
Abstract
After CNS injury, axonal regeneration is limited by myelin-associated inhibitors; however, this can be overcome through elevation of intracellular cyclic AMP (cAMP), as occurs with conditioning lesions of the sciatic nerve. This study reports that expression of secretory leukocyte protease inhibitor (SLPI) is strongly upregulated in response to elevation of cAMP. We also show that SLPI can overcome inhibition by CNS myelin and significantly enhance regeneration of transected retinal ganglion cell axons in rats. Furthermore, regeneration of dorsal column axons does not occur after a conditioning lesion in SLPI null mutant mice, indicating that expression of SLPI is required for the conditioning lesion effect. Mechanistically, we demonstrate that SLPI localizes to the nuclei of neurons, binds to the Smad2 promoter, and reduces levels of Smad2 protein. Adenoviral overexpression of Smad2 also blocked SLPI-induced axonal regeneration. SLPI and Smad2 may therefore represent new targets for therapeutic intervention in CNS injury.
Introduction
CNS myelin proteins, such as myelin associated glycoprotein (MAG), Nogo, and oligodendrocyte myelin glycoprotein (OMgp), contribute to regenerative failure after spinal cord injury by inhibiting axonal growth (McKerracher et al., 1994; Mukhopadhyay et al., 1994; Chen et al., 2000; GrandPré et al., 2000; Prinjha et al., 2000; Wang et al., 2002). One effective strategy for countering these effects has been to manipulate gene expression within neurons and thereby confer resistance to myelin-associated inhibitors. The prototypical example of this is the conditioning lesion effect, in which transection of the sciatic nerve 7 d before a dorsal column lesion significantly enhances regeneration of dorsal root ganglion (DRG) central processes (Neumann and Woolf, 1999). Subsequent studies have established that elevation of intracellular cyclic AMP (cAMP) levels and CREB-mediated transcription are required for the conditioning lesion effect (Neumann et al., 2002; Qiu et al., 2002; Gao et al., 2004). To identify genes that are transcribed in response to elevation of cAMP, we performed a microarray analysis, which revealed significantly increased expression of secretory leukocyte protease inhibitor (SLPI).
SLPI is an 11.7 kDa serine protease inhibitor belonging to the family of whey acidic protein motif-containing proteins (Thompson and Ohlsson, 1986; Eisenberg et al., 1990). It is commonly found in the secretions lining the surfaces of the oral mucosa, bronchial epithelium, and urogenital tract (Thompson and Ohlsson, 1986; Fritz, 1988; Sallenave et al., 1994). Little is known about the function of SLPI in the nervous system; however, two studies have found that SLPI expression is increased after cerebral ischemia. SLPI was strongly induced in neurons, astrocytes, and microglia after middle cerebral artery occlusion (MCAO) in the rat (Wang et al., 2003), and similar increases in SLPI levels were reported in the sera of human stroke patients (Iłzecka and Stelmasiak, 2002). More importantly, adenoviral expression of SLPI in the cerebral cortex before MCAO significantly reduced infarct size, which suggests that SLPI may be neuroprotective (Wang et al., 2003). This hypothesis is supported by a recent study by Ghasemlou et al. (2010), which reported that treatment with SLPI leads to improved locomotor recovery, decreased lesion volume, and reduced myelin loss 1 week after spinal cord contusion.
Here we describe a new role for SLPI in axonal regeneration. We report that administration of exogenous SLPI overcomes MAG inhibition for several neuronal populations in vitro. We have also found that regeneration of transected dorsal column axons does not occur after a conditioning lesion in SLPI null mutant mice (Slpi−/−), which suggests that SLPI is an essential component of the conditioning lesion effect. In addition, we present evidence that myelin-associated inhibitors induce phosphorylation of the transforming growth factor β (TGFβ) signaling protein Smad2 and that SLPI can reduce levels of Smad2 protein in neurons. Last, we show that administration of SLPI enhances regeneration of injured retinal ganglion cell axons in vivo and that this effect can be blocked by overexpression of Smad2.
Materials and Methods
All animal procedures were approved by the Institutional Animal Care and Use Committee of Hunter College, City University of New York, and the Protocol Management and Review Committee of the University of Manitoba. The experiments were performed in accordance with all institutional and national regulations.
Neuronal preparations.
For cortical or hippocampal neurons, cortices and hippocampi were dissected from postnatal day 1 (P1) Long–Evans rat pups of both sexes and incubated twice with 0.5 mg/ml papain in plain Neurobasal-A media (Invitrogen). Cell suspensions were layered on an Optiprep density gradient (Sigma) and centrifuged at 2000 × g for 15 min. The purified neurons were then collected and counted.
For cerebellar granule neurons (CGNs), cerebellar cortex was isolated from P5–P6 rats of both sexes and treated with papain and soybean trypsin inhibitor as described above. After tritutation, cells were diluted in Sato's media and counted.
For DRG neurons, DRGs were isolated from P5–P6 rats of both sexes and treated with 0.015% collagenase in Neurobasal-A media for 45 min at 37°C. This was followed by a second incubation in collagenase for 30 min at 37°C, with the addition of 0.1% trypsin and 50 μg/ml DNase I. Trypsin was inactivated with DMEM containing 10% dialyzed fetal bovine serum, and the ganglia were triturated in Sato's media.
Microarray analysis and quantitative real-time PCR.
For the RNA preparations, P21–P23 Long–Evans rats of both sexes were anesthetized with isoflurane, and the right sciatic nerve was transected at the midpoint of the thigh. Animals were killed 18 h later, and the lesioned and unlesioned L4 and L5 DRGs were collected and snap frozen. P5 DRG neurons were also prepared as described and incubated for 18 h at 37°C in the presence or absence of 1.5 mm dibutyryl cAMP (dbcAMP). In both cases, the cells were homogenized in TRIzol (Invitrogen), and RNA was purified using the RNeasy RNA isolation kit (Qiagen). Microarray hybridization and quantitative real-time PCR were then performed as described previously (Cao et al., 2006). The full results of the microarray can be viewed on the following website: http://genome.rutgers.edu/slpi/.
Endpoint PCR.
Neonatal rat CGNs, DRGs, and cortical neurons were treated with 1 mm dbcAMP (Calbiochem) and incubated for 18 h at 37°C. RNA was then isolated using the RNeasy RNA isolation kit (Qiagen). For the conditioning lesion experiments, P28 Long–Evans rats of both sexes received unilateral sciatic nerve lesions and were killed 24 h later. The lesioned and unlesioned lumbar DRGs (L2–L5) were removed and homogenized in TRIzol. RNA was extracted using chloroform, precipitated with isopropanol in the presence of linear polyacrylamide, and solubilized in RNase free water.
RNA was reverse transcribed using oligo-dT and AccuScript High Fidelity RT (Stratagene), and the resulting cDNA was amplified using PfuUltra High Fidelity DNA polymerase (Stratagene). The following primers were used for SLPI and glyceraldehyde-3-phosphate dehydrogenase (GAPDH): SLPI forward, 5′-CCTGCCTTCACCATGAAGT-3′; SLPI reverse, 5′-CCAAATGTCAGGAATCAGAC-3′; GAPDH forward, 5′-ATGGTGAAGGTCGGTGTGAACG-3′; and GAPDH reverse, 5′-TGGTGAAGACGCCAGTAGACTC-3′. Densitometric measurements were made using NIH ImageJ software.
Intrathecal delivery of SLPI.
Osmotic minipumps with a flow rate of 0.5 μl/h (model 1007D; Alzet) were filled with either sterile saline or solutions of recombinant human SLPI (R&D Systems) in sterile saline at concentrations of 0.25, 0.5, and 1 μg/μl. After equilibrating overnight at 37°C, pumps were attached to a cannula and implanted into P28 Long–Evans rats anesthetized with isoflurane. A laminectomy was performed between L5 and L6, and the cannula was inserted under the dura mater so that the tip rested on the dorsal spinal cord between L4 and L5. Animals were killed 24 h later, and the lumbar DRGs (L2–L5) were collected and processed as described above.
Neurite outgrowth assay.
Monolayers of control or MAG-expressing Chinese hamster ovary (CHO) cells were prepared in eight-well chamber slides as described previously (Mukhopadhyay et al., 1994). Alternatively, suspensions of purified CNS myelin (1–2 μg/well) were plated in chamber slides and desiccated overnight. Purified P1 cortical, P5–P6 CGN, or P5–P6 DRG rat neurons were diluted to 35,000 cells/ml in Sato's media and treated with either 1 mm dbcAMP (Calbiochem) or recombinant human SLPI at one of the following concentrations: 1, 2, 5, or 10 μg/ml. Neurons from intrathecal delivery experiments received no additional treatment. Neurons were incubated for 14–18 h at 37°C and immunostained using a monoclonal anti-βIII tubulin antibody (Tuj1; Covance) and Alexa Fluor 568-conjugated anti-mouse IgG (Invitrogen). For quantification, images were taken, and the length of the longest neurite for each neuron was measured using MetaMorph software (Molecular Devices).
Conditioning and dorsal column lesions.
Slpi−/− mice of both sexes (Ashcroft et al., 2000; gift from Dr. Sharon Wahl, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD) were bred homozygously, whereas C57BL/6 and 129/SvJ mice (The Jackson Laboratory) of both sexes were mated to generate wild-type mice of the appropriate background strain. The right sciatic nerve was lesioned in C57BL/6 × 129/SvJ (n = 11) and Slpi−/− (n = 10) mice. Seven days later, these animals were anesthetized and a laminectomy was performed at the mid-thoracic level. The dorsal column of the spinal cord was then transected at T8–T10 to a depth of ∼1 mm. Another set of C57BL/6 × 129/SvJ (n = 5) and Slpi−/− (n = 7) mice received only dorsal column lesions. At 5 weeks after surgery, 2 μl of 1% cholera toxin B subunit (CTB; List Biological) was injected into the right sciatic nerve of each animal, and the animals were transcardially perfused with 4% paraformaldehyde (PFA) 3 d later. Spinal cord sections were immunostained using goat anti-CTB antibody (1:2000; List Biological), biotinylated donkey anti-goat IgG (1:200; List Biological), and avidin–biotin complex (Vector Laboratories). To visualize CTB, the sections were reacted in a solution of 0.05% diaminobenzidine tetrahydrochloride, 0.04% nickel chloride, and 0.015% hydrogen peroxide.
Individual images of the spinal cord sections were taken under bright-field optics and combined into photomontages using Adobe Photoshop (Adobe Systems). The photomontages were then analyzed using NIH Image J software. Pixel thresholding was performed to identify CTB-labeled axons, and 10,000 μm2 blocks were drawn at distances 100, 200, and 300 μm rostral and caudal to the lesion site. The area within each block was then measured to determine the area occupied by CTB-labeled axons at that particular point. A minimum of two sections were measured for each animal.
Nuclear localization of SLPI.
P5–P6 rat CGN were treated in suspension with 1, 5, or 10 μg/ml recombinant human SLPI and incubated for 1 h at 37°C. Nuclear and cytoplasmic fractions were then isolated using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific). Fifteen percent gels were used for SDS-PAGE, and proteins were transferred to nitrocellulose at 50 V for 45 min. Membranes were treated with goat anti-human SLPI affinity-purified IgG (1:2000; R&D Systems) and horseradish peroxidase (HRP)-conjugated anti-goat IgG (1:5000; Santa Cruz Biotechnology). Membranes were subsequently stripped and reprobed using rabbit anti-phospho-CREB (1:1000; Cell Signaling Technology) and rabbit anti-actin (1:1000; Sigma).
To visualize nuclear localization of SLPI, recombinant human SLPI was labeled with fluorescein using the Fluorescein-EX Protein Labeling kit (Invitrogen). The labeling reaction was performed according to the instructions of the manufacturer, and the protein was concentrated using Centricon centrifugal filter devices (Ultracel YM-3; Millipore). The final protein concentration was ∼2 μg/μl. To test internalization, P6 rat CGNs and DRG neurons were prepared and treated in suspension with 10 μg/ml fluorescein-tagged SLPI (fSLPI) or an equivalent volume of unconjugated fluorescein from the labeling kit. Cells were then plated in poly-l-lysine (PLL)-coated chamber slides and incubated for 1 h at 37°C. Cells were then fixed with 4% PFA and immunostained for βIII tubulin as described.
To test internalization of fSLPI in vivo, 10 μg of fSLPI (2 μg/μl) or an equivalent volume of unconjugated fluorescein was injected into the vitreous chamber of the eye in adult male Fischer rats. Animals were transcardially perfused with 4% PFA 4 h later. Eyes were sectioned sagittally, and the sections were immunostained for βIII tubulin as described and counterstained with DAPI to visualize cell nuclei. Multichannel images were taken under fluorescence optics.
Neurite outgrowth assay using SLPI-conjugated beads.
Recombinant human SLPI was covalently bound to carboxylated green fluorescent microspheres (6 μm diameter; Polysciences) using a carbodiimide labeling kit (Polysciences). P5–P6 rat DRG neurons were diluted to 35,000 cells/ml in Sato's media and treated with 1 mm dbcAMP, 10 μg/ml SLPI, or a volume of beads containing 10 μg/ml SLPI protein. Cells were plated on CHO cell monolayers and incubated for 15 h at 37°C. Neurons were immunostained for βIII tubulin, and neurite outgrowth was quantified as described above.
siRNA experiments.
Accell SMARTpool rat Smad2 siRNA, Accell Green Non-Targeting siRNA, and Accell Non-Targeting siRNA (Dharmacon) were reconstituted to 100 μm using 1× siRNA buffer (Dharmacon). To assess transfection efficiency, P6 CGNs and P1 rat cortical neurons were prepared as described previously and diluted in supplemented Neurobasal-A media (Invitrogen). Neurons were plated in PLL-coated eight-well chamber slides at a density of 75,000 cells per well. At 24 h later, the culture media in each well was replaced with 100 μl of 1 μm Accell Green Non-Targeting siRNA in Accell siRNA delivery media (Dharmacon). The neurons were fixed after an additional incubation of 24, 48, or 72 h and immunostained for βIII tubulin as described. Transfection efficiency for this carboxyfluorescein-conjugated siRNA was 95–100% for P6 CGNs (data not shown).
To assess Smad2 knockdown, P6 rat CGNs were diluted in supplemented Neurobasal-A media and plated in PLL-coated 60 mm tissue culture dishes at a density of ∼1.2 million cells per plate. Cells were incubated at 37°C overnight, and then the media were replaced with 2 ml of 1 μm Smad2 siRNA, nontargeting siRNA, or delivery media alone. Cells treated with the Smad2 siRNA were lysed 24, 48, or 72 h later in 50 μl of 1× RIPA lysis buffer (Millipore), whereas cells treated with delivery media alone or nontargeting siRNA were lysed 24 and 48 h later, respectively. Samples were analyzed by Western blotting for Smad2/3 as described.
For the neurite outgrowth assays, chamber slides were coated with CNS myelin as described, and P6 rat CGNs were plated at 75,000 cells per well in supplemented Neurobasal media. The slides were incubated at 37°C for 8 h, and the culture media was replaced with 100 μl of either 1 μm Smad2 siRNA or delivery media. Cells were fixed after an additional incubation of 24, 48, or 72 h, and immunostained for βIII tubulin. Neurite outgrowth was then quantified and analyzed as described.
Chromatin immunoprecipitation.
P5–P6 rat CGNs were treated with 10 μg/ml recombinant human SLPI for 1 h at 37°C and then crosslinked with 1% PFA. Chromatin immunoprecipitation was performed using the EZ-ChIP chromatin immunoprecipitation kit (Millipore) and the following antibodies: rabbit anti-acetyl-histone H3 (Millipore), goat anti-human SLPI affinity-purified IgG (R&D Systems), and goat anti-biotin (Sigma). Immunoprecipitated and non-immunoprecipitated DNA was amplified using PCR Master Mix (Promega) and the following primers for the tumor necrosis factor ɑ (TNFɑ) (Dai et al., 2005), fatty acid synthase (FASN) (Teran-Garcia et al., 2007), and Smad2 promoters: TNFα forward, 5′-TGATGCCTGGGTGTCCCCAAC-3′; TNFα reverse, 5′-TGGTGTCCTCGCTGAGTTCTG-3′; FASN forward, 5′-GGCATCACCCCACCGACG-3′; FASN reverse, 5′-GCTCCCTCTAGGCCGCGC-3′; Smad2 forward, 5′-GGCTTGCTGGACTAACTTGC-3′; and Smad2 reverse, 5′-ACCCGCAGTTTGATTTTGAC-3′. In humans, the Smad2 promoter region contains multiple Sp-1 binding sites and is found within a CpG island 5′ to the first Smad2 exon (Takenoshita et al., 1998). We identified a similar region 5′ to the first Smad2 exon in the rat genome using National Center for Biotechnology Information Map Viewer and used Primer3 software (Rozen and Skaletsky, 2000) to generate primer sequences.
Smad2 downregulation experiments.
P6 CGNs, P6 DRG neurons, and P1 cortical neurons were prepared from Long–Evans rat pups of both sexes and diluted to a concentration of ∼750,000 cells/ml in supplemented Neurobasal-A media. P6 CGNs and P1 cortical neurons were also prepared from age-matched SLPI null mutant and wild-type mouse pups (C57BL/6 × 129/SvJ) of both sexes and diluted to the same concentration in supplemented Neurobasal-A media. Cells were then treated with 1 mm dbcAMP, incubated for 18 h at 37°C, and lysed in 50 μl of 1× RIPA buffer.
To assess the effects of a conditioning lesion on Smad2 expression, P28 Long–Evans rats (n = 3), 8-week-old SLPI null mutant mice (n = 5), and age-matched wild-type mice (n = 4) of both sexes received transections of the right sciatic nerve and were killed 24 h later. The lesioned and unlesioned lumbar DRGs were collected and lysed in 50 μl of 1× RIPA buffer.
Proteins were separated on 10% polyacrylamide gels and transferred to nitrocellulose at 75 V for 1 h. Membranes were incubated successively with rabbit anti-Smad2/3 (1:1000; Cell Signaling Technology) and HRP-conjugated anti-rabbit IgG (1:2000; Cell Signaling Technology). Membranes were reacted with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Membranes were then stripped using Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) and reprobed with rabbit anti-actin (1:1000; Sigma). Densitometric measurements were made using NIH Image J software.
Smad2 phosphorylation assays.
For experiments examining the effect of myelin-associated inhibitors and SLPI on Smad2 phosphorylation, P6 rat CGNs (3 million cells per plate) were treated with 20 μg/ml MAG-Fc, a soluble form of MAG (Tang et al., 1997), or 1 μg/ml Nogo-AP, a soluble form of the extracellular domain of Nogo (Nogo-66) conjugated to alkaline phosphatase (Koprivica et al., 2005; gift from Dr. Zhigang He, Harvard University, Cambridge, MA) for 30 min at 37°C. In the same experiment, P6 rat CGNs were treated with 10 μg/ml recombinant human SLPI (R&D Systems) for 1 h at 37°C, and this was followed by the addition of 20 μg/ml MAG-Fc or 1 μg/ml Nogo-AP for 30 min. Cells were lysed in 50 μl of 1× RIPA buffer, and Western blotting was performed as described above. Membranes were probed successively for pSmad2 [rabbit anti-phospho Smad2 antibody (Ser465/467); 1:1000; Cell Signaling Technology], Smad 2/3, and actin. Densitometric measurements were made using NIH Image J software.
Optic nerve crush experiments.
In the first set of surgeries, adult male Fischer rats (200–250 g) were anesthetized with isoflurane and placed in a stereotaxic frame. The right optic nerve was exposed and crushed with fine forceps for 10 s. The animals then received a single 5 μl of intravitreal injection of either sterile saline (n = 7) or recombinant human SLPI (2 μg/μl in sterile saline; n = 7). Lens injury was also induced (Leon et al., 2000). Animals were transcardially perfused with 4% PFA after a 14 d postsurgical survival period.
For the adenoviral injection experiments, monolayers of HEK 293 cells were infected with high-titer adenoviruses expressing either red fluorescent protein (RFP) or full-length rat Smad2 (Uemura et al., 2005; gift from Dr. Rebecca G. Wells, University of Pennsylvania, Philadelphia, PA). At 2 d later, the cells were collected and lysed by subjecting them to three freeze/thaw cycles. A 1:10 volume of 5% sodium deoxycholate was then added to release the virus from the cells, and genomic DNA was digested with DNase I. To purify the adenoviruses, saturated CsCl was added to the viral suspensions, and the suspensions were centrifuged at 35,000 rpm for 16–20 h at 4°C in an SW40 Beckman ultracentrifuge. The viral band was then collected, and the ultracentrifugation was repeated. The viral band was then dialyzed against three changes of ∼100 vol of 10 mm Tris-HCl and 1 mm Mg, pH 8.0. Aliquots were prepared and stored at −80°C, and viral titers were determined by measuring the total viral DNA. The titer for the Smad2 adenovirus was 5.6 × 107 pfu/μl, and the titer of the RFP virus was 7.96 × 1012 pfu/μl.
Adult male Fischer rats (200–250 g) received a single 5 μl intravitreal injection of either RFP or Smad2 adenovirus in the right eye. At 2 d later, the right optic nerve was exposed and crushed with fine forceps for 10 s. The animals then received a single 5 μl intravitreal injection of either sterile saline (RFP, n = 2; Smad2, n = 4) or recombinant human SLPI (2 μg/μl in sterile saline; RFP, n = 2; Smad2, n = 6). Lens injury was not induced in these animals. Animals were transcardially perfused with 4% PFA after a 14 d postsurgical survival period.
Optic nerve sections were immunostained using anti-growth associated protein-43 (GAP-43) polyclonal antibody (gift from Dr. Larry Benowitz, Harvard University, Cambridge, MA) and FITC-conjugated rabbit anti-sheep IgG (1:500; Jackson ImmunoResearch). Images were taken on a fluorescence microscope using MetaMorph software (Molecular Devices). Quantification of axonal density was performed using NIH ImageJ software. Pixel thresholding was used to identify GAP-43-positive axons within the optic nerve images, and 250,000 μm2 blocks (500 × 500 μm) were drawn on the images. These blocks encompassed the following distances from the lesion site: 0–500, 500–1000, 1000–1500, and 1500–2000 μm. The thresholded pixels in each block were then counted to determine the area occupied by GAP-43-positive axons within that segment of the nerve, and these measurements were reported in square micrometers.
Statistical analyses.
All analyses were performed using GraphPad Prism software, and data are represented as mean ± SEM. Statistical significance was assessed using paired one-tailed Student's t tests to compare two groups, and one-way ANOVAs with Bonferroni's post hoc tests to compare between three or more groups.
Results
SLPI expression is upregulated in response to elevation of intracellular cAMP levels
To identify cAMP- and conditioning lesion-responsive genes with potential roles in axonal regeneration, RNA was isolated from P5 DRGs treated with 1 mm dbcAMP for 18 h and DRGs from P21–P23 rats that received unilateral lesions of the sciatic nerve. These RNAs were hybridized to a custom microarray consisting of 5000 sequences that were 65–70 nt in length. Genes whose expression changed more than twofold under both conditions were compared using a one-tailed Student's t test (*p < 0.05), and, of the genes that reached this level of significance, 11 were upregulated and four were downregulated. The full results of the microarray can be viewed on the following website: http://genome.rutgers.edu/slpi/. SLPI expression was increased 3.9-fold in the microarray, making it one of the most highly upregulated genes in the analysis. Quantitative real-time PCR was then performed using the same samples, and this yielded an 8.5-fold increase in SLPI mRNA expression.
To confirm this result, P5–P6 DRGs and CGNs, as well as P1 cortical neurons, were treated with 1 mm dbcAMP. RNA was isolated 18 h later, cDNA was synthesized, and the samples were amplified using primers for SLPI. SLPI mRNA expression was increased ∼2- to 2.5-fold in neurons from each population after treatment with dbcAMP (Fig. 1A). In untreated neurons, SLPI mRNA was present, but levels were relatively low (Fig. 1A). To confirm that SLPI expression is also upregulated in response to a sciatic nerve lesion, P28 rats received unilateral sciatic nerve lesions, and the lesioned and unlesioned lumbar DRGs were collected 24 h later. Endpoint PCR analysis of the resulting cDNA revealed that SLPI mRNA levels were significantly increased by an average of 2.2-fold in the lesioned DRG (Fig. 1B). These observations demonstrate that SLPI expression is upregulated after a sciatic nerve lesion and that elevation of intracellular cAMP stimulates the production of SLPI mRNA in several diverse neuronal populations.

SLPI mRNA expression is increased in response to elevation of intracellular cAMP, and exogenous SLPI overcomes inhibition by MAG and myelin in vitro. A, Reverse transcription (RT)-PCR analysis of P5–P6 CGNs, P5–P6 DRG neurons, and P1 cortical neurons treated with 1 mm dbcAMP for 18 h. B, RT-PCR analysis of lesioned and unlesioned lumbar DRGs isolated from P28 rats 24 h after a unilateral sciatic nerve lesion. Graph represents the average densitometric measurements for lesioned and unlesioned DRGs from three individual animals (measured in arbitrary units) ± SEM. C–E, Untreated P5–P6 DRG neurons plated on monolayers of control (C) or MAG-expressing (D) CHO cells and SLPI-treated DRG neurons plated on MAG-expressing CHO cells (E). F–H, Untreated P1 cortical neurons plated on monolayers of control (F) or MAG-expressing (G) CHO cells and SLPI-treated cortical neurons plated on MAG-expressing CHO cells (H). I–K, Untreated P1 cortical neurons (I) and neurons treated with either 1 mm dbcAMP (J) or 10 μg/ml SLPI (K) plated on CNS myelin. L–N, DRG neurons from P28 rats that received intrathecal delivery of sterile saline plated on monolayers of control (L) or MAG-expressing CHO cells (M) and DRG neurons from P28 rats that received intrathecal delivery of SLPI plated on monolayers of MAG-expressing CHO cells (N). Neurite outgrowth was measured from a minimum of 200 neurons for each treatment. Graphs represent the average length of the longest neurite per neuron (depicted as percentage of control) ± SEM (*p < 0.05; **p < 0.01, ***p < 0.001).
SLPI overcomes inhibition by MAG and myelin in vitro
Our laboratory has demonstrated previously that the products of several cAMP-regulated genes can overcome MAG inhibition in an in vitro neurite outgrowth assay (Cai et al., 2002; Cao et al., 2006). To determine whether SLPI is capable of mediating a similar effect, P1 cortical and P5 DRG neurons were treated with increasing concentrations of recombinant human SLPI and plated on monolayers of control or MAG-expressing CHO cells. Neurite outgrowth was strongly inhibited by MAG for both DRG and cortical neurons, but after treatment with SLPI, inhibition was completely blocked (Fig. 1C–H). Neurite outgrowth was not significantly increased when SLPI-treated DRG and cortical neurons were plated on control CHO cells (data not shown), which indicates that SLPI specifically overcomes inhibition by myelin-associated inhibitors and does not enhance growth on a permissive substrate. P1 cortical neurons were also plated on substrata of purified CNS myelin, and treatment with 5 or 10 μg/ml SLPI significantly increased neurite outgrowth (Fig. 1I–K). These results demonstrate that SLPI can overcome inhibition by not only MAG but all myelin-associated inhibitors.
We next tested whether infusion of SLPI directly into the CSF could enhance the growth capacity of DRG neurons and allow them to overcome inhibition by MAG in vitro. Osmotic minipumps attached to catheters were filled with saline or SLPI at concentrations of 0.25, 0.5, and 1 μg/μl and implanted into the lumbar cisterns of P28 rats, where they remained for 24 h. The lumbar DRG neurons were then harvested and used in our neurite outgrowth assay without additional SLPI treatment. Neurons from rats that received intrathecal delivery of either 0.5 or 1 μg/μl SLPI were able to fully overcome inhibition, but saline-treated neurons were strongly inhibited by MAG (Fig. 1L–N). These observations indicate that SLPI reaches the DRGs when delivered intrathecally, remains biologically active in CSF, and induces molecular changes that allow neurons to overcome inhibition by MAG when subsequently cultured in the absence of SLPI.
SLPI expression is required for the conditioning lesion effect
Having shown that SLPI mRNA expression is significantly increased in response to a sciatic nerve lesion, we wanted to determine whether SLPI is an essential component of the conditioning lesion effect. Age-matched adult Slpi−/− mice (Ashcroft et al., 2000) and wild-type mice of the appropriate background strain (C57BL/6 × 129/SvJ) received unilateral transections of the sciatic nerve, and 7 d later these mice received dorsal column lesions at T8. At 5 weeks later, the dorsal column axons were labeled with CTB.
In wild-type mice that received only a dorsal column lesion, we observed robust labeling of dorsal column axons, which extended up to the site of injury but not beyond it (Fig. 2A). Conversely, wild-type mice that received a conditioning lesion before the dorsal column lesion showed clear regeneration of dorsal column axons into and beyond the site of injury (Fig. 2B). The extent of this regeneration is similar to what has been reported previously for wild-type mice that received conditioning lesions of the sciatic nerve (Cafferty et al., 2004; Cao et al., 2006), but it should be noted that the conditioning lesion effect in mice is far less robust than that observed in rats (Neumann and Woolf, 1999). When we examined the spinal cords of Slpi−/− mice that received only a dorsal column lesion, it was immediately apparent that no axons had crossed the lesion site (Fig. 2C). Moreover, it appeared that the CTB-labeled axons had retracted a substantial distance from the epicenter of the lesion. Slpi−/− mice that received conditioning lesions before the dorsal column lesion were remarkably similar. In most Slpi−/− mice that received conditioning lesions, there were few axons in the immediate vicinity of the injury, and none of these axons appeared to regenerate across the lesion site (Fig. 2D).

Transected dorsal column axons fail to regenerate after a conditioning lesion in Slpi−/− mice. Representative images of spinal cords from wild-type mice that received a dorsal column lesion only (A, WT DCL), wild-type mice that received a conditioning lesion before the dorsal column lesion (B, WT DCL + CL), Slpi−/− mice that received a dorsal column lesion only (C, Slpi−/− DCL), and Slpi−/− mice that received a conditioning lesion before the dorsal column lesion (D, Slpi−/− DCL + CL). Axons that have regenerated beyond the lesion site (*) are indicated by the arrowheads. E, Quantification of axonal density at points 100, 200, and 300 μm rostral and caudal to the lesion site. Graph depicts average axonal density (square micrometers) ± SEM (**p < 0.01, ***p < 0.001). Scale bar, 100 μm.
To verify our qualitative observations, axonal density was quantified at points 100, 200, and 300 μm rostral and caudal to the lesion site. When we compared axonal densities rostral to the lesion site for wild-type and Slpi−/− mice that received conditioning lesions, we found that axonal density was significantly greater for wild-type mice at both 100 and 200 μm rostral to the lesion (Fig. 2E). This indicates that axonal regeneration was impaired in Slpi−/− mice, which means that the conditioning lesion failed to enhance the regenerative capacity of Slpi−/− neurons. We therefore conclude that expression of SLPI is an essential component of the conditioning lesion effect.
Nuclear localization of SLPI is required for reversal of inhibition by MAG
Having established that SLPI can overcome inhibition by MAG and myelin, we then investigated its mechanism of action. In addition to its well-characterized ability to inhibit serine proteases (Thompson and Ohlsson, 1986; Eisenberg et al., 1990), SLPI has been shown to localize to the nuclei of leukocytes, in which it regulates gene expression (Taggart et al., 2005; Ghasemlou et al., 2010). These findings prompted us to test whether SLPI also localizes to the nuclei of neurons. We treated P6 CGNs with 1, 5, or 10 μg/ml recombinant human SLPI for 1 h and then performed subcellular fractionation to isolate cytoplasmic and nuclear fractions. As the neurons were treated with increasing concentrations of SLPI, we detected corresponding amounts of exogenous SLPI in both the cytoplasmic and nuclear fractions (Fig. 3A), which suggests that SLPI had localized to the nuclei of CGNs. No SLPI was present in lysates from untreated neurons (Fig. 3A). Taggart et al. (2005) showed that fSLPI could be visualized in the nuclei of monocytes, and so, to confirm our Western blot results, we labeled SLPI with fluorescein and used it to treat P6 DRGs and CGNs. After 1 h at 37°C, there was no visible fluorescence in DRG neurons treated with unconjugated fluorescein, but in DRG neurons treated with 10 μg/ml fSLPI, we observed a clear fluorescent signal that was particularly strong within the nucleus (Fig. 3B). Strong nuclear fluorescence was also detected in CGNs treated with fSLPI, and we confirmed the nuclear localization of fSLPI in both types of neurons through colocalization with Hoechst (data not shown). To determine whether SLPI is internalized by neurons in vivo, we performed intravitreal injections of fluorescein or fSLPI in adult rats and examined the retina 4 h later. In the animals that received injections of fSLPI, there was clear fluorescence within the retinal ganglion cell layer, but we did not observe any signal in the retina after injection of fluorescein alone (Fig. 3C).
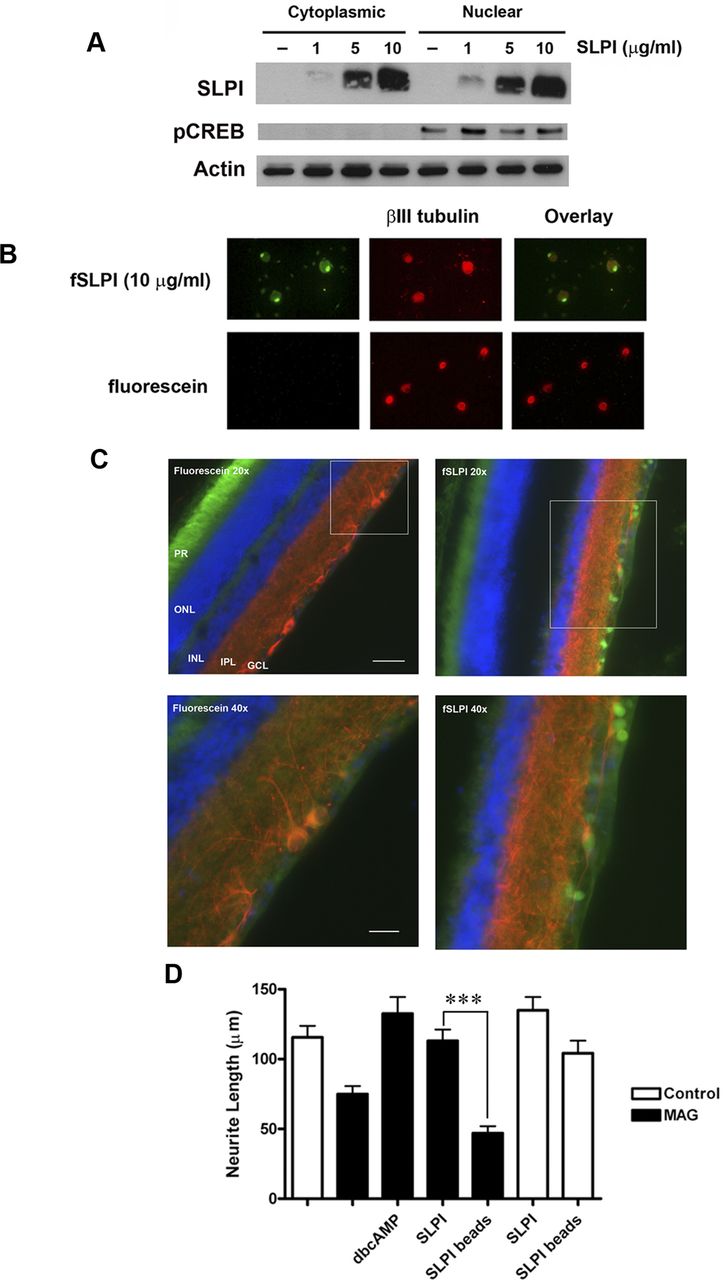
Nuclear localization of SLPI is required to overcome inhibition by myelin. A, Western blot of P5–P6 CGN treated with 1, 5, or 10 μg/ml SLPI for 1 h and subjected to subcellular fractionation. Membranes were probed for pCREB to demonstrate extraction of nuclear proteins. B, Representative images of P5–P6 DRG neurons treated with 10 μg/ml fSLPI or fluorescein alone for 1 h and counterstained for βIII tubulin. C, Representative images of retinas from adult Fischer rats that were killed 4 h after receiving intravitreal injections of 10 μg of fSLPI or unconjugated fluorescein. Sections were immunostained for βIII tubulin (red) and counterstained with DAPI (blue). PR, Photoreceptor layer; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars: 20×, 100 μm; 40×, 100 μm. D, Quantification of neurite outgrowth for P5–P6 DRG neurons treated with SLPI-conjugated beads and plated on CHO cell monolayers. Neurite outgrowth was quantified from a minimum of 200 neurons per condition. Graph depicts average neurite length ± SEM (***p < 0.001).
Our next goal was to determine whether internalization of SLPI is necessary for its ability to overcome MAG inhibition. To prevent entry of SLPI into the cells, we conjugated SLPI to carboxylated beads measuring 6 μm in diameter. P6 DRG neurons were treated with dbcAMP, SLPI, or SLPI-conjugated beads and plated on monolayers of control or MAG-expressing CHO cells. Neurons treated with dbcAMP or SLPI were able to fully overcome inhibition by MAG, but when SLPI was conjugated to beads, neurite outgrowth in the presence of MAG was significantly reduced (Fig. 3D). However, it is important to note that neurite outgrowth was unaffected for neurons that were treated with SLPI-conjugated beads and plated on control CHO cells (Fig. 3D). This suggests that the reduced neurite outgrowth observed with SLPI-conjugated beads on MAG-expressing CHO cells was attributable to the loss of SLPI internalization as opposed to toxicity or steric hindrance caused by the beads. This leads us to conclude that internalization of SLPI is required for its ability to reverse MAG inhibition, and having observed that fSLPI localizes predominantly to the nucleus, we hypothesize that this is where SLPI mediates its effect.
SLPI binds to the Smad2 promoter in CGN
Within the nuclei of monocytes, SLPI binds to the promoters for TNFα and interleukin-8 (IL-8), blocking nuclear factor-κB-mediated transcription and leading to a decrease in TNFα and IL-8 levels (Taggart et al., 2005). This raises the possibility that SLPI may downregulate the expression of genes that mediate inhibition of neurite outgrowth by myelin, and our experiments have focused on Smad2, an intermediate in the TGFβ signaling pathway. Stegmüller et al. (2008) have shown that siRNA knockdown of Smad2 enhances neurite outgrowth on myelin, which indicates that this protein is required for inhibition of neurite outgrowth by myelin, and we have confirmed these findings in our own siRNA experiments (Fig. 4). Interestingly, SLPI null mutant mice display elevated levels of active TGFβ in the epidermis after cutaneous injury (Ashcroft et al., 2000), which suggests that SLPI may modulate TGFβ signaling. We therefore tested whether SLPI can interact with the Smad2 promoter. For the chromatin immunoprecipitation experiments, P6 CGNs were treated with 10 μg/ml SLPI for 1 h at 37°C, and chromatin was extracted after crosslinking with PFA. After sonication, chromatin was immunoprecipitated using antibodies to histone H3, biotin, or recombinant human SLPI. Histone H3 and biotin serve as positive and negative controls, respectively, for the immunoprecipitation (Fig. 5). Endpoint PCR was then performed using primers specific for the TNFα promoter, the FASN promoter, or the Smad2 promoter.

Smad2 knockdown overcomes inhibition by CNS myelin. A, Western blot of P6 CGNs treated with Smad2 siRNA or nontargeting (NT) siRNA. B, Quantification of neurite outgrowth for P6 CGNs plated on CNS myelin and treated with Smad2 siRNA. C, Representative images of control (C) and siRNA-treated P6 CGNs at 24, 48, and 72 h after the addition of Smad2 siRNA. Neurite outgrowth was quantified from a minimum of 200 neurons per condition. Graph depicts average neurite length ± SEM (***p < 0.001).

SLPI binds to the Smad2 promoter. P5–P6 CGNs were treated with 10 μg/ml SLPI for 1 h and crosslinked with 1% PFA. Chromatin was isolated and immunoprecipitated (IP) using antibodies to histone H3 (positive control), SLPI, and biotin (negative control). DNA was then amplified using primers for the TNFα promoter, the FASN promoter, and the Smad2 promoter.
As mentioned, SLPI binds strongly to the TNFα promoter (Taggart et al., 2005), and so amplification with the TNFα primer served as our positive control. Samples from untreated neurons displayed only a small amount of PCR product, but amplification of chromatin immunoprecipitated from SLPI-treated neurons produced a strong band (Fig. 5), which indicates that the exogenous SLPI had bound to the TNFα promoter. FASN is a liver enzyme with no known role in inflammation or axonal growth, and so we selected this as our negative control. There was no amplification of the FASN promoter after SLPI immunoprecipitation in either control or SLPI-treated samples (Fig. 5), which shows that SLPI does not bind to this promoter. Taggart et al. (2005) reported that SLPI binds to the promoters for TNFα and IL-8 but not IL-10, and together with our data, this suggests that SLPI interacts only with specific promoters. When we immunoprecipitated for SLPI and amplified with primers for the Smad2 promoter, we observed only a weak signal in untreated samples, but samples from neurons that were treated with SLPI yielded a large amount of PCR product (Fig. 5). This indicates that SLPI binds to the Smad2 promoter in CGNs, and this interaction may inhibit transcription of the Smad2 gene.
Elevation of intracellular cAMP suppresses Smad2 expression in an SLPI-dependent manner
To further investigate the role of Smad2 in myelin-mediated inhibition, we tested whether Smad2 is downregulated in response to elevation of intracellular cAMP and whether SLPI is involved in this process. P1 cortical neurons, P6 CGNs, and P6 DRG neurons were treated with 1 mm dbcAMP for 18 h and analyzed by Western blotting. Smad2 is expressed in all three types of neurons, but after treatment with dbcAMP, levels of Smad2 were significantly reduced (Fig. 6A). When compared with untreated neurons, total Smad2 declined by an average of 60% for CGNs and DRG neurons and 45% for cortical neurons (Fig. 6A). To determine whether this effect of cAMP was SLPI dependent, we prepared P6 CGNs from SLPI null mutant and wild-type mice (C57BL/6 × 129/SvJ) and treated them with 1 mm dbcAMP for 18 h. Like rat neurons, wild-type mouse CGNs treated with dbcAMP showed significant reductions in the amount of total Smad2 (Fig. 6B). However, in SLPI null mutant CGNs, there was no significant difference in Smad2 levels between dbcAMP-treated neurons and untreated neurons (Fig. 6B).

Elevation of intracellular cAMP suppresses Smad2 in an SLPI-dependent manner. A, Western blots of P6 CGNs, P1 cortical neurons, and P6 DRG neurons treated with 1 mm dbcAMP for 18 h. B, Western blots of P6 CGNs isolated from wild-type (WT) and Slpi−/− mouse pups and treated with 1 mm dbcAMP for 18 h. C, Western blots of lysates prepared from lesioned and unlesioned lumbar DRGs isolated 24 h after a unilateral sciatic nerve lesion in P28 rats, wild-type mice, and Slpi−/− mice. Graphs depict densitometric measurements expressed as percentage of control ± SEM (*p < 0.05, **p < 0.01, ****p < 0.0001). All membranes were probed successively for Smad2/3 and actin. C, Control.
We also performed unilateral sciatic nerve lesions in P28 Long–Evans rats to determine whether the resulting increase in cAMP reduces Smad2 levels in vivo. When compared with the unlesioned ganglia, total Smad2 in the lesioned ganglia was significantly reduced (Fig. 6C). To ascertain the importance of SLPI in this effect, we performed unilateral sciatic nerve lesions in wild-type and SLPI null mutant mice. For wild-type mice, levels of Smad2 were significantly lower in the lesioned ganglia (Fig. 6C). In SLPI null mutant mice, levels of Smad2 in lesioned ganglia were not significantly different from those in unlesioned ganglia (Fig. 6C), which suggests that lesion-induced downregulation of Smad2 is SLPI dependent. Together, these findings provide evidence that the expression of SLPI is required for cAMP-mediated downregulation of Smad2.
Myelin-associated inhibitors induce phosphorylation of Smad2
The observation that Smad2 is required to mediate the inhibitory effects of myelin led us to consider whether myelin-associated inhibitors activate the TGFβ signaling pathway and induce phosphorylation of Smad2. We therefore treated P6 rat CGN with either MAG-Fc (20 μg/ml) or Nogo-AP (1 μg/ml). The lysates were analyzed by Western blotting using an antibody that recognizes Smad2 only when it is phosphorylated at serines 465 and 467. These residues are directly phosphorylated by the active type I TGFβ receptor, and phosphorylation at these sites is required for Smad2 to form a signaling complex with Smad4 (Abdollah et al., 1997). We observed some basal phosphorylation of Smad2 in CGN, but within 30 min of MAG or Nogo treatment, levels of pSmad2 had significantly increased (Fig. 7A,B).

Myelin-associated inhibitors induce phosphorylation of Smad2, and SLPI reduces pSmad2 levels in the presence of myelin-associated inhibitors. A, Western blots of P6 CGNs treated with 10 μg/ml SLPI for 1 h and then exposed to 20 μg/ml MAG-Fc for 30 min. B, Western blots of P6 CGNs treated with 10 μg/ml SLPI for 1 h and then exposed to 1 μg/ml Nogo-AP for 30 min. Graphs depict densitometric measurements expressed as percentage of control ± SEM (*p < 0.05). Membranes were probed successively for pSmad2, Smad2/3, and actin. C, Control.
These observations raise the possibility that pSmad2 is part of a common signaling mechanism for myelin-associated inhibitors, and this prompted us to investigate whether SLPI could counteract this effect. P6 CGNs were treated with 10 μg/ml recombinant human SLPI for 1 h, followed by treatment with either 20 μg/ml MAG-Fc or 1 μg/ml Nogo-AP for an additional 30 min. Remarkably, when MAG or Nogo were added to SLPI-treated neurons, pSmad2 levels were not significantly different from those observed in untreated neurons (Fig. 7A,B), which suggests that MAG- or Nogo-mediated induction of Smad2 phosphorylation did not occur in the presence of SLPI. Levels of total Smad2 were not significantly different when we compared untreated neurons and neurons treated with MAG or Nogo (Fig. 7A,B), which indicates that exposure to myelin-associated inhibitors does not lead to increased Smad2 expression in CGNs. However, in neurons treated with SLPI and myelin-associated inhibitors, we observed a clear, but not statistically significant, reduction in total Smad2 (Fig. 7A,B). These data demonstrate that myelin-associated inhibitors activate the TGFβ signaling pathway, leading to significant increases in Smad2 phosphorylation. They also show that exogenous SLPI can block MAG- and Nogo-induced phosphorylation of Smad2 and suppress Smad2 protein expression in CGNs. This supports our hypothesis that SLPI is responsible for the downregulation of Smad2 that occurs in response to elevation of cAMP. We therefore propose that SLPI lowers pSmad2 levels in MAG- and Nogo-treated neurons by reducing the amount of Smad2 protein that is available for phosphorylation.
SLPI promotes regeneration of retinal ganglion cell axons in vivo
Having observed that SLPI could overcome inhibition by myelin-associated inhibitors in vitro and reduce Smad2 levels within neurons, the next logical course of action was to test whether SLPI could enhance axonal regeneration in an in vivo model of CNS injury. For these experiments, we selected the optic nerve crush model, which has been used effectively in many studies of CNS axonal regeneration (Leon et al., 2000, Yin et al., 2006, Deng et al., 2009). In our first series of surgeries, adult rats received unilateral crushes of the optic nerve, followed by a single intravitreal injection of either 10 μg of SLPI or sterile saline. Animals were killed 2 weeks later, and the optic nerve sections were immunostained for GAP-43. It is important to note that lens injury was induced in animals from both treatment groups and that lens injury has been shown to enhance regeneration of retinal ganglion cell axons (Leon et al., 2000). We did observe some axonal regeneration in saline-treated animals, but the response was modest, with few axons extending beyond the lesion site (Fig. 8A). In contrast, injection of SLPI produced extensive regeneration of retinal ganglion cell axons (Fig. 8A). To quantify axonal regeneration in the optic nerves of these animals, we measured the density of GAP-43-positive axons at 500 μm intervals. When axonal density was compared for saline- and SLPI-treated animals, we found that axonal density was significantly higher for SLPI-treated animals at each point that was measured (Fig. 8B). This indicates that administration of SLPI significantly improves regeneration of retinal ganglion cell axons and that many of these axons extended at least 2 mm beyond the site of injury.

SLPI promotes regeneration of retinal ganglion cell axons. A, Representative images of optic nerves from rats that received intravitreal injections of sterile saline or 10 μg of SLPI after crush injury. Axons that have regenerated beyond the lesion site (*) are indicated by the arrows. B, Quantification of axonal density in regions 0–500, 500–1000, 1000–1500, and 1500–2000 μm distal to the lesion site. Graph depicts average axonal density (square micrometers) ± SEM (**p < 0.01, ***p < 0.001). Scale bar, 100 μm.
Based on these results and those of our previous experiments, we then hypothesized that SLPI enhances axonal regeneration by reducing Smad2 expression and that SLPI mediates this effect at the level of the nucleus by binding to the Smad2 promoter. If this hypothesis is correct, then SLPI-mediated axonal regeneration should be abolished by ectopic overexpression of Smad2. To achieve this, we used a rat Smad2 adenovirus that produces robust overexpression of functional Smad2 in hepatic stellate cells (Uemura et al., 2005). When tested in P1 cortical neurons, this Smad2 adenovirus produced a ninefold increase in total Smad2 after 2 d (data not shown). Adult rats received intravitreal injections of either the Smad2 adenovirus or a control RFP-expressing adenovirus, and optic nerve crushes were performed 2 d later. Immediately after the optic nerve crush, the animals received a single intravitreal injection of either 10 μg of SLPI or sterile saline and were killed 2 weeks later. These animals had no lens injury. When the optic nerve sections were immunostained for GAP-43, it was immediately apparent that there was little axonal regeneration in animals that received either the RFP virus and saline, or the Smad2 virus and saline (Fig. 9A). In animals that received the RFP virus and SLPI, regenerating axons were clearly visible within the nerves, and these axons extended up to 1 mm beyond the site of injury (Fig. 9A). However, when SLPI was administered after injection of the Smad2 virus, no axonal regeneration was observed (Fig. 9A).

SLPI-mediated regeneration of retinal ganglion cell axons is abolished by ectopic overexpression of Smad2. A, Representative images of optic nerves from rats that received intravitreal injections of RFP or Smad2 adenovirus, followed by crush injury and intravitreal injection of sterile saline or 10 μg of SLPI. Axons that have regenerated beyond the lesion site (*) are indicated by the arrows. B, Quantification of axonal density in regions 0–500 and 500–1000 μm distal to the lesion site. Graph depicts average axonal density (square micrometers) ± SEM (****p < 0.0001). Scale bar, 100 μm.
Quantification of axonal density in these animals confirmed that axonal regeneration was significantly increased in animals that received the RFP virus and SLPI and that this response was abolished in animals that received the Smad2 virus and SLPI (Fig. 9B). We therefore conclude that overexpression of Smad2 blocks the ability of SLPI to promote axonal regeneration in vivo. This raises the possibility that reducing Smad2 levels through administration of SLPI or other agents may be an effective strategy for enhancing axonal regeneration in the CNS.
Discussion
The complex nature of spinal cord injuries makes it difficult to identify potential therapeutic agents, but the conditioning lesion model gives us the opportunity to do exactly that by providing a scenario in which axons can regenerate under adverse conditions. Using this model, we have now identified SLPI as a new and promising means of promoting axonal regeneration in the adult mammalian CNS. Not only is SLPI an essential component of the conditioning lesion effect, but it can also promote CNS axonal regeneration in vivo when administered exogenously. In addition, we have shown that the effects of SLPI on regeneration can be abolished by overexpression of Smad2 and provided the first evidence that myelin-associated inhibitors induce phosphorylation of Smad2. The phosphorylation of Smad2 by myelin-associated inhibitors represents not only a new signaling pathway for these inhibitors but also a novel target for therapeutic intervention to promote axonal regeneration after injury.
There has been enormous interest in elucidating the underlying mechanism of the conditioning lesion effect, but to date, the genes responsible have remained elusive. Our data indicate that SLPI is necessary for the conditioning lesion effect, but it is not yet known whether it is sufficient. Like SLPI, both activating transcription factor 3 (ATF3) and the cytokine IL-6 are significantly increased after a sciatic nerve lesion (Tsujino et al., 2000; Cao et al., 2006). Neurite outgrowth was significantly increased when DRG neurons from ATF3 transgenic mice were cultured on permissive substrates, but these neurons were not able to overcome inhibition by myelin in vitro (Seijffers et al., 2007). Furthermore, regeneration of dorsal column axons in these mice was not improved (Seijffers et al., 2007). These findings indicate that increasing the intrinsic growth capacity of neurons is not sufficient to overcome inhibition by myelin and replicate the conditioning lesion effect. Cafferty et al. (2004) performed conditioning lesions in IL-6 null mutant mice and reported that both neurite outgrowth and regeneration of dorsal column axons were impaired. In contrast, a similar study performed by our laboratory reported that the extent of regeneration was equivalent in wild-type mice and IL-6 null mutants, which led us to conclude that IL-6 is sufficient but not necessary for the conditioning lesion effect (Cao et al., 2006). Recently, we reported that upregulation of arginase I-mediated polyamine synthesis contributes to the conditioning lesion effect and that polyamines can promote axonal regeneration in vivo (Deng et al., 2009). However, we have not yet established whether activation of this pathway is sufficient to mimic the conditioning lesion effect.
Spinal cord injury is most commonly associated with axonal damage, but there is also widespread inflammation, cell death, and demyelination. Recently it was reported that wild-type mice treated with exogenous SLPI after spinal cord contusion displayed a significant improvement in locomotor function as early as 3 d after injury, an improvement that was maintained for the 28 d duration of the experiment (Ghasemlou et al., 2010). Although axonal regeneration was not specifically examined in that study, an increase in serotonergic innervation of ventral motor neurons was reported (Ghasemlou et al., 2010). The authors attribute the functional recovery and the increased innervation to the significant increases in tissue sparing, improved motor neuron survival, and decreased expression of TNFα that were observed (Ghasemlou et al., 2010). Now we show that SLPI also has a direct effect on neurons and promotes axonal growth in an inhibitory environment. Together, these studies demonstrate that SLPI mediates a unique combination of pro-regenerative and neuroprotective effects that enhance the growth capacity of axons and create a more favorable environment within the CNS.
Chondroitin sulfate proteoglycans (CSPGs) expressed by reactive astrocytes also contribute to the inhibitory environment of the injured CNS (Asher et al., 2000; Schachtrup et al., 2010; Hellal et al., 2011), and a recent spinal cord injury study has shown that injury-induced expression of CSPGs was reduced after administration of taxol (Hellal et al., 2011). This effect was attributed to the ability of taxol to inhibit nuclear translocation of Smad2 (Hellal et al., 2011). This finding adds to a growing body of evidence that TGFβ signaling contributes to astroglial scarring (Asher et al., 2000; Schachtrup et al., 2010), and our observation that CNS myelin induces phosphorylation of Smad2 now raises intriguing questions about the role of TGFβ signaling in myelin-mediated inhibition. Our experiments show that Smad2 is phosphorylated at serines 465 and 467, which are directly phosphorylated by the active type I TGFβ receptor (Abdollah et al., 1997). This indicates that myelin proteins activate the TGFβ receptor complex, but it is not known how this would occur (Fig. 10). Given the tremendous structural differences between MAG, Nogo, and TGFβ, and the fact that the type II TGFβ receptor binds TGFβ very specifically (Massagué, 2000), it is unlikely that myelin-associated inhibitors bind directly to the type II TGFβ receptor. Expression of active TGFβ is strongly upregulated in neurons and glia within 2 d of spinal cord injury (Buss et al., 2008), and this could lead to activation of the receptor in vivo; however, it is more likely that this upregulation of TGFβ occurs as part of the acute inflammatory response (Buss et al., 2008) and does not involve myelin-associated inhibitors. The He laboratory has shown that MAG, Nogo, and OMgp can transactivate the epidermal growth factor receptor via an unknown mechanism (Koprivica et al., 2005), and so, it is possible that the binding of MAG or Nogo to the Nogo receptors could also lead to transactivation of the type II TGFβ receptor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic of myelin-mediated activation of the TGFβ signaling pathway and downregulation of Smad2 by SLPI. A, Myelin-associated inhibitors activate the TGFβ receptor complex through as yet undefined means. The activated receptor complex phosphorylates Smad2, which is essential for inhibition of neurite outgrowth by myelin. B, SLPI, either exogenously applied or produced in response to elevation of cAMP, localizes to the nucleus of the neuron and binds to the Smad2 promoter, inhibiting transcription and reducing the amount of Smad2 protein within the neuron. Thus, less Smad2 is available for phosphorylation, which allows the neuron to extend neurites in the presence of myelin.
There is now substantial evidence that Smad2 protein is required to mediate inhibition by myelin, but what is the role of pSmad2 in this process? The Bonni laboratory has provided insight into this by describing a connection between pSmad2, the transcriptional corepressor SnoN, and the E3 ubiquitin ligase Cdh1–anaphase-promoting complex (Cdh1–APC). In previous studies, they showed that Cdh1–APC negatively regulates axonal growth in the cerebellum by ubiquitinating SnoN (Konishi et al., 2004; Stegmüller et al., 2006). They subsequently demonstrated that pSmad2 interacts with SnoN within the nucleus and presents it to Cdh1–APC for ubiquitination, which leads to impaired neuritogenesis (Stegmüller et al., 2008). Knockdown of either Cdh1 or Smad2 enhanced neurite outgrowth on myelin substrates (Konishi et al., 2004; Stegmüller et al., 2008), and they have therefore proposed that the Cdh1–APC–SnoN pathway plays a role in myelin-mediated inhibition of axonal growth (Konishi et al., 2004; Stegmüller et al., 2008). Our data support and expand this hypothesis, because myelin-induced phosphorylation of Smad2 would increase SnoN degradation and thereby inhibit neurite outgrowth. It should be noted that the Cdh1–APC–SnoN pathway has been studied primarily in the context of embryonic and early postnatal development (Konishi et al., 2004; Stegmüller et al., 2006, 2008), and so it would be interesting to explore the function of this pathway in the mature CNS, particularly after injury.
The primary function of pSmad2, together with pSmad3 and Smad4, is to regulate transcription by associating with DNA binding cofactors and recruiting transcriptional coactivators or corepressors (Massagué et al., 2005). This is a highly complex process affecting hundreds of genes (Massagué et al., 2005), and the role of transcription in myelin-mediated inhibition is unknown; however, we should consider the possibility that myelin-induced activation of the Smad complex modulates transcription in a way that leads to inhibition of axonal growth. Deletion of phosphatase and tensin homolog activates the mammalian target of rapamycin (mTOR) pathway and promotes axonal regeneration in the optic nerve (Park et al., 2008). Because the mTOR pathway facilitates protein translation, it was proposed that protein synthesis is essential for axonal regeneration (Park et al., 2008). It is therefore possible that myelin-associated inhibitors suppress protein synthesis by activating Smad2 and inhibiting the transcription of genes that are required to promote growth.
Interestingly, Smad signaling through the bone morphogenic protein (BMP) pathway appears to have the opposite effect. The binding of BMPs to their cognate receptors leads to phosphorylation of Smad1, Smad5, and Smad8 (Massagué et al., 2005). Like Smad2 and Smad3, these Smads form complexes with Smad4 and translocate to the nucleus in which they act as transcriptional regulators (Massagué et al., 2005). A recent study by Parikh et al. (2011) has reported that pSmad1 plays an essential role in axonogenesis during embryonic development and enhances the growth capacity of adult DRG neurons. In addition, they also show that intrathecal injection of a BMP4 adeno-associated virus leads to activation of Smad1 and increased axonal regeneration, even when administered after a dorsal hemisection (Parikh et al., 2011). This indicates that Smad1 signaling enhances axonal regeneration, which suggests that pSmad1 may modulate the expression of pro-regenerative genes. It therefore appears that the TGFβ–Smad signaling pathway restricts axonal regeneration, whereas the BMP–Smad signaling pathway enhances it, and it is possible that the fate of a regenerating axon may ultimately be decided by the competing transcriptional events mediated by these two pathways.
Transcriptional regulation may also be central to the ability of SLPI to overcome inhibition by myelin. In HEK 293 cells, it has been shown that, once pSmad2 performs its function in the nucleus, it is ubiquitinated and degraded by the proteasome (Lo and Massagué, 1999; Seo et al., 2004), and so it is likely that new Smad2 protein must be synthesized to sustain myelin-mediated inhibition in neurons. By binding to the Smad2 promoter, SLPI would prevent de novo transcription of the Smad2 gene, and this would ultimately lead to a decrease in the amount of total Smad2 protein within the neuron (Fig. 10). Levels of pSmad2 would also be reduced because there is less protein available for phosphorylation, and this loss of Smad2 function would allow the neuron to overcome inhibition by myelin. In addition to Smad2, SLPI can also downregulate expression of the proinflammatory cytokine TNFα (Taggart et al., 2005; Ghasemlou et al., 2010), which is strongly upregulated after spinal cord injury and has been implicated in both neuronal and glial apoptosis (Lee et al., 2000; Pearse et al., 2004; Ghasemlou et al., 2010). Thus, it appears that SLPI may be capable of downregulating a variety of genes that contribute to the pathophysiology of spinal cord injury. If these genes could be identified, it would greatly advance our understanding of the mechanisms underlying regenerative failure and potentially provide new targets for pharmacological intervention.
Footnotes
- Received November 15, 2012.
- Revision received January 10, 2013.
- Accepted January 16, 2013.
This work was supported by National Institutes of Health/National Institute of Neurological Disorders (NIH/NINDS) Stroke Specialized Neuroscience Research Program Grant 3U54NS041073, NIH/NINDS Grant RO1NS037060, and NIH/National Center for Research Resources Infrastructure Grant RR003037 from the Research Centers in Minority Institutions Program at Hunter College. S.S.H. is the recipient of a postdoctoral fellowship from the Christopher and Dana Reeve Foundation. M.M.S. is the recipient of a Ruth L. Kirschstein National Research Service Award (F32) (NIH/NINDS). We thank Saranna Belgrave and Dr. Wilfredo Mellado for their outstanding technical support. We also thank Dr. Sharon Wahl of the National Institute for Dental and Craniofacial Research (NIH, Bethesda, MD) for providing the SLPI null mutant mice and Drs. Larry Benowitz and Barbara Lorber of Harvard University (Cambridge, MA) for providing instruction in the optic nerve crush surgery and the GAP-43 antibody. The Nogo-AP fusion protein construct was generously provided by Dr. Zhigang He of Harvard University. We also thank Dr. Rebecca G. Wells of the University of Pennsylvania (Philadelphia, PA) for providing the Smad2 adenovirus.
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: Dr. Marie T. Filbin, Department of Biological Sciences, Hunter College, City University of New York, 695 Park Avenue, New York, NY 10065, filbin{at}genectr.hunter.cuny.edu; or Dr. Sari S. Hannila at her present address, Department of Human Anatomy and Cell Science, University of Manitoba, 745 Bannatyne Avenue, Winnipeg, Manitoba, Canada R3E 0J9, hannila{at}cc.umanitoba.ca
- Copyright © 2013 the authors 0270-6474/13/335138-14$15.00/0