

Abstract
The accumulation of intracellular β amyloid (Aβ) may be one of the factors leading to neuronal cell death in Alzheimer's disease (AD). Using a pyrazole called CNB-001, which was selected for its ability to reduce intracellular Aβ, we show that the activation of the eIF2α/ATF4 arm of the unfolded protein response is sufficient to degrade aggregated intracellular Aβ. CNB-001 is a potent inhibitor of 5-lipoxygenase (5-LOX), decreases 5-LOX expression, and increases proteasome activity. 5-LOX inhibition induces eIF2α and PERK (protein kinase R-like extracellular signal-regulated kinase) phosphorylation, and HSP90 and ATF4 levels. When fed to AD transgenic mice, CNB-001 also increases eIF2α phosphorylation and HSP90 and ATF4 levels, and limits the accumulation of soluble Aβ and ubiquitinated aggregated proteins. Finally, CNB-001 maintains the expression of synapse-associated proteins and improves memory. Therefore, 5-LOX metabolism is a key element in the promotion of endoplasmic reticulum dysfunction, and its inhibition under conditions of stress is sufficient to reduce proteotoxicity both in vivo and in vitro.
Introduction
A major participant in Alzheimer's disease (AD) pathology may be the accumulation of intracellular amyloid-β (Aβ; Oakley et al., 2006; LaFerla et al., 2007; Steele and Gandy, 2011). However, the molecular mechanisms leading to this form of proteotoxicity are not known, nor are the pathways that promote the clearance of intracellular amyloid understood. We have used a drug discovery approach for AD based in part upon the ability of compounds to remove intracellular Aβ (Liu et al., 2008; Chen et al., 2011; Schubert and Maher, 2012). A library of hybrid compounds between curcumin and the neurotrophic molecule cyclohexyl bisphenol A was screened in toxicity assays that represent multiple degenerative pathways involved in aging and AD, including intracellular amyloid toxicity. One synthetic curcumin derivative, called CNB-001, was identified that has the ability to stimulate the removal of intracellular Aβ (Liu et al., 2008). Experiments were then performed using CNB-001 to gain insight into the molecular basis of intraneuronal amyloid clearance.
Several studies have shown that aberrant endoplasmic reticulum (ER) stress is associated with AD, and that reduced ER function enhances Aβ toxicity (Kaneko et al., 2010; Lee do et al., 2010; Lee et al., 2010; Kögel et al., 2012). In addition, the disruption of 5-lipoxygenase (5-LOX) and some phospholipases reduces AD pathology, and 5-LOX is elevated in AD (Qu et al., 2000; Firuzi et al., 2008; Sanchez-Mejia et al., 2008; Oliveira et al., 2010). However, no mechanistic link between the two has been identified. It is shown here that CNB-001 activates the neuroprotective eIF2α/ATF4 arm of the unfolded protein response (UPR), leading to the clearance of intracellular Aβ aggregates, and that 5-LOX inhibition by CNB-001 accounts for this activity. In an AD mouse model, CNB-001 also induces eIF2α phosphorylation and ATF4 expression, and reduces soluble Aβ1–42 and ubiquitinated insoluble proteins in the hippocampus.
Materials and Methods
Intracellular amyloid toxicity.
The growth and induction of intracellular amyloid toxicity in MC65 cells was performed exactly as described previously (Maezawa et al., 2006; Liu et al., 2008). PERK−/− mouse embryonic fibroblast (MEFs) were a gift from Antonis Koromilas (McGill University, Montreal, Quebec, Canada). Primary E18 cortical neurons were used 7 d after plating (Liu et al., 2008). For nuclear extraction and ATF4 measurements, MEFs were grown in 60 mm dishes for 24 h and the nuclei were harvested as described previously (Lewerenz and Maher, 2009).
huAPPswe/PS1ΔE9 transgenic mice.
The APP/PS1 transgenic mice (line 85) have been well characterized (Jankowsky et al., 2007). At 3 months of age, male transgenic mice and their wild-type littermates (8–10 per group) were fed a diet with and without CNB-001 (500 ppm). Mouse body weights and food consumption were measured weekly, and there were no significant differences between the groups. The mice were tested for behavior and killed at 9 months.
Animal behavior.
The Y-maze assay in Figure 11B was performed as described previously (Chen et al., 2011), and the acute toxicity assay was performed by PsycoGenics, as described in the figure legend.
Aβ ELISA.
Soluble and RIPA-insoluble hippocampal fractions were prepared and analyzed for Aβ as described previously (Chen et al., 2011).
Proteasome assay.
To assay the effects of the different treatments on proteasome activity, cells from the same density cultures as used for the cell death assays were treated with varying concentrations of the proteasome inhibitors for the indicated time periods. The cells were then scraped into assay buffer (50 mm HEPES, pH 7.8, 10 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 250 mm sucrose, and 5 mm DTT), sonicated, and centrifuged at 16,000 × g for 10 min at 4°C. The supernatants were assayed for proteasome activity in the same buffer supplemented with 2 mm ATP using the chymotrypsin activity substrate Suc-LLVY-AMC (7-amino-4-methylcoumarin; Biomol), the trypsin activity substrate Z-ARR-AMC (Calbiochem), or the caspase-like activity substrate Z-LLE-AMC (Calbiochem) all at 100 μm in a final volume of 250 μl. The mixture was incubated at 37°C for 60 min, and then the released fluorogenic AMC was measured at 360 nm excitation and 460 nm emission using a fluorometric plate reader. Relative fluorescence units were normalized to protein values determined using the Bradford protein assay kit (Pierce).
SDS-PAGE and immunoblotting.
All immunoblotting was done on cultured cells or RIPA-soluble hippocampal homogenates, and the protein amount was quantified exactly as described previously (Chen et al., 2011). For aggregated proteins, the entire gel was scanned. The primary antibodies are listed below.
Primary antibodies.
The following primary antibodies were used: Pro-BDNF (monoclonal, 32 kDa; BD Biosciences); 5-LOX (polyclonal, 78 kDa; StressMarq Biosciences; beta amyloid (monoclonal; Wako); Homer 1 (polyclonal, 40 kDa; Sigma); anti-actin 1:40,000 (polyclonal, 45 kDa; Enzo Life Sciences); clusterin (goat polyclonal, 35–40 kDa; Santa Cruz Biotechnology); synapsin-1, 1:10,000 (monoclonal, 80 kDa; Cayman Chemicals); phospho S603 synapsin 1 (polyclonal, 80 kDa; Invitrogen); protein kinase R-like extracellular signal-regulated kinase (PERK; polyclonal, 150,000 kDa; Sigma); phospho-T980 PERK (polyclonal, 150,000 kDa; Cell Signaling Technology); ubiquitin (monoclonal; Bioscience Research Reagents); heat shock protein 90 (HSP90; polyclonal, 90 kDa; Cell Signaling Technology); eIF2α (polyclonal, 38 kDa; Cell Signaling Technology); phospho-eIF2α (polyclonal; Cell Signaling Technology); and ATF4 (polyclonal, 55 kDa; Santa Cruz Biotechnology).
Immunofluorescence of MC65 cells.
Immunofluorescence labeling and confocal microscopy for Aβ-immunoreactive aggregates were performed according to published protocols (Maezawa et al., 2004).
Transfection of MC65 cells.
MC65 cells were plated at a density of 5 × 105 cells/well in 35 mm culture dishes and grown for 24 h. Cells were transfected with 0.5 μg of expression plasmid for GADD34, washed twice with PBS, and incubated for 48 h in OptiMEM with or without tetracycline (tet) and CNB-001, 0.5 μm. Aβ aggregates were measured by Western blot using the antibody 6E10.
mRNA assays.
Total RNA was extracted using the RNeasy kit (Qiagen). One microgram of total RNA was used for reverse transcription reaction, and mRNA was converted to cDNA using oligo dT primers and Superscript II reverse transcriptase (Invitrogen). For quantitative PCR (qPCR) analysis, 1 μl of total cDNA was diluted in 6 μl of H2O, and 3 μl of this dilution was used in each reaction.
qPCR procedures.
Primers for qPCR were designed using Primer3 software to yield a 75–150 bp product, 20 bp long and a melting temperature of 60°C. PCRs were prepared in Microamp optical 96-well reaction plates (Applied Biosystems). Three microliters of a 1:6 dilution of total cDNA was mixed with 5 μl of SYBR green PCR master mix 2× (Roche) and 1 μm each primer pair in a total volume of 10 μl. Reactions were run in an ABI Prism 7900 Sequence Detector (Applied Biosystems), and results were analyzed with SDS2.3 software (Applied Biosystems) for Ct calculations. Calculations of ΔΔCt values were performed following specifications of the manufacturer. All assays were performed at least three times and were normalized to β-actin or GAPDH in the same samples.
Data analysis.
Statistical analysis of qPCR data was performed using GraphPad Prism 5 software. One-way ANOVA and Bonferroni post-test were used to compare multiple datasets. For Western blots, protein levels were normalized to actin levels. ANOVA assays with the Tukey post hoc test were used to determine differences between means for Western blot and qPCR analysis. Two-way ANOVAs with repeated measures were used with behavioral data, and Tukey or Dunnett post hoc tests were used for multiple comparisons. Significance was rejected at the 0.05 level.
Results
CNB-001 increases the clearance of intracellular Aβ
To determine how CNB-001 alters the metabolism of intracellular Aβ, MC65 cells were used that conditionally express the C99 fragment of the amyloid precursor protein (APP) under the control of a tet promoter (Sopher et al., 1994). Upon the removal of tet, MC65 cells express C99, which is cleaved to Aβ by γ-secretase. Aβ polymers accumulate within the cells, leading to cell death within 4 d (Sopher et al., 1994; Maezawa et al., 2006). The addition of γ-secretase inhibitors blocks cell death but allows the accumulation of the nontoxic C99 protein (Maezawa et al., 2006; Hong et al., 2007). Figure 1, A and B, shows that when MC65 cells were induced to generate Aβ in the presence of CNB-001 Aβ accumulation is inhibited and the cells were protected from cell death with an IC50 of ∼500 nm. Cells do not accumulate monomeric Aβ, and C99 does not polymerize (Maezawa et al., 2004, 2006; Hong et al., 2007).

CNB-001 is neuroprotective against intracellular Aβ toxicity. A, MC65 cells were induced to make the C-terminal fragment of APP by the removal of tetracycline, followed by the addition of increasing amounts of CNB-001. Cell death was determined 4 d later by the MTT assay and confirmed visually. The data are plotted as the percentage viable relative to controls of uninduced cells, ±SEM, n = 3. B, Intracellular Aβ in MC65 cells was assayed at 1 d following tetracycline removal in the presence (+) or absence (−) of 1 μm CNB-001. Aβ aggregates and C99 (∼13 kDa) were detected with the 6E10 antibody. C, Cells were induced for 1 d to make C99, followed by the addition of tet and γ-secretase inhibitor 10 (SI; 5 μm; Calbiochem) in the presence or absence of 1 μm CNB-001. One day later, the cells were harvested by the addition of sample buffer and immunoblotted for Aβ. Data are presented as the percentage of Aβ expressed in the induced cells without tet or SI. D, MC65 cells were plated on coverslips and incubated with or without tet plus or minus CNB-001 for 2 d. Amyloid aggregates were visualized using the 6E10 antibody by confocal microscopy. E–I, CNB-001 induces the deposition of Aβ and HSP90 in the SAM. MC65 cells were either induced (−tet) or uninduced (+tet) for 24 h. The cells were then dissociated and replated on new dishes followed by the addition of a γ-secretase inhibitor and tet to all cultures. One micromolar CNB-001 was added where indicated (+CNB-001). After another 24 h, the cultures were separated into cells, culture medium (secreted), and SAM. The three fractions were then immunoblotted for Aβ and APP (antibody 6E10), HSP90, or actin. E, Cells, anti-Aβ. F, SAM, anti-Aβ. G, Secreted, anti-Aβ. H, I, Quantitation of HSP90 in the SAM (H) and secreted fractions (I). **p < 0.01; ***p < 0.001, n = 3–4. Throughout the text, n is the number of experiments performed to generate the statistics.
To determine whether CNB-001 has the ability to remove preformed intracellular Aβ aggregates, cells were induced to make Aβ for 24 h, followed by the addition of tet and a γ-secretase inhibitor to block further C99 synthesis and Aβ production. CNB-001 was then added for an additional 24 h. Figure 1C shows that CNB-001 enhanced the removal of preformed aggregates. Figure 1D shows by immunocytochemistry that CNB-001 prevented the accumulation of Aβ in cytoplasmic inclusions (Maezawa et al., 2006). However, the site of C99 cleavage within the cell to Aβ is not known. It was then asked, what is the mechanism of Aβ polymer clearance?
Following the initiation of cellular stress caused by misfolded or aggregated proteins, cells mount a multifaceted stress response, including attempts to secrete the protein or degrade it via activation of the ubiquitin–proteasome pathway, direct lysosomal degradation, autophagy, or a combination of these mechanisms. The mechanism by which Aβ is reduced by CNB-001 was initially explored by examining its presence in the extracellular compartments following drug treatment for 24 h. These compartments include soluble (secreted) proteins and extracellular insoluble substrate attached material (SAM). SAM is composed of proteins such as fibronectin, collagen, and proteoglycans that are attached to the culture dish, and can be regarded as a surrogate for the extracellular matrix that surrounds all cells.
SAM is experimentally defined as the material left on the culture dish after cells are removed by EGTA (Schubert, 1977). To determine whether CNB-001 causes the release of Aβ into the extracellular space, MC65 cells were induced to accumulate Aβ for 24 h, dissociated, and replated on new tissue culture dishes in the presence of tet and a γ-secretase inhibitor plus or minus CNB-001 for an additional 24 h. Figure 1E shows that intracellular C99 and Aβ aggregates were reduced by CNB-001. Furthermore, Aβ was only found in the SAM following exposure to CNB-001 (Fig. 1F). Aβ was not detected secreted in the culture medium (Fig. 1G); APP was only in the medium, and CNB-001 did not reduce APP levels inside or outside of cells (Fig. 1E,G). It follows that CNB-001 can stimulate the deposition of Aβ in the extracellular space associated with matrix proteins.
HSPs are induced in response to stress and to maintain aggregation-prone proteins in a properly folded state to prevent their accumulation. In some cases, HSPs are secreted (Song and Luo, 2010). HSP90 has the ability to directly bind Aβ and inhibit the early stages of amyloid aggregation (Evans et al., 2006), and HSP90 and Aβ are cosecreted under conditions of stress (Goldsbury et al., 2008). We therefore examined the levels of HSP90 in the intracellular, SAM, and the soluble secreted protein compartments of MC65 cells with or without CNB-001 treatment. Figure 1, E, F, and H, shows that CNB-001 greatly enhanced the deposition of HSP90 in SAM but had no effect on intracellular HSP90. In contrast to the results with SAM, CNB-001 decreased the accumulation of HSP90 in the soluble medium fraction (Fig. 1G,I). These data are consistent with the observation that CNB-001 enhanced the deposition of Aβ from cells into the extracellular space, perhaps in conjunction with HSP90. However, quantifying the amount of Aβ in the SAM indicated that only 5–10% of the protein depleted from cells by CNB-001 was in this fraction. We therefore asked, what is the mechanistic basis for the removal of the majority of the aggregated Aβ?
Intracellular Aβ is degraded via lysosomal and proteasomal pathways
Besides secretion, there are multiple alternatives for the removal of intracellular aggregated proteins, including direct proteolysis through the proteasome and various pathways leading to the lysosome, including autophagy. Autophagy is enhanced in the AD brain, perhaps as a protective mechanism (Nixon, 2007), and the activation of autophagy in fly brains removes aggregated protein and extends lifespan (Simonsen et al., 2008). However, no evidence for the induction of autophagy by CNB-001 could be found as defined by electron microscopy, changes in p62 or LC3, or inhibition by rapamycin (data not shown). Therefore, the effect of CNB-001 on protein degradation directly via proteasomes or lysosomes was examined.
MC65 cells were induced to synthesize C99 for 24 h followed by the addition of tet and the γ-secretase inhibitor to block further C99 and Aβ production. Different combinations of CNB-001, the proteasome inhibitor lactacystin, or the lysosome inhibitor chloroquine were then added, and the accumulation of aggregated Aβ was determined 48 h later. Figure 2A shows that CNB-001 effectively stimulated the removal of Aβ from cells, as did the proteasome inhibitor lactacystin. Chloroquine blocked the clearance of Aβ by lactacystin and reduced the clearance by CNB-001, and the combination of chloroquine and lactacystin completely inhibited Aβ removal by CNB-001. Cell viability after 4 d could not be determined because of the toxicity of lactacystin. Figure 2B shows that CNB-001 increases all three proteasomal protease activities. Therefore, CNB-001 stimulates Aβ degradation through both the proteasome and the lysosome, and the experimental inhibition of the proteasome pathway redirects clearance through lysosomes. Similar dual-pathway mechanisms of protein turnover have been reported (Komatsu et al., 2006), as have examples where the inhibition of the proteasome redirects proteins to lysosomes (Zhu et al., 2010).

CNB-001 stimulates Aβ clearance through both the proteasome and lysosome. A, MC65 were induced to make C99 for 24 h, followed by the indicated additions of tet, γ-secretase inhibitor 10 (5 μm), chloroquine (50 μm), lactacystin (1 μm), or CNB-001 (1 μm). Two days later, the cells were immunoblotted for Aβ or actin. B, MC65 cells were treated for 48 h in either the presence or absence of tet and the indicated compounds, and the three proteasome activities were measured. BW, BW B70C (0.5 μm); 001, CNB-001 (1 μm); MK, MK886 (2 μm), n = 4. C, MC65 cells were either uninduced (C+ only) or induced (C− and all others) to make Aβ in the presence of BW (0.5 μm), MK (2 μm), or 001 (1 μm), and cell viability was determined 4 d later. D, Western blots of cells treated as in C with anti-Aβ 48 h following the removal of tet. *p < 0.05; ***p < 0.001, n = 4.
The inhibition of 5-LOX leads to Aβ degradation
Because of the structural similarity of CNB-001 to several LOX inhibitors, the ability of CNB-001 to inhibit the three human LOX isoforms was examined. CNB-001 inhibits 5-LOX with an IC50 of ∼70 nm (performed by Ricerca Biosciences; data not shown). There is no inhibition of human 12-LOX or 15-LOX below 5 μm. It was then asked whether the selective 5-LOX inhibitor BW B70C and the 5-LOX activating protein (FLAP) inhibitor MK886 could mimic the effects of CNB-001 on Aβ clearance and proteasome activity and inhibit cell death. Figure 2B shows that BW B70C and MK886 activated the proteasome, and both 5-LOX inhibitors blocked the death of induced MC65 cells (Fig. 2C). Figure 2D shows that they also prevented Aβ accumulation. Therefore, the inhibition of 5-LOX by three molecularly distinct molecules and two mechanisms is sufficient for the removal of intracellular Aβ and the maintenance of MC65 viability.
The activity and metabolic stability of 5-LOX is largely determined by its subcellular distribution, with the stable active state associated with the nucleus. Localization is determined by phosphorylation. The phosphorylation of S523 leads to exit from the nucleus, while the phosphorylation of S271 leads to nuclear localization and an increase in activity (Luo et al., 2005). Figure 3A shows that the accumulation of Aβ within MC65 cells leads to increased 5-LOX expression, and Figure 3B shows that CNB-001 increased the phosphorylation of S573 and decreased S271 phosphorylation, and there was a time-dependent loss of 5-LOX from the nucleus in the presence of CNB-001.

A, CNB-001 increases 5-LOX expression in MC65 cells following C99 induction. B, CNB-001 modifies the phosphorylation of 5-LOX. MC65 cells were grown without tet for 24 h, 1 μm CNB-001 added to the cultures, and the phosphorylation of the protein at S523 and S271 was determined as a function of time by Western blotting. Cells were exposed to 1 μm CNB-001, and the disappearance of 5-LOX (n5LOX) from the nuclear fraction followed as a function of time. t5-LOX, Total 5-LOX; nActin, nuclear actin. C, Fibroblasts in which the PERK gene is deleted or control fibroblasts with PERK were exposed to 1 μm CNB-001 (001) or the indicated concentrations of BW B70C (BW) and apomorphine (Apo). The expression of PERK was monitored by Western blotting 6 h later. D, PERK phosphorylation in induced MC65 cells as a function of time following exposure to 1 μm CNB-001. The data are presented as the ratio of phospho to total PERK. *p < 0.05, n = 3.
CNB-001 activates one arm of the UPR
The accumulation of proteins within cells can trigger the UPR, leading to increased protein degradation. Three response pathways participate in the UPR. These are mediated by PERK, ATF6, and inositol-requiring enzyme 1α (IRE1α). HSP90 is a surrogate marker for the UPR (Kunte et al., 2012), and CNB-001 strongly induces the synthesis of this protein (Fig. 1F). To determine whether the UPR contributes to the effect of CNB-001 on cell survival, several components of each arm were assayed using both Western blotting and quantitative RT-PCR (qRT-PCR). The following shows that CNB-001 primarily activates one arm of the UPR, that this pathway is sufficient to remove aggregated Aβ and promote cell survival, and that it can also be activated by the inhibition of 5-LOX.
Since we have shown that the phosphorylation of eIF2α leads to neuroprotection from oxidative stress-induced programmed cell death (Tan et al., 2001b) and eIF2α is involved in learning and memory (Trinh et al., 2012), the role of eIF2α phosphorylation in CNB-001-mediated protection was examined first. To compare the effect of CNB-001 to that of a bona fide UPR inducer, cells were also treated with tunicamycin. Figure 4A shows that in uninduced cells (+tet) both CNB-001 and tunicamycin caused a transient increase in eIF2α phosphorylation, while in induced cells CNB-001 caused a more sustained phosphorylation of eIF2α (−tet; Fig. 4B). When MC65 cells were grown with or without tet for 24 h, the basal level of eIF2α phosphorylation was decreased by 75% in induced cells (Fig. 4C). When added after 24 h, CNB-001 dose-dependently increased eIF2α phosphorylation in induced cells while CNB-001 decreased eIF2α phosphorylation in uninduced (+tet) cells at this longer time point (Fig. 3C). Therefore, the effect of CNB-001 on eIF2α phosphorylation is dependent upon whether or not the cells have accumulated a toxic peptide.
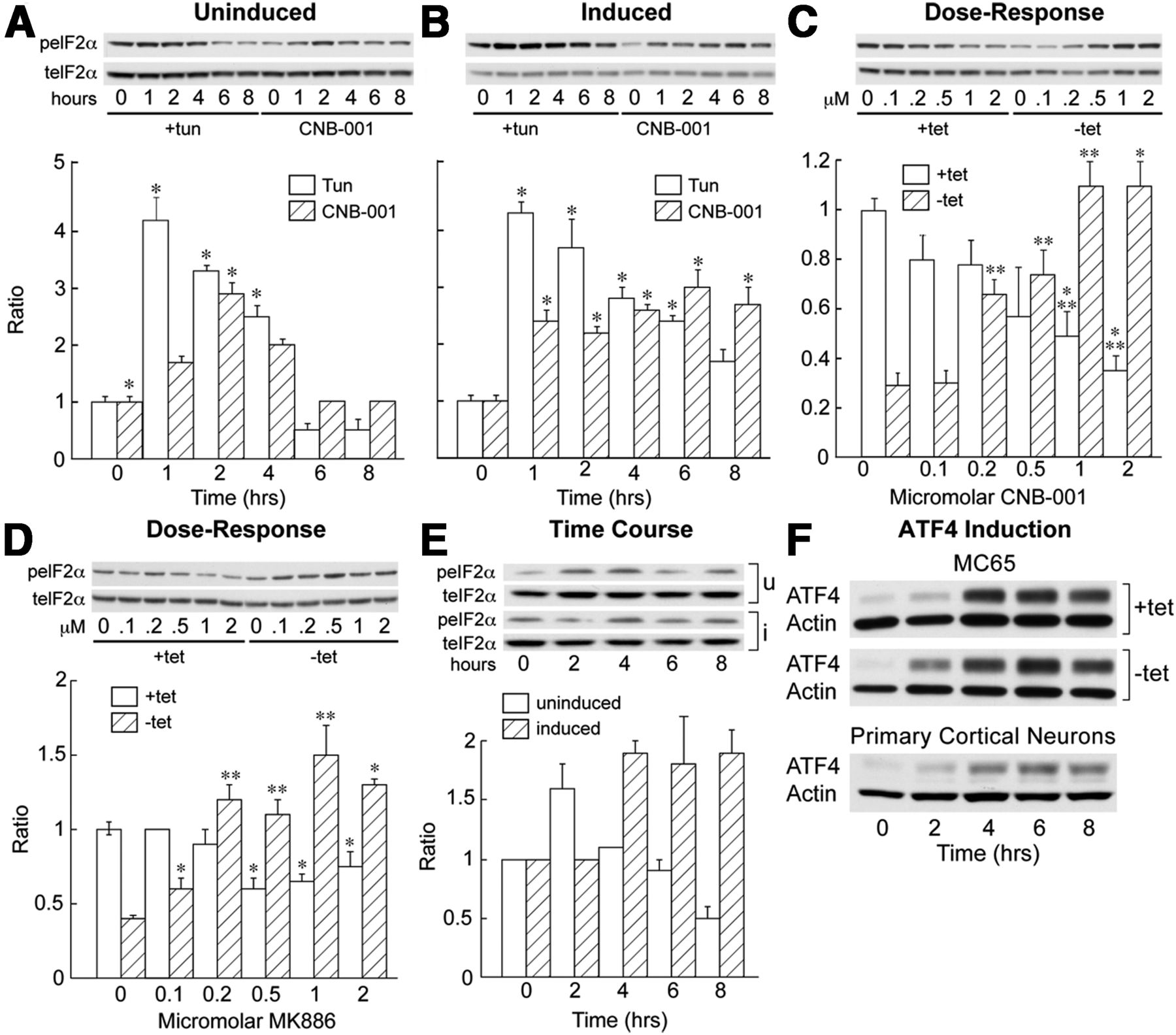
CNB-001 and 5-LOX inhibition activates eIF2α. A, Uninduced MC65 cells were treated with 5 μg/ml tunicamycin as a control or 1 μm CNB-001, and the phosphorylation of eIF2α on serine 51 compared with total eIF2α as a function of time. B, The same experiment was performed on cells previously induced to make C99 and Aβ for 20 h before the addition of CNB-001. C, Cells were grown with tet (+tet) or without tet (−tet) for 24 h, increasing amounts of CNB-001 were added for an additional 24 h, and the phosphorylation of eIF2α was monitored as described in A. D, MC65 cells were induced (−tet) or not induced (+tet) for 24 h, followed by an addition of increasing concentrations of MK886, and eIF2α phosphorylation was assayed 24 h later. E, MK886 (1 μm) was added to induced or uninduced MC65 cells, and the phosphorylation of eIF2α was followed as a function of time. F, CNB-001 induces the expression of ATF4 in MC65 cells and primary cortical neurons. CNB-001 (1 μm) was added to 7-d-old cultures of E18 rat cortical neurons and ATF4 expression monitored as a function of time. *p < 0.05; **p < 0.01.
It was then asked whether the inhibition of 5-LOX is also sufficient to cause the phosphorylation of eIF2α. Figure 4D shows there was a dose-dependent increase in eIF2α phosphorylation by the FLAP inhibitor MK886, and Figure 4E shows the time course of this increase. Similar results were obtained with BW B70C, which, like CNB-001, is a direct 5-LOX enzyme inhibitor (data not shown). Figure 5 shows that CNB-001 does not induce eIF2α phosphorylation in astrocytes from 5-LOX knock-out mice, while it does in wild-type and 12/15-LOX knock-out astrocytes. Finally, overnight treatment with CNB-001 lowered the expression of 5-LOX in MC65 cells (Fig. 6B).

CNB-001 does not elevate eIF2α phosphorylation in 5-LOX knock-out mouse astrocytes. Astrocytes were prepared from newborn wild-type, 5-LOX, or 12/15 LOX knock-out mice (Jackson Laboratories) using standard procedures. Confluent cultures were treated with 1 μm CNB-001 or tunicamycin, and eIF2α phosphorylation was followed as a function of time. ON, Overnight; peIF2α, phospho-eIF2α; teIF2α, total eIF2α. *p < 0.01 relative to control without drugs. n = 3.

A, MC65 cells were exposed to 500 nm CNB-001 or 5 μg/ml tunicamycin (Tun) and Western blotted for actin, BiP, CHOP, and ATF4 at 6 and 8 h. B, 5-LOX was assayed in uninduced MC65 cells following a 20 h exposure to the indicated amounts of CNB-001 (n = 4). C, The splicing of XBP-1 was assayed by RT-PCR at the indicated times. Only tunicamycin increased splicing. D, The phosphorylation of IRE1α was monitored following exposure to 500 nm CNB-001 or 5 μg/ml tunicamycin. E, MC65 cells were induced for 2 d, and the RIPA-insoluble fraction blotted with anti-ubiquitin or actin. *p < 0.01; ***p < 0.001. n = 4–10.
The phosphorylation of eIF2α increases the translation of the transcription factor ATF4 to activate the expression of several UPR target genes. ATF4 is also involved in the reduction of oxidative stress and the maintenance of glutathione levels (Lewerenz and Maher, 2009). Figures 4F and 6A show that CNB-001 increased the expression of ATF4 in a time-dependent manner in both MC65 cells and primary cortical neurons. In contrast, the downstream, pro-death protein CHOP is only induced weakly by CNB-001 relative to tunicamycin (Fig. 6A).
To determine whether CNB-001 also modulates the other arms of the UPR, IRE1α activation was measured using an antibody against S724 of IRE1α. While tunicamycin increased IRE1α phosphorylation, CNB-001 did not (Fig. 5D). The activation of IRE1α stimulates the deletion of a 26 nt intron from XBP-1 mRNA. Tunicamycin induced this splicing event, but CNB-001 did not (Fig. 6C). The third arm of the UPR is mediated by ATF6. Tunicamycin increased the expression of ATF6 in the nucleus, but CNB-001 did not as defined by Western blotting (data not shown). BiP is a recognized target of ATF6, and it was not increased by CNB-001 (Fig. 6A). Therefore, while eIF2α phosphorylation and ATF4 expression are upregulated by CNB-001, the ATF6 and IRE1α arms of the UPR are poorly activated.
To verify the Western blotting data, we examined the CNB-001-dependent expression of several ATF4-mediated gene transcripts by qPCR (Fig. 7). There was increased mRNA expression of xCT/SLC7A11, ATF3, ASNS, and GADD34/PPP1R15A. We also observed a transient increase in CHOP/DDIT3, HERP/HERPUD1, and ATF4 (Fig. 8) mRNA, but there was only a limited increase in the expression of the CHOP protein (Fig. 6A). Finally, as predicted by the Western blot results, there were no significant changes in the mRNA levels of genes classically upregulated by the ATF6 or IRE1α arms of the UPR (Fig. 8).

CNB-001 activates the eIFα arm of the UPR. MC65 cells were either induced (+tet) or uninduced (−tet) for 24 h. At this time, 1 μm CNB-001 was added, and mRNA was harvested at 0, 2, 4, 8, 12, and 24 h followed by qRT-PCR for the expression of the indicated genes. T indicates incubation with tunicamycin 5 μg/ml for 8 h as a positive control. Analysis was done by ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001.

CNB-001 does not activate the ATF6 or the IRE1α arms of the UPR. MC65 cells were either induced (+tet) or uninduced (−tet) for 24 h. At this time, 1 μm CNB-001 was added, and mRNA was harvested at 0, 2, 4, 8, 12, and 24 h followed by qPCR for the expression of the indicated genes. T indicates incubation with tunicamycin 5 μg/ml for 8 h as positive control. Genes in the top row are associated with the eIF2α/ATF4 arm, and the remaining six with the ATF6 and IRE1α arms of the UPR. Analysis was done by ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001.
Four kinases stimulate eIF2α phosphorylation. In the UPR, the phosphorylation of eIF2α is mediated by PERK. To determine whether PERK plays a role in the induction of eIF2α phosphorylation and ATF4 expression by CNB-001, fibroblasts from PERK knock-out mice were used. CNB-001 was added to the cultures and the expression of ATF4 was monitored. Figure 3C shows that the deletion of PERK led to dramatically decreased ATF4 expression relative to wild-type fibroblasts following exposure to CNB-001. PERK itself is activated by phosphorylation, and Figure 3D shows that CNB-001 caused the phosphorylation of PERK in a time-dependent manner. Together, these data demonstrate that the upregulation of the neuroprotective eIF2α/ATF4 pathway by CNB-001 is likely to be initiated by PERK.
eIF2α phosphorylation is sufficient for Aβ clearance
Three experiments were performed to determine whether the phosphorylation of eIF2α alone is sufficient to clear intracellular Aβ. In the first experiment, the dephosphorylation of eIF2α was induced and the effects on Aβ clearance and cell survival examined. GADD34 recruits the serine/threonine protein phosphatase PP1 to eIF2α and stimulates its dephosphorylation. When overexpressed in MC65 cells, GADD34 decreased eIF2α phosphorylation by ∼80% (Fig. 9A) and promoted the death of transfected cells within 2 versus 4 d for control cells (Fig. 9B). Overexpression of GADD34 also caused increased intracellular Aβ accumulation in induced MC65 cells (Fig. 9A). Salubrinal inhibits the dephosphorylation of the α-subunit of eIF2α (Lewerenz and Maher, 2009). Therefore, if the phosphorylation of eIF2α is sufficient to clear intracellular Aβ and protect cells from intracellular Aβ toxicity, salubrinal should promote both activities. Figure 9, C and D, shows that this was indeed the case. The third experiment was to ask whether another compound that induces eIF2α phosphorylation but does not inhibit 5-LOX is able to promote Aβ clearance. J147 is a potent neuroprotective compound (Chen et al., 2011), but a very poor 5-LOX inhibitor, with an IC50 over 4 μm (data not shown). Fifty nanomolar J147 caused the phosphorylation of eIF2α, cleared intracellular Aβ, and prevented cell death (Fig. 9E,F). Together, these data suggest that the phosphorylation of eIF2α is sufficient to remove intracellular Aβ and promote cell viability.

eIF2α phosphorylation, intracellular Aβ, and viability. MC65 cells were exposed to the following conditions, and eIF2α phosphorylation, intracellular Aβ, and cell viability were determined. A, Cells were transfected with an expression plasmid for GADD34 or empty plasmid. After 24 h, cells were induced to make Aβ and 1 μm CNB-001 was added. eIF2α phosphorylation was determined 24 h later (top). Intracellular Aβ levels were determined after 2 d (A, bottom), and cell viability was determined after 3 d (B). C, D, Induced MC65 cells were exposed to 20 μm salubrinal and intracellular Aβ, eIF2α phosphorylation was determined after 2 d (C), and cell viability was determined on day 4 (D). E, Induced MC65 cells were exposed to 50 nm J147 for 2 d, and eIF2α phosphorylation and intracellular Aβ levels were determined. F, Cell viability was assayed at day 4. ***p < 0.001. n = 3–4.
Apomorphine activates eIF2α and removes intracellular Aβ
We have shown that apomorphine is very neuroprotective against oxidative stress-induced nerve cell death in the absence of intracellular Aβ, and it does so by the activation of the dopamine D4 receptor (Ishige et al., 2001). Apomorphine also prevents the accumulation of intracellular Aβ in AD transgenic mice (Himeno et al., 2011). To determine whether apomorphine stimulates eIF2α phosphorylation and clears intracellular Aβ from MC65 cells, MC65 cells were treated with apomorphine, and eIF2α phosphorylation followed as a function of time (Fig. 10A) and after 24 h (Fig. 10B). Apomorphine increased the phosphorylation of eIF2α and also PERK (Fig. 3B). Figure 10, C and D, shows that apomorphine also cleared intracellular Aβ aggregates and prevented cell death. The possibility that CNB-001 directly activated the D4 receptor in addition to inhibiting 5-LOX was ruled out because the D4 antagonist U1011958 did not block the effect of CNB-001 on Aβ clearance and 10 μm CNB-001 did not inhibit direct agonist binding to the dopamine receptors D4.2, D4.4, or D4.7 (performed by Ricerca Biosciences).

Apomorphine (Apo) induced eIF2α phosphorylation, clears Aβ polymers, and blocks cell death. A, Induced MC65 cells were exposed to 1 μm apomorphine for the indicated amounts of time and were immunoblotted for phospho- and total eIF2α. B, MC65 cells were grown without tet for 24 h followed by the addition of the indicated amounts of apomorphine for an additional 24 h, and were immunoblotted as in A. C, MC65 cells were incubated with (+) or without (−) tet for 24 h in the presence of the indicated amounts of apomorphine or CNB-001, and were immunoblotted with anti-Aβ (6E10). D, Cell viability was determined 4 d later under the same conditions as in C. *p < 0.05. n = 3.
Efficacy of CNB-001 in AD transgenic mice
Since CNB-001 stimulates LTP and rodent memory (Maher et al., 2010), CNB-001 was initially assayed in mice in an acute Aβ toxicity assay using a contextual memory-conditioning paradigm. CNB-001 completely reversed the memory loss caused by Aβ1–42 (Fig. 11A). While this memory-enhancing effect is likely to be too rapid to be due to relief from intracellular amyloid accumulation, it may be due to the rapid activation of the BDNF signaling pathways by CNB-001 (Liu et al., 2008; Figure 11E,F).

CNB-001 reverses cognitive deficits and maintains synaptic proteins in an AD mouse model. A, CNB-001 blocks Aβ disruption of contextual memory. Cannula were bilaterally implanted in the dorsal hippocampi of mice, and CNB-001 500 mg/kg or gavage solution alone was orally administered 30 min before training and 15 min before training; Aβ1–42 (500 nm) was bilaterally injected. Animals were subjected to the standard fear conditioning training paradigm and were assayed 24 h later. The data are shown as the time frozen (in seconds) with gavage alone, Aβ1–42 alone, or Aβ1–42 plus CNB-001 (performed by Psychogenics). B, 9-month-old hAPP/Swe/PS1ΔE9 mice were fed 500 ppm CNB-001 (∼25 mg/kg/d) from 3 months of age and were assayed for memory in the Y-maze. A lower number of alterations indicates reduced memory of previous explorations. The hippocampi from the mice in B were isolated and assayed for HSP90 in the 100,000 × g RIPA-insoluble fraction (there is no change in the soluble fraction; C); phosphosynapsin-1 (RIPA soluble; D); Pro-BDNF (soluble; E); Homer 1 (soluble; F); clusterin (soluble; G); ATF4 (H). Statistics are done by one-way ANOVA with 8–10 animals per group, 4 or 5 animals per group are shown. *p < 0.05; **p < 0.01; ***p < 0.001.
We next determined the efficacy of CNB-001 in preventing behavioral impairment in a transgenic mouse strain expressing mutated human APP and PS-1, the APPswe/PS1ΔE9 line (line 85; Jankowsky et al., 2005). This strain develops amyloid plaques at ∼6 months of age and behavioral deficits by 9 months of age. AD mice and nontransgenic littermates were placed on a diet containing CNB-001 at 500 ppm (∼25 mg/kg/d) starting at 3 months of age, and control AD and nontransgenic mice received the identical chow without CNB-001. At 9 months of age, the mice were tested in a behavioral paradigm to assay memory and then killed for biochemical analysis. The Y-maze was used to assess working memory as the spontaneous tendency to alternate free choices in entering the three arms of the maze. The groups of animals did not differ in the overall number of arm entries, suggesting that activity levels were not altered by CNB-001 treatment. When control mice were given a choice of entering the arms in the Y-maze, they tended to alternate their choices spontaneously. Nine-month-old AD transgenic mice, however, had less of a tendency than control mice to alternate the arms on successive choices (Fig. 11B), which is characteristic of animals with damage to the hippocampus. In contrast, in AD mice fed CNB-001 the behavior approached that of the nontransgenic controls. Because CNB-001 increases the UPR in cultured cells, it was asked whether the same occurs in mice.
In cultured cells, CNB-001 increases the phosphorylation of eIF2α, increases ATF4 and HSP90 expression, and promotes the clearance of aggregated proteins, all markers for UPR activation. Figure 11, C and H, shows that HSP90 and ATF4 were elevated by CNB-001 in the hippocampal extracts of AD mice relative to untreated AD mice, and Figure 12A shows that CNB-001 dramatically increased eIF2α phosphorylation in both control and AD mice. Our antisera were unable to detect any PERK phosphorylation in the lysates due to low expression levels of the protein. 5-LOX was reduced by CNB-001 in both wild-type and AD mice (Fig. 12F).

CNB-001 induces hippocampal eIF2α phosphorylation, and reduces soluble Aβ1–42 and ubiquitinated proteins in AD mice. A, Hippocampal lysates from AD mice fed 500 ppm CNB-001 were immunoblotted with anti-phospho and total eIF2α. C, Control; A, AD; C+, control plus CNB-001; A+, AD + CNB-001. *p < 0.05; **p < 0.01; ***p < 0.001. B–D, Aβ1–40 and Aβ1–42 levels were measured by ELISA in the RIPA-soluble and the RIPA-insoluble 100,000 × g pellet of hippocampal lysates of 10-month-old mice fed CNB-001 or control diet for 7 months. B, Insoluble pellet. C, RIPA-soluble supernatant, Aβ1–40. D, RIPA-soluble fraction, Aβ1–42. **p < 0.01. n = 10 per group. E, RIPA-insoluble protein was immunoblotted with anti-ubiquitin. A reduction in ubiquitinated particulate protein was seen in 9-month-old mice that had been fed CNB-001 at 500 ppm in chow for the previous 6 months. F, 5-LOX expression was monitored in the hippocampus of AD transgenic mice fed CNB-001 for 6 months (see Fig. 11). **p < 0.01; ***p < 0.001.
There are a number of biochemical parameters that covary with memory improvement in drug-treated, cognitively deficient rodents. Among these is synapsin-1, an actin-binding protein that is involved in synaptic vesicle recycling and is activated by phosphorylation by CaMKII at serine 603. Since CNB-001 activates CaMKII (Maher et al., 2010), we assayed the phosphorylation of synapsin-1 in the hippocampi of the animals. Figure 11D shows that CNB-001 increased synapsin-1 phosphorylation. Figure 11E shows that CNB-001 increased pro-BDNF levels in AD mice. BDNF directly regulates the expression of several genes involved in synaptic spine formation. Homer 1 is an actin binding protein that is induced by BDNF (Sato et al., 2001). Homer 1 was increased in AD brain and was further increased by CNB-001 (Fig. 11F). Finally, clusterin is a secretory protein that is implicated in AD pathology, including a role in the proapoptotic aspect of the ER stress response (Kögel et al., 2012). It was elevated in line 85 AD transgenic mice and reduced by CNB-001 (Fig. 11G).
CNB-001 reduces soluble Aβ1-42 and ubiquitinated protein aggregates
The therapeutic target for AD drugs has historically been the removal of amyloid plaques, but more recently it has been argued that lower-molecular-weight soluble polymers made up primarily of Aβ1–42 are responsible for much of the AD pathology (Krafft and Klein, 2010). Indeed, in the line 85 AD mice that we used, soluble Aβ, perhaps of intraneuronal origin, is the primary contributor to cognitive decline, not insoluble Aβ (Zhang et al., 2011). There were abundant amyloid plaques in the line 85 AD transgenic animals at 9 months, but there were no significant differences between drug-treated and control animals. There was also no change in Aβ levels in the RIPA-insoluble fraction of AD mice treated with CNB-001, relative to control AD mice (Fig. 12B), nor were RIPA-soluble Aβ1–40 levels significantly lowered (Fig. 12C). However, CNB-001 caused a 50% reduction in the levels of soluble Aβ1–42 (Fig. 12D).
Since the accumulation of aggregated ubiquitinated proteins in the brain is associated with normal aging and their removal extends lifespan in flies (Simonsen et al., 2008), it was asked whether the removal of protein aggregates by CNB-001 is unique to Aβ or extends to other aggregates. Figure 12E shows that in 9-month-old wild-type mice that were fed CNB-001 at 500 ppm in their food for 6 months there was a 50% reduction of ubiquitinated protein aggregates in their hippocampi. There was also a reduction of ubiquitinated proteins in many AD mice, but this was not statistically significant due to variation between individual animals. When MC65 cells were treated with CNB-001 for 2 d, there was also a reduction of RIPA-insoluble ubiquitinated proteins (Fig. 6E). Together, these data support the argument that the eIF2α arm of the UPR is activated by CNB-001 in AD mice in a manner similar to the response in cultured cells (Fig. 13).

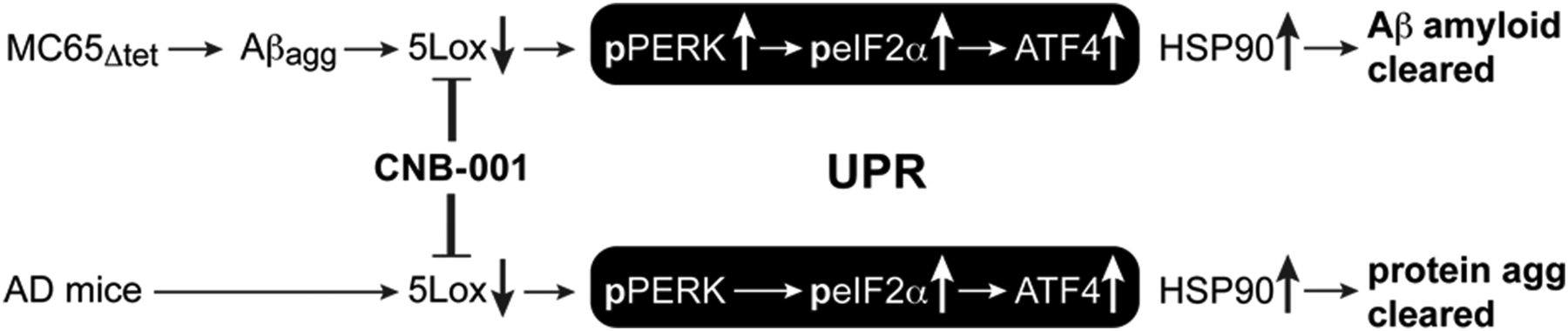
Schematic representation of the effect of CNB-001 on the metabolism of aggregated proteins both in vitro and in vivo.
Discussion
The above data show that the stimulation of eIF2α phosphorylation is sufficient to reduce the amount of intracellular Aβ polymers through a combination of proteasomal and lysosomal degradation, and that the inhibition of 5-LOX can induce this response. The accumulation of intracellular Aβ is likely to be a major cause of nerve cell death in AD. This conclusion is supported by a number of observations: (1) in humans, intracellular Aβ accumulation precedes plaques; (2) nerve cell death occurs in mutant PS1 transgenic mice that have extensive intraneuronal Aβ, but no plaques (Chui et al., 1999); (3) there is intraneuronal Aβ accumulation well before extracellular amyloid in several different AD models (Wirths et al., 2001; Billings et al., 2005; Oakley et al., 2006; Tampellini et al., 2010); (4) the reduction in intracellular Aβ also reduces soluble Aβ in vivo (Oddo et al., 2006); and (5) the removal of intracellular Aβ by apomorphine in 3XFAD mice before there are plaques improves memory and reduces AD pathology (Himeno et al., 2011). Our results show that CNB-001 and apomorphine both reduce intracellular Aβ in cultured cells and stimulate eIF2α phosphorylation, suggesting that they share a common mechanism for Aβ clearance.
Aβ degradation occurs in MC65 cells treated with CNB-001 in the absence of ultrastructural or biochemical evidence for autophagy. A combination of proteasomal and lysosomal degradation of Aβ without the involvement of autophagy has been reported (Chakrabortee et al., 2012). The data presented here suggest that proteasomal and lysosomal degradation pathways associated with the UPR are directly responsible for the removal of Aβ by CNB-001, a result supported in vivo by the strong phosphorylation of eIF2α by CNB-001-elevated ATF4 and HSP90, and the reduction of soluble Aβ1-42 and protein aggregates. Additionally, CNB-001 induces the deposition of a small fraction of the intracellular amyloid load as Aβ polymers in the extracellular matrix compartment. The ability of a compound to enhance Aβ secretion is desirable, as the impaired ability to secrete Aβ and reduce intraneuronal Aβ may promote AD pathogenesis (Tampellini et al., 2011).
Neuronal 5-LOX expression increases with age and is highly elevated in AD brains, and AD transgenic mice lacking 5-LOX have reduced levels of Aβ (Qu et al., 2000; Firuzi et al., 2008). Mice deficient in phospholipase A2- or D2-derived arachidonic acid, the major 5-LOX substrate, also have reduced AD-associated memory deficits (Sanchez-Mejia et al., 2008; Oliveira et al., 2010). Both line 85 AD mice and control mice have a high level of 5-LOX, which is reduced by >40% following a diet containing CNB-001. The ability of 5-LOX to mediate eIF2α phosphorylation and the UPR in the context of Aβ clearance may explain the multiple observations linking arachidonic acid metabolism to AD pathology.
The phosphorylation of eIF2α frequently leads to the induction of ATF4. CNB-001 increases ATF4 expression both in vivo and in AD mice, and is neuroprotective. Furthermore, ATF4 induction via the UPR requires PERK phosphorylation, and knockdown of PERK enhances extracellular Aβ neurotoxicity in neurons (Lee do et al., 2010) and also decreases multiple aspects of cognitive function (Trinh et al., 2012). We show here that the induction of ATF4 by CNB-001 requires PERK. Importantly, MC65 cells making Aβ respond differently to CNB-001 in terms of the UPR than uninduced cells (Fig. 4). This observation strongly suggests that the molecular responses to the inhibition of 5-LOX are directly related to the level of cellular stress caused by intracellular Aβ.
The response of transgenic AD mice to CNB-001 confirms several aspects of the cell culture data. These include a reduction of aggregated protein, the higher expression of ATF4 and HSP90, and the reduced expression of 5-LOX. In addition, the phosphorylation of eIF2α is greatly enhanced both in vivo and in vitro. We have shown that the phosphorylation of the UPR component eIF2α is very neuroprotective against multiple aspects of oxidative stress (Tan et al., 2001a). HSP90 is a marker for some aspects of the UPR, and there is a decrease in HSP90 in AD mice relative to control AD mice that is restored to control levels by CNB-001. Chaperones such as HSP90 bind amyloidogenic proteins and may affect their aggregation process. These chaperones not only colocalize with the pathological lesions of AD, but may also be involved in the clearance of Aβ from the brain via phagocytosis or active transport across the blood–brain barrier (Wilhelmus et al., 2007).
BDNF is a neurotrophic growth factor that is required for learning and memory as well as the overall maintenance and survival of CNS neurons, and several observations link BDNF to the UPR. Pro-BDNF is reduced in AD brain (Michalski and Fahnestock, 2003), and CNB-001 increases BDNF mRNA in rat models of traumatic brain injury (Wu et al., 2011). Here we show that pro-BDNF was increased by CNB-001 in control mice and to a similar extent in AD mice. CNB-001 also increased the downstream target of BDNF, Homer 1, in control and AD mice. CNB-001 causes the phosphorylation of synapsin-1, a synaptic spine protein whose phosphorylation is mediated indirectly by BDNF. The activation of the PERK/eIF2α arm of the UPR can lead to the elevation of death-promoting protein CHOP. BDNF is known to block CHOP induction (Chen et al., 2007), but activate some components of the UPR (Hayashi et al., 2007). CNB-001 activates the BDNF signaling pathway in the absence of the BDNF receptor (Liu et al., 2008) as well as stimulates BDNF and pro-BDNF accumulation in the brain (Wu et al., 2011; Fig. 12E). Therefore, the stimulation of the BDNF signaling pathway may explain why CNB-001 strongly induces the PERK/eIF2α arm of the UPR but not CHOP.
Finally, clusterin, also known as apolipoprotein J, is greatly increased in AD brain as well as with aging (Wu et al., 2012). Clusterin expression was significantly elevated in AD mice and reduced to control levels by CNB-001, another indication that CNB-001 is normalizing brain physiology. Together, these data show that CNB-001 enhances the expression of pathways involved in neuroprotection, Aβ clearance, as well as learning and memory.
In conclusion, the intracellular accumulation of toxic polymers is prevented by the 5-LOX inhibitor CNB-001. In cultured MC65 cells, CNB-001 not only protects from cell death but also induces the degradation of Aβ aggregates as well as their secretion to the extracellular matrix. The accumulation of aggregated Aβ in MC65 cells leads to elevated 5-LOX. 5-LOX metabolites lower components of the eIF2α arm of the UPR, resulting in the accumulation of Aβ aggregates and cell death. The inhibition of 5-LOX by CNB-001 leads to a reduction of 5-LOX protein and the activation of the PERK/eIF2α/ATF4 arm of the UPR, resulting in degradation of Aβ aggregates through either the proteasome or lysosome and cell survival. In mice, CNB-001 significantly reduces 5-LOX, and elevates eIF2α phosphorylation and ATF4 expression, resulting in a reduction in overall insoluble aggregated protein in the hippocampus. This information is shown schematically in Figure 13. Finally, CNB-001 reduces the accumulation of soluble Aβ1–42 and improves memory in animal models of AD. These data show that the pharmacological manipulation of 5-LOX and the PERK/eIF2α/ATF4 arm of the UPR may be useful for the treatment of AD and other neurodegenerative diseases caused by proteotoxicity.
Footnotes
This research was supported by grants from The National Institutes of Health (Grant R01AG035055; D.S.), the Fritz B. Burns Foundation (D.S. and P.A.M.), and the Alzheimer's Association (D.S.).
The authors declare no competing financial interests.
- Correspondence should be addressed to David Schubert, 10010 North Torrey Pines Road, La Jolla, CA 92037-1002. schubert{at}salk.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}