by nicotine was also significantly stronger in LII–III interneurons than in pyramidal neurons in LVI (p = 0.02). Lower nicotine levels (100 nm) also had a desensitizing effect on β2* responses of LVI pyramidal neurons (Fig. 4F; 43.21 ± 8.5% remaining charge, n = 5, p < 0.01), but less compared with layer II–III interneurons (p = 0.04). These data show that layer-specific interference with cholinergic signaling also holds true for lower concentrations of nicotine.

Differential desensitization of β2*-mediated nAChR currents in layer VI. A, nAChR modulation of layer VI microcircuitry. On the right, in gray shading, the recording setup for the different experiments is displayed. B, Morphological staining of an adolescent LVI pyramidal neuron. Scale bar, 250 μm. On the right example traces are shown of acetylcholine-induced β2* nAChRs of layer VI neurons before and after exposure to nicotine (300 nm, 10 min). C, Average current responses of β2* nAChR currents during baseline and during application of nicotine (300 nm, 10 min). Nicotine strongly reduces current responses of LVI interneurons. D, Same as in C but now for LVI pyramidal neurons. E, Same as in D but now for adolescent mice. F, Summary bar graph showing desensitization of β2* nAChRs in the PFC. nAChR currents in LVI interneurons were strongly desensitized (Student's t test, p < 0.01) in contrast to nAChR currents in LVI pyramidal neurons, which remain partially available for activation ($p < 0.01). nAChR currents in adolescent layer VI pyramidal neurons desensitized (p < 0.01) to a similar degree as in juvenile mice (p = 0.64). β2* nAChRs currents of LVI interneurons desensitized more than LV interneurons ($p < 0.01). In addition, β2* nAChR currents of LII–III interneurons desensitized stronger than layer VI pyramidal neurons ($p = 0.02). 100 nm nicotine also strongly desensitized β2* nAChRs of adolescent LVI pyramidal neurons (*p < 0.01), but less compared with layer II–III ($p = 0.04).
Involvement of α5 nAChR subunit explains layer-specific interference of nicotine with cholinergic signaling
The level of desensitization of β2*nAChR-mediated ACh-induced currents differed in different PFC layers. Layer VI pyramidal neurons express the accessory α5 nAChR subunit, which protects β2* nAChRs from complete desensitization (Kassam et al., 2008; Grady et al., 2012). We hypothesized that β2* nAChRs expressed by neuron types that showed stronger desensitization did not contain the α5 nAChR subunit. To investigate this, we first used galantamine, an allosteric modulator that potentiates β2* nAChRs containing α5 subunits, but not β2* nAChRs lacking the α5 subunit (Kassam et al., 2008; Kuryatov et al., 2008). We applied acetylcholine (1 mm) with a puff electrode for 30 s and repeated this procedure after 10 min exposure to galantamine (1 μm) to test for possible potentiation in adolescent animals (Fig. 5A). ACh-induced β2*-mediated currents in layer II–III interneurons were not potentiated by galantamine exposure (Fig. 5A,B; n = 6, 30.4 ± 10.1 *10∧-9 vs 30.5 ± 7.2 *10∧-9 C, p = 0.97). In contrast, β2* nAChR currents in layer VI pyramidal neurons were potentiated after application of galantamine (Fig. 5A,B; n = 10, 64 ± 12 *10∧-9 vs 93.2 ± 14.6 *10∧-9 C, p = 0.01). These data suggest that a layer-specific receptor composition of β2* nAChRs exists in the prefrontal cortex. β2* nAChRs in layer II–III do not contain α5 subunits, whereas β2* nAChRs on layer VI pyramidal neurons do contain α5 subunits.

Galantamine does not potentiate LII–III β2* nAChRs. A, The effect of galantamine on β2* nAChRs was tested on LII–III interneurons and LVI pyramidal neurons. ACh was applied for 30 s before (black traces) and after galantamine (1 μm, pink traces) was washed in for 10 min. The bottom show the average response for LII–III NFS interneurons (n = 6) and layer VI pyramidal neurons (n = 10). The effect on the ACh-induced currents was assessed by calculating the total charge during the 30 s ACh application. B, Galantamine potentiates β2* nAChR currents on layer VI pyramidal neurons (Student's t test, p < 0.01), but not in LII–III interneurons (p = 0.97).
We next tested the hypothesis that the nAChR α5 subunit determines the different layer-specific degree of desensitization. We targeted layer II–III interneurons and layer VI pyramidal neurons in the PFC of adolescent α5-null mice and their wild-type littermates (P34-P43). LVI pyramidal neurons lacking the α5 subunit showed a faster and stronger degree of desensitization of ACh-induced currents by nicotine than wild-type LVI neurons (Fig. 6A). After 2 min of exposure to nicotine, desensitization of ACh-responses was significantly stronger in the α5 knock-out neurons compared with wild-type neurons (Fig. 6C; wild type vs knock-out; 75.2 ± 2.9 vs 53.7 ± 5.9, p = 0.01). After 10 min, β2* nAChRs of layer VI pyramidal neurons were completely desensitized, while pyramidal neurons in wild-type mice remained partially available for ACh activation (Fig. 6C; 23.9 ± 2.1% vs 8.1 ± 4.5%, p = 0.01). In layer II—III, however, the degree of desensitization was not affected by the absence of the α5 subunit at any time point (Fig. 6B; wild type vs knock-out; 54.9 ± 10% vs 65.4 ± 6.1%, p = 0.40 after 2 min nicotine and 15.0 ± 4.9% vs 11.6 ± 5.4%, p = 0.65 after 10 min of nicotine). These data confirm that α5 subunits are not expressed by LII–III neurons and therefore show a stronger degree of desensitization of β2* nAChR currents by smoking concentrations of nicotine.

Expression of a5 nAChR subunits explains layer-specific desensitization of β2* nAChR currents by nicotine. A, Recording setup of experiment and example traces of nAChR currents in PFC LVI pyramidal neurons and in LII–III interneurons of wild-type and α5-null littermates before and after exposure to nicotine (10 min, 300 nm). B, Average response of β2* nAChRs on LII–III NFS interneurons to ACh stimulation (1 mm) in wild-type and α5-null adolescent mice. The degree of desensitization was not different for any time point in the absence of the α5 subunit (p > 0.05 for all time points). C, Average response of β2* nAChRs on LVI pyramidal neurons to ACh stimulation (1 mm) in wild-type and α5 knock-out adolescent mice. The degree of desensitization in the absence of the α5 subunit was faster (at 2 min, Student's t test, p = 0.01) and stronger (at 10 min, p = 0.01).
Nicotine limits nAChR-mediated neuronal activation to layer VI pyramidal neurons
Nicotine strongly affects cholinergic activation of β2* nAChRs in a layer-specific manner. Therefore, we asked the question to what extent neuronal activation by ACh in the different layers would be affected by the presence of smoking nicotine concentrations. To test this, we used two-photon imaging of fura-2 loaded PFC slices and bath applied nicotine (300 nm) for 10 min before bath applying ACh (Fig. 7A). Bath application of ACh mainly affects action potential firing in neurons by activating β2* nAChRs (Poorthuis et al., 2013). Nicotine application increased neuronal activity in layer V and VI of the PFC (layer V, Fisher's LSD post hoc test, p < 0.01, layer VI, p = 0.04; Fig. 7B,C). In LII–III, after application of nicotine, subsequent application of ACh did not increase neuronal activity and the number of activated cells per slice was similar as control conditions (Fig. 7C,D, p = 0.82). In layer V, neurons were activated (p < 0.01) by low concentrations nicotine and subsequent application of ACh slightly increased this activity (p < 0.05, Fig. 7C,D). In layer VI, application of ACh in the presence of nicotine prominently increased neuronal activity (p < 0.001, Fig. 7C,D). To address the question whether the remaining activation of neurons in deep layers were pyramidal neurons or interneurons, we identified from the high resolution z-stacks imaged neurons as pyramidal neurons or interneurons (Fig. 7E). Nicotine application strongly reduced activation of interneurons in the PFC (p = 0.039, Fig. 7G). The effect of nicotine on pyramidal neurons was layer specific. Layer VI pyramidal neurons were the only cell type that still showed an increase in activation upon ACh application in the presence of nicotine (p < 0.001, Fig. 7F). Pyramidal neurons in layer II–III and layer V and PFC interneurons showed no significant subsequent activation by ACh (Fig. 7F,G, p > 0.05). Thus, nicotine concentrations experienced by smokers results in the loss of ACh modulation of pyramidal and interneurons in LII–III and LV. In the presence of nicotine, only layer VI pyramidal neurons will respond to fast ACh signaling.

Nicotine limits nAChR-mediated neuronal activation to LVI pyramidal neurons. A, Example of an experiment using network calcium imaging. Contours of Fura2-AM loaded neurons were detected after which traces from these neurons were extracted. Shown are calcium events before, during, and after the application of nicotine (300 nm) and ACh (1 mm). B, Rasterplot of the activity of all neurons in a slice during the experiment. C, Average percentage of active cells per slice per minute. Nicotine (300 nm) increased activity in layers V and VI (Fisher's LSD post hoc test; layer V: p = 0.0012; layer VI: p = 0.038; significant effects indicated with #) but not in layer II/III (p = 0.71). Subsequent application of ACh (1 mm) only resulted in a significant increase of the percentage of active cells in layer VI (p < 0.0001; layer II/III: p = 0.82; layer V: p = 0.11; significant effect indicated with @). D, Nicotine preapplication (300 nm) reduced the activation by subsequent ACh (1 mm) application (all layers: p = 0.002). This effects was significant for layer V (p = 0.00004) but not for layer II/III (p = 0.4) or layer VI (p = 0.09). Despite this, there remained a significant activation in layer V and VI (aCSF layer V: p = 0.000001; aCSF layer VI: p = 0.001; nicotine layer V: p = 0.032; nicotine layer VI: p = 0.035), whereas activation in layer II/III remained nonsignificant (aCSF layer II/III: p = 0.22; nicotine layer II/III: p = 0.74). E, Projection of z-stack showing the morphology of imaged neurons. F, Nicotine (300 nm) desensitized the response to ACh (1 mm) in layer V pyramidal neurons (LV vs LVI: p = 0.0036; without nicotine: p = 0.0019; with nicotine preapplication: p = 0.08), whereas layer VI pyramidal neurons remain responsive (LV vs LVI: p = 0.66; without nicotine: p < 0.001; with nicotine preapplication: p < 0.001). G, Nicotine (300 nm) desensitized the responses of interneurons to ACh (1 mm) throughout all layers (p = 0.039). H, The absence of desensitization of layer VI pyramidal neurons is dependent on the α5 nAChR subunit. Mice lacking this subunit have a desensitized response to ACh (1 mm) after nicotine preapplication in both layer V and layer VI (layer V: p = 0.40; layer VI: p = 0.64), whereas their WT littermate controls still show significant activation by ACh in layer VI (layer V: p = 0.55; layer VI: p = 0.004). The interaction between genotype and layer was significant (p = 0.027) and the activation in layer VI of the littermate controls was significantly bigger than the activation in layer VI of the α5-null mice (p = 0.004) and in layer V of the WT animals (p = 0.001).
To test whether the remaining activation of layer VI neurons depended on the presence of the α5 subunit we imaged slices from α5 knock-out and wild-type littermates. As shown in the previous experiment, there was a stronger activation of layer VI compared with layer V in wild-type mice (Fig. 7H, p < 0.01). In α5-null mice, ACh did not increase activity in layer VI in the presence of nicotine (Fig. 7H, p = 0.64), and ACh-induced activity was strongly reduced in PFC layer VI of α5-null mice compared with wild-type mice (Fig. 7H, p < 0.01). Hence, these data show that exposure to low concentrations of nicotine limits neuronal activation by cholinergic signaling through β2* nAChRs in the PFC to layer VI pyramidal neurons that express α5 subunits.
Discussion
In this study we showed that nicotine strongly reduces cholinergic activation of the PFC network and that this effect is cell type and layer specific and depends on nAChR subunit expression. Cholinergic responses mediated by β2* nAChRs desensitize after 10 min exposure to smoking concentrations of nicotine (300 nm). In contrast, α7 nAChRs remained available for cholinergic signaling throughout the PFC circuitry. β2* nAChR currents in interneurons in LII–III and LVI were completely desensitized by nicotine. β2* nAChR currents in LV interneurons were less compromised by nicotine exposure, just as β2* nAChR currents in LVI pyramidal neurons. Also, β2* nAChRs on thalamic terminals activating layer V pyramidal neurons were strongly desensitized by nicotine. A similar degree of desensitization was found in adolescent animals, a developmental time period in which the PFC is vulnerable for long-term adaptations induced by nicotine (Counotte et al., 2011b; Goriounova and Mansvelder, 2012a). Layer-dependent desensitization of β2* nAChR currents in adolescent mice was caused by the presence or absence of α5 subunits. In conclusion, nicotine greatly reduced cholinergic activation and altered the balance of cholinergic signaling through nAChRs in the PFC neuronal network depending on nAChR subunit composition.
Cigarette smoking leads to a prolonged presence of nicotine levels in the brain that reach 300–600 nm for minutes (Matta et al., 2007). Smoking of one cigarette leads to nearly complete β2* nAChR receptor saturation in humans (Brody et al., 2006). Sustained exposure to low levels of nicotinic agonists rapidly desensitizes nicotinic receptors (Fenster et al., 1997; Picciotto et al., 2008). Whether smoking nicotine concentrations influence nAChRs by desensitization in circuits involved in attention behavior was not known. We find that nicotine rapidly decreases responsiveness of β2* nAChRs in the PFC, while leaving α7 nAChRs intact. Because of coapplication of ACh and nicotine, we cannot rule out agonist competition at the receptor binding site, however, the persistent reduced responsiveness of β2* nAChRs (>45 min) after the presence of nicotine suggests that nicotinic receptors indeed were desensitized. An alternative explanation could be that nicotinic receptors were internalized (St. John and Gordon, 2001). However, the responses did recover after an hour, suggesting recovery from desensitization. The subunit specificity of receptor desensitization observed is similar to that seen in the ventral tegmental area where nicotine desensitizes β2* nAChRs on GABAergic interneurons, but not α7 nAChRs on glutamatergic terminals and dopamine neurons (Mansvelder et al., 2002; Wooltorton et al., 2003). Hence, whereas α7 nAChRs display rapid desensitization kinetics after being activated by rapid increases in agonists, they do not desensitize upon the prolonged presence of smoking concentrations of nicotinic agonist. These separate processes, referred to as “classical” and “high-affinity” desensitization (Giniatullin et al., 2005), thus operate in the PFC as well suggesting that α7 nAChRs remain available for activation by fast cholinergic transients (Parikh et al., 2007).
The desensitizing properties of β2* nAChRs are heterogeneous. The accessory α5 subunit plays a critical role in determining whether β2* nAChRs remain available for cholinergic signaling (Bailey et al., 2010; Grady et al., 2012). In the cortex, α5 subunits are preferentially expressed by neurons in deep layers (Winzer-Serhan and Leslie, 2005). Expression of α5 subunits is lower in superficial layers (Winzer-Serhan and Leslie, 2005), but still α5 could be located on NFS interneurons, which constitute a small number of cells in the PFC modulated by β2* nAChRs (Poorthuis et al., 2013). In the PFC, α5 and β2 subunits coassemble in LVI pyramidal neurons (Bailey et al., 2010). We find that the presence of α5 subunits does not extend to NFS interneurons in layer VI, which show a higher and complete degree of desensitization after nicotine exposure. However, it has been reported that some cortical interneurons express β2 and α4 subunits in combination with α5 subunits (Porter et al., 1999). We find that β2-mediated responses in LV interneurons show similar levels of desensitization as responses by LVI pyramidal neurons, suggesting that they may also express α5 subunits.
Exposure to nicotine during adolescence has perturbing effects on attention performance in later life (Counotte et al., 2011a). We investigated the effect of nicotine on cholinergic signaling in the juvenile (P14–P21) and adolescent mouse (P34–P43). Although β2*, but not α7, nAChR receptor expression changes with age (Kassam et al., 2008; Counotte et al., 2012), we find similar percentage of β2* nAChR desensitization in both age groups. Receptor desensitization and strong interference with cholinergic signaling by concentrations of nicotine experienced by smokers may be the first step in a cascade of events leading to molecular, cellular, and functional changes in the PFC. After adolescent nicotine exposure, the nicotinic receptor subunits α4 and β2 are strongly upregulated, whereas α7 and α5 subunit expression remains unchanged (Counotte et al., 2012). One may hypothesize that the strong desensitization of receptors containing the β2 subunit induces the upregulation following adolescent exposure as an adaptive strategy to maintain cholinergic signaling through these receptors. Similarly, the lack of desensitization of α7 nAChRs and the limited desensitization of α5 containing nAChRs do not trigger the upregulation. Indeed, after repeated nicotine exposure during adolescence, cholinergic control over GABAergic inhibition in LII–III is increased (Counotte et al., 2012), suggesting an augmentation of functional nicotinic receptors. Whether nAChR upregulation in the PFC after nicotine exposure during adolescence is cell type and layer specific remains to be investigated. An increase in number of nAChRs at neuronal surfaces after prolonged nicotine exposure is probably mediated by several posttranslational mechanisms (Goriounova and Mansvelder, 2012a; Govind et al., 2012). Ultimately, compensatory mechanisms secondary to altered cholinergic signaling might lead to reduced mGluR levels and consequently alters synaptic learning rules and attention behavior (Counotte et al., 2011a, b; Goriounova and Mansvelder, 2012b).
Although acute exposure to nicotine has been shown to enhance attention performance in rats under some circumstances (Hahn et al., 2003; Levin et al., 2006), nicotine has been found to decrease attention performance in mice (Bailey et al., 2010). Our integrative network approach shows that nicotine concentrations seen by smoking limits nAChR-induced action potential firing to layer VI pyramidal neurons. What could be the functional consequence of this shift in cortical computation? Fast cholinergic transients are important for cue detection and attention behavior (Parikh et al., 2007, 2010). Nicotine exposure strongly abolishes control over GABAergic circuitry in the PFC. Nicotinic receptor activation of interneurons has been shown to modulate pyramidal neuron activity and increases the threshold for induction of spike-timing dependent synaptic plasticity in cortex and hippocampus (Ji et al., 2001; Couey et al., 2007). Cholinergic signaling might therefore increase the signal-to-noise ratio in the PFC. When nicotine is present in the PFC, this mechanism is absent and might lead to compromised information processing. At the behavioral level, a lack of functional β2* nAChRs has been shown to lead to a hyperactive medial prefrontal cortex and altered social and exploratory behavior (Avale et al., 2011; Bourgeois et al., 2012), suggesting that the PFC network is disinhibited in the absence of this receptor. Supporting this, genetic deletion of β2* nAChRs also leads to impaired attention behavior, which depends on β2 subunits in the medial PFC (Guillem et al., 2011).
Nicotine-induced desensitization also reduced nAChR-mediated control over excitatory elements in layer V and VI. In the absence of nicotine, activity of pyramidal neurons in layer V is strongly enhanced by glutamate release induced by β2* nAChRs on axonal terminals originating in the medial dorsal thalamus (Lambe et al., 2003; Parikh et al., 2008; Poorthuis et al., 2013). The reduction in cholinergic nAChR-mediated control over this circuitry in the presence of nicotine might compromise cue-induced cholinergic transients and hence signal detection during attentional tasks (Parikh et al., 2010). Cholinergic induced activity of layer VI pyramidal neurons is also reduced. Part of the output neurons in layer VI form a thalamocortical loop (Kassam et al., 2008) and are important for regulating sensory presentations in the cortex (Olsen et al., 2012). Therefore, a decrease in cholinergic control of this circuitry might interfere with optimal attention performance (Bailey et al., 2010). In conclusion, nicotine leads to strong interference with cholinergic control over β2* nAChRs in the PFC that might compromise attention behavior in the short term, and leads to maladaptive changes of PFC circuitry that leads to altered attention behavior in the long term.
Footnotes
Funding was received from the Netherlands Organization for Scientific Research (NWO; 917.76.360 and 912.06.148), ERC StG “BrainSignals,” the Dutch Fund for Economic Structure Reinforcement (FES, 0908 “NeuroBasic PharmaPhenomics project”), EU 7th Framework Programme (HEALTH-F2-2009-242167 “SynSys”), and VU University Amsterdam. We thank Hans Lodder, Brendan Lodder, and Dr. Christiaan de Kock for technical assistance, and Dr. Uwe Maskos for providing the nicotinic receptor knock-out mice.
The authors declare no competing financial interests.
- Correspondence should be addressed to Huibert D. Mansvelder, De Boelelaan 1085, 1081HV Amsterdam, The Netherlands. h.d.mansvelder{at}vu.nl





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}