
Abstract
Peripheral nociceptors are excited by the activation of membrane receptors and ion channels. The heat-sensitive TRPV1 ion channel responds to various noxious chemical and thermal stimuli, causing pain and itch. Here, we show that TRPV1 is coexpressed with PKCβII in a subset of mouse sensory neurons and that, in these neurons, TRPV1 binds directly to PKCβII, leading to the activation and translocation of PKCβII. Activated PKCβII, in turn, significantly increases the responsiveness of TRPV1 by phosphorylating Thr705. The heat sensitivity of TRPV1 is almost eliminated by either knocking down PKCβII or mutating Thr705; however, neither of these manipulations affects the potentiation of TRPV1 caused by the activation of PKCε. PKCβII thus acts as an auxiliary subunit of TRPV1 by forming a population-dependent TRPV1 ion channel complex controlling the sensitivity of TRPV1 and setting the threshold for pain and itch.
- auxiliary subunit
- pain
- phosphorylation
- PKC
- TRP ion channels
- TRPV1
Introduction
The responsiveness of animals to noxious stimuli depends on the sensitivity of nociceptors in peripheral nerve endings. Many ion channels in nociceptive neurons serve as sensors or transducers conveying noxious information to the CNS. TRPV1 is such an ion channel that responds to diverse pain-producing agents, inflammatory mediators, and metabolites such as bradykinin and protons (Basbaum et al., 2009). TRPV1 is also targeted directly by toxins derived from tarantula spider venom, producing pain and inflammation (Siemens et al., 2006). Interestingly, excitation of TRPV1 by histamine in a subpopulation of dorsal root ganglion (DRG) neurons mediates the sensation of itch instead of pain (Shim et al., 2007). In addition to being targeted by these painful and itching chemicals, TRPV1 is best known for its direct activation by noxious heat (Caterina et al., 1997). The basal sensitivity of TRPV1 in response to these noxious stimuli is thus critical for the latency and intensity of elicited pain and itch; however, it is little known about how the basal sensitivity of TRPV1, and in particular its thermal sensitivity, is determined.
It is well known that different populations of TRPV1+ neurons exhibit differential sensitivities to heat even though they respond similarly to the agonist capsaicin; conversely, not all capsaicin-responding neurons are heat sensitive (Kirschstein et al., 1997; Vyklický et al., 1999). Furthermore, there are many discrepancies between heat- and capsaicin-evoked currents in DRG neurons, leading to speculation that different TRPV1 channel complexes may have been formed in different populations of neurons (Nagy and Rang, 1999b); however, no such TRPV1 complexes have ever been identified. Membrane lipids are negative regulators of the basal sensitivity of TRPV1 (Cao et al., 2013), but it is unknown whether different populations of neurons have different levels of phosphoinositide lipids giving rise to different sensitivities of TRPV1. Collectively, the mechanisms causing varied thermal sensitivity of TRPV1 are still poorly understood.
In contrast, the mechanisms underlying the modulation of TRPV1 sensitivity in disease conditions such as inflammation are relatively well understood. During inflammation, the thermal threshold of TRPV1 is reduced significantly, leading to inflammatory thermal hyperalgesia through a mechanism involving sensitization of TRPV1 via phosphorylation of the channel by activated PKA or PKCε (Huang et al., 2006). Sensitization of TRPV1 also requires the scaffolding protein AKAP79/150, which anchors both PKA and PKC adjacent to TRPV1 (Jeske et al., 2008; Schnizler et al., 2008; Zhang et al., 2008; Jeske et al., 2009). However, it is unknown whether this mechanism may also determine the basal sensitivity of TRPV1.
Here, we report that the basal sensitivity of TRPV1 is determined mainly by PKCβII, but not by PKCε. TRPV1 is sufficient to activate PKCβII through direct binding to the kinase domain of PKCβII. Activated PKCβII then phosphorylates TRPV1 at the residue Thr705, leading to an increased sensitivity of TRPV1. Coexpression of PKCβII and TRPV1 was found in a subpopulation of DRG neurons, where they form a population-dependent TRPV1 ion channel complex. This population of TRPV1+ neurons may represent a subset of hypersensitive nociceptive neurons.
Materials and Methods
Cell culture and transfection
HEK293 cells and HEK 293 Tet-on advanced cells (Clontech) were maintained in DMEM containing 10% FBS, 100 UI/ml ampicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine in a humidified incubator containing 5% CO2. For the culture of HEK 293 Tet-on cells, 500 μg/ml G-418 (Sigma) was also included in the medium. Cells were transfected with the polyfect transfection reagent (QIAGEN), as described previously (Zhang et al., 2012), and used for electrophysiology 2 d after transfection. For Western blotting, cells were transfected with TurboFect reagent (Fermentas) according to the manufacturer's instructions.
DRG neurons were isolated from neonatal Wistar rats (<7 d), wild-type C57BL/6 mice, and TRPV1-null mice of either sex, and cultured as described previously (Zhang et al., 2008). Briefly, DRG were collected and dissociated with collagenase IV (Worthington). Dissociated neurons were then cultured in the DMEM containing 50 ng/ml NGF (Promega) and 1.2 μg/ml cytosine-d-arabinofuranoside. in some experiments, neurons were transfected with the Amaxa rat neuron nucleofector kit using a nucleofection device according to the manufacturer's instructions.
cDNA constructs
GFP-PKCβII was kindly provided by Dr. Yusuf Hannun (Feng et al., 1998). PKCβII was a kind gift from Dr. Alexandra Newton (University of California at San Diego). To induce PKCβII expression in the Tet-On expression system, the coding cDNA for PKCβII or its kinase domain was incorporated into the pTRE2 vector (Clontech) through BamHI and XbaI. The GST-coupled TRPV1 N-terminal and C-terminal fragments were constructed and used as described previously (Zhang et al., 2005). The GST-coupled C1C2 domain and the kinase domain from PKCβII were constructed by incorporating PKCβII fragments into the GST-pcNDA3 mammalian expression vector. The human PKCβII shRNA was constructed by ligating synthesized oligos (targeting sequence: 5′-GGTCATGCTTTCAGAACGAAA-3′) into the siRNA expression vector pGSU6-GFP (Genlantis) though BamHI and NotI according to the manufacturer's instructions. The corresponding scrambled sequence was 5′-ATAGGATACGGACGTCATATC-3′. The targeting sequence for rat PKCβII was 5′-GGTACCAAGGCTCCAGAAGAA-3′. The human PKCε shRNA was constructed similarly by targeting the following sequence: 5′-GGTCATGTTGGCAGAACTCAA-3′. The corresponding scrambled sequence was 5′-ATAGCCAGAACGGTTGTCTAG-3′. Successfully transfected cells were identified using the coexpressed gene reporter GFP from the shRNA vector. All other mutations were generated using the Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer's instructions. All constructs were confirmed by DNA sequencing.
Protein biochemistry
GST pull down.
The GST-coupled TRPV1 N-terminal and C-terminal fragments were expressed and purified from BL21 cells, as described previously (Zhang et al., 2008). Purified protein fragments were then incubated with pure PKCβII (Merck). For pull down of TRPV1 by PKCβII fragments, HEK293 cells expressing GST-coupled PKCβII protein fragments and TRPV1 were solubilized in a lysis buffer containing 10 mm Tris, 1.0% NP-40, 150 mm NaCl, 1.0 mm EDTA, and 1.0 mm EGTA plus protease inhibitor mixture (Roche). GST-binding proteins were then pulled down by glutathione-agarose (Sigma), followed by extensive washing. Proteins interacting with GST-coupled protein fragments were analyzed on a 10% SDS-PAGE gel.
Immunoprecipitation.
Coimmunoprecipitation was performed as described previously (Zhang et al., 2008). TRPV1 from DRG lysates was precipitated by anti-TRPV1 antibody (Santa Cruz Biotechnology) and then isolated by protein A-agarose. To investigate the interactions between TRPVs (TRPV1, TRPV2, TRPV3, and TRPV4) and PKCβII, HEK 293 cells transfected with cDNAs coding for TRPVs-V5 and PKCβII were solubilized in the lysis buffer, followed by incubation with anti-V5 antibody (Invitrogen) and protein A-agarose (Santa Cruz Biotechnology). Agarose-associated proteins were then dissociated with a sample buffer and analyzed on SDS-PAGE gels.
In vitro PKC phosphorylation assay.
Phosphorylation of the purified GST-coupled TRPV1 C-terminal fusion protein by PKCβII was conducted in a 30 μl of kinase buffer containing 25 mm Tris-HCl, pH 7.5, 0.5 mm DTT, 10 mm MgCl2, 0.6 mm CaCl2, 100 μm ATP, and 1 μg of purified GST-C-TRPV1 or GST-C-T705A TRPV1 protein fragments plus 25 ng of PKCβII (Enzo Life Sciences). To determine whether TRPV1 enhances PKCβII activity, PKCβII was also precipitated by the anti-GFP antibody (Santa Cruz Biotechnology) from HEK293 cell lysate containing GFP-PKCβII either alone or together with TRPV1 (see Fig. 4H). Immunoprecipitated PKCβII was washed three times with the lysis buffer and twice with the kinase buffer and then incubated with 30 μl of kinase buffer in the presence of 1 μg of GST-C-TRPV1 fusion protein. The reactions were incubated at 30°C for 60 min and terminated by boiling in sample buffer, followed by analysis on SDS-PAGE gels. Phosphorylation of fusion proteins was detected using the anti-phosphothreonine antibody (Cell Signaling Technology).
Western blotting.
Protein samples were resolved on 10% SDS-PAGE gels and then transferred to a PVDF membrane. Blots were incubated with primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies (GE healthcare). Proteins on the blot were visualized by enhanced chemiluminescence (Pierce). The PKCβII and PKCε antibodies were obtained from Santa Cruz Biotechnology, the tubulin antibody was from Sigma, and the GST antibody was from GE Healthcare.
Immunocytochemistry
Cultured DRG neurons were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. After blocking with 0.1% gelatin, cells were incubated with mouse anti-TRPV1 (Santa Cruz Biotechnology) and rabbit anti-PKCβII antibodies, followed by secondary antibodies (Alexa Fluor 594 anti-mouse and Alexa Fluor 488 anti-rabbit; both from Life Technologies). Cells were mounted onto a slide for examination by confocal microscopy. The localization of PKCβII was examined visually by two independent experimenters. A distinct, ring-like staining of PKCβII (see Fig. 1B) was considered to indicate localization at the plasma membrane; otherwise, localization was considered to be cytoplasmic.
Electrophysiology
Whole-cell patch clamping was performed as described previously (Zhang et al., 2008; Zhang et al., 2012). Electrodes were fabricated from glass capillaries using a micropipette puller (Sutter Instruments). The electrodes had resistances of 2–4 MΩ when filled with internal solution containing the following (in mm): 140 KCl, 2.0 MgCl2, 5.0 EGTA, 10 HEPES, pH 7.4, with KOH. Cells were perfused with a bath solution containing the following (in mm): 140 KCl, 1.0 MgCl2, 1.8 CaCl2, 10 HEPES, pH 7.4, with KOH. TRPV1 inward currents were measured at a holding potential of −60 mV. To determine temperature-induced activation of TRPV1, the temperature of the perfusion solution was controlled by a Peltier device (CV Scientific), generating a ramp of temperatures from 21° up to 48°C. The I–V relationship of TRPV1 channels was revealed using a ramp protocol starting at −120 mV, followed by a 650 ms linear ramp-up to 160 mV. To examine TRPV1 activation by depolarization, 0 mV of initial holding potential was applied followed by steps of 100 ms voltage pulses ranging from −140 mV to +200 mV in 20 mV increments, with a final step to +60 mV. The half-maximal activation voltage (V1/2) was obtained by the fitting normalized channel conductance (G/Gmax)-voltage relationship to a Boltzmann equation as follows: G/Gmax = G0/Gmax + 1/(1 + exp[−(Vm − V1/2)/k]). Series resistance was routinely ≤10.0 MΩ and was compensated >75%. For neuronal recordings, we chose small rat DRG neurons (<25 μm). All recordings were made at room temperature (24°C) unless otherwise stated with an Axopatch 200B patch clamp amplifier (Axon) associated with pClampex version 10.2 software (Molecular Devices). Signals were analog filtered using a 1 kHz low-pass Bessel filter.
Statistics
All data are presented as mean ± SEM. Differences between groups were evaluated by one-way ANOVA with Bonferroni's post hoc test unless otherwise stated. Results were considered significant at p < 0.05.
Results
TRPV1 causes the translocation of PKCβII in a subset of DRG neurons
PKC comprises a family of important signaling kinases involved in many biological processes. It is mainly located in the cytoplasm. The translocation of PKC to the cell membrane is a hallmark of PKC activation and is elicited by a concerted cascade of second signaling messengers such as diacylglycerol and calcium (Parker and Murray-Rust, 2004). We investigated the expression of PKCβII and PKCε in DRG neurons. Consistent with a previous report (Cesare et al., 1999), PKCε was mainly expressed in the cytoplasm in all DRG neurons (Fig. 1A). Similar observations were also made for PKCβII in a proportion of DRG neurons (38%; Fig. 1B). However, PKCβII was expressed on the plasma membrane in a subpopulation of small DRG neurons (17%; Fig. 1B–E). Interestingly, this latter subpopulation of DRG neurons always coexpressed TRPV1 (Fig. 1B,D). In contrast, TRPV1 was not expressed in most of the neurons (87%) in which PKCβII was localized in the cytoplasm (Fig. 1B,D). The membrane localization of PKCβII is thus highly correlated with coexpression of TRPV1 in DRG neurons.

TRPV1 causes the translocation of PKCβII in DRG neurons. A, Example of double immunostaining of PKCε and TRPV1 in DRG neurons. PKCε was expressed in the cytoplasm in all DRG neurons regardless of whether TRPV1 was coexpressed. Scale bars, 10 μm. B, Sample images of immunolocalization of TRPV1 (middle) and PKCβII (left) in DRG neurons. On the right are bright-field images of neurons. Scale bars, 10 μm. C, Distribution of TRPV1- and PKCβII-expressing DRG neurons. n = 470 cells. D, Summary of percentages of neurons with membrane and cytosol localization in neurons expressing only TRPV1 (n = 178 cells) and neurons expressing both TRPV1 and PKCβII (n = 80 cells). E, F, Histogram distribution of membrane and cytoplasm-localized PKCβII as a function of cell diameter in neurons isolated from wild-type (WT) mice (E; n = 321 cells) and from TRPV1-null mice (F; n = 502 cells).
To investigate whether TRPV1 drives the translocation of PKCβII to the cell membrane, we performed colocalization studies in neurons isolated from mice lacking the TRPV1 gene. As anticipated, the membrane expression of PKCβII was almost absent from DRG neurons deficient in TRPV1 (Fig. 1F), supporting the idea that TRPV1 does indeed cause the translocation of PKCβII in a subset of DRG neurons.
TRPV1 binds directly to PKCβII
The close relationship between coexpression of TRPV1 and translocation of PKCβII can also be recapitulated in an in vitro expression system. Figure 2A shows that PKCβII was expressed in the cytoplasm of HEK293 cells in the absence of TRPV1, but was translocated to the membrane in cells coexpressing TRPV1. These data suggest that PKCβII may bind directly to TRPV1, leading to the translocation of PKCβII. Indeed, PKCβII can be coprecipitated by TRPV1 from both DRG neurons and HEK293 cells expressing both proteins (Fig. 2B,C). Binding of PKCβII to TRPV1 is twice as strong as both control and binding to other members of TRPV family (TRPV2, TRPV3, and TRPV4; Fig. 2C). To determine whether the interaction between TRPV1 and PKCβII is direct or indirect, we used the GST-coupled cytoplasmic fragments of TRPV1 to pull down purified PKCβII. Figure 2D shows that PKCβII binds only to the TRPV1 N terminus, not to a control GST tag or to the TRPV1 C terminus. In contrast, neither the N terminus nor the C terminus of TRPV1 bound directly to PKCε in a similar experiment (Fig. 2E). The lack of direct binding of PKCε to TRPV1 is consistent with the finding that PKCε requires the scaffolding protein AKAP79/150 to mediate an indirect interaction with TRPV1 (Zhang et al., 2008). These data demonstrate that TRPV1 binds directly to PKCβII, but not to PKCε, leading to the translocation of PKCβII.

TRPV1 binds directly to PKCβII. A, Localization of PKCβII (left) and TRPV1 (middle) in HEK 293 cells expressing GFP-PKCβII and TRPV1. On the right is a merged image. Scale bars, 10 μm. B, PKCβII was coprecipitated by TRPV1 in DRG neurons. TRPV1 was precipitated by anti-TRPV1 or a control IgG followed by the detection with anti-PKCβII (top blot). The blot was stripped and reprobed with anti-TRPV1 antibody (bottom blot). Molecular weight is shown on the right and is the same in all other blots. C, TRPV proteins were precipitated by anti-V5 from HEK293 cell lysates expressing PKCβII together with different TRPV channels (all tagged with V5) as indicated, and associated PKCβII detected by anti-PKCβII antibody (top blot). The same blot was stripped and reprobed for different TRPV channels by anti-V5 antibody (middle blot). The bottom blot shows the expression of PKCβII in total cell lysates (TCL) from all groups. Note that TRPV2, TRPV3, and TRPV4 have the same nonspecific background bindings to PKCβII as observed in control (lane 1), whereas the binding to TRPV1 is twice as high as in the other groups. PKCβII binding was normalized to the corresponding total expression of PKCβII, giving rise to the following relative amount of PKCβII binding: 0.45 for control, 1.04 for TRPV1, 0.49 for TRPV2, 0.48 for TRPV3, and 0.55 for TRPV4. D, PKCβII binds to the TRPV1 N terminus. GST-coupled TRPV1 N-terminal and C-terminal fragments were purified and incubated with purified PKCβII. Bound PKCβII was detected (top blot). The blot was stripped and reprobed with anti-GST to show equal levels of TRPV1 fragments (bottom blot). E, PKCε does not bind directly to TRPV1. Experiments were performed as in D except that HEK293 cell lysate expressing PKCε was included for the binding assay. F, TRPV1 binds to the kinase domain (KD) from PKCβII. The GST-coupled C1C2 domain (1∼340) and the KD domain (341∼73) from PKCβII were incubated with cell lysate expressing TRPV1. Associated TRPV1 was detected with anti-V5 antibody (top blot). The blot was stripped and reprobed with anti-GST to show similar expression of all GST fragments. G, GST-coupled kinase domain of PKCβII binds to TRPV1, but not to TRPV2, TRPV3, or TRPV4 (all tagged with V5) in a similar GST pull-down assay as in F. All blots were repeated at least three times, with similar results.
We then investigated further which regions on PKCβII are responsible for its binding to TRPV1. PKCβII contains two important structural domains, the C1C2 domain and the catalytic kinase domain. Both domains are essential for the activation of PKCβII. We constructed protein fragments for these two domains and coupled them to a GST tag for a GST pull-down assay. TRPV1 was pulled down by the kinase domain, but not by the C1C2 domain of PKCβII nor by a control GST tag (Fig. 2F). Binding to the kinase domain of PKCβII is specific to TRPV1 because none of other TRPVs (TRPV2, TRPV3, or TRPV4) was pulled down by the same domain of PKCβII (Fig. 2G), which is consistent with the results of the coimmunoprecipitation experiment (Fig. 2C). These experiments demonstrate that TRPV1 binds directly to the kinase domain of PKCβII, an event that may expose the kinase domain, leading to the translocation and thus activation of PKCβII.
PKCβII causes the downregulation of TRPV1 by phosphorylating the channel
We further examined the expression of TRPV1 and PKCβII in HEK293 cells coexpressing the two proteins. Surprisingly, coexpression of PKCβII caused the downregulation of TRPV1 (Fig. 3A). Similar TRPV1 downregulation was also observed with PKCα, another member of the conventional PKC family. However, other members of PKC family, such as PKCδ or PKCε, had no effect on TRPV1 expression (Fig. 3A). Reciprocally, the downregulation of TRPV1 was accompanied by the reduced expression of PKCα and PKCβII (Fig. 3A,B), but no expression changes in PKCδ or PKCε were observed upon coexpression with TRPV1 (Fig. 3A,B).

Downregulation of TRPV1 and PKCβII. A, Expression of TRPV1 was downregulated by PKCα and PKCβII, but not by PKCδ or PKCε. HEK 293 cells were transfected with TRPV1–V5 together with different PKC isoforms (all tagged with GFP at their N terminus) as indicated. The expression of TRPV1 was detected with anti-V5 antibody (top blot). The blot was stripped and reprobed for PKCs with anti-GFP, followed by further stripping and reprobing with antitubulin to show similar expression of tubulin and thus similar sample loading in all cases (bottom blot). B, Downregulation of PKCα and βII by TRPV1. HEK 293 cells were transfected with different GFP-tagged PKC isoforms as indicated together with (+) or without (−) TRPV1. The expression of all PKCs was detected by anti-GFP (top blot). The blot was stripped and reprobed with antitubulin to show similar expression in all groups (bottom blot). C, Time course of downregulation of TRPV1 by PKCβII. TRPV1–V5, and PKCβII-pTRE2 were cotransfected in Tet-On HEK 293 cells. Twenty-four hours after transfection, PKCβII was induced by DOX for different times, as indicated. The expression of TRPV1 (top blot) and PKCβII (middle blot) was detected by anti-V5 and anti-PKCβII, respectively. The blot was stripped and reprobed with antitubulin showing similar expression in all cases. D, E, Proteolytic enzymes (D) and organelles (E) are not involved in PKCβII-induced downregulation of TRPV1. HEK-293 cells were transfected with TRPV1–V5 or together with PKCβII, as indicated. Transfected cells were pretreated with Z-VAD (50 μm; to inhibit caspase), calpeptin (5 μm; to inhibit calpain), phenanthroline (Phe, 2 mm; to inhibit metallo-protease), MG132 (1 μm), and lactacystin (5 μm; to inhibit proteasome) for 20 h before solubilization. TRPV1 was detected by anti-V5 (top blot) followed by stripping and reprobing with antitubulin (bottom blot). F, Downregulation of TRPV1 by PKCβII was prevented by TRPV1 blockers. Tet-On HEK-293 cells were cotransfected with TRPV1–V5 and PKCβII-pTRE2. PKCβII was induced 24 h after transfection for an additional 24 h with (+) or without (−) ruthenium red RR (10 μm) or BCTC (10 μm), as indicated. The blot was probed for TRPV1 with anti-V5 (top blot), followed by stripping and reprobing with anti-PKCβII. The blot was then further stripped and reprobed with antitubulin to show equal sample loading in all cases (bottom blot). G, Downregulation of PKCβII by TRPV1 was prevented by RR. HEK 293 cells expressing TRPV1–V5 and PKCβII were treated with RR (10 μm) as indicated. The top blot shows the detection of PKCβII. The blot was stripped and reprobed with antitubulin (bottom blot). H, TRPV1 F641L protein expressed from HEK293 cells was probed with anti-V5 (top blot), followed by stripping and reprobing with antitubulin (bottom blot). All blots were repeated at least three times, with similar results. Molecular weights are shown on the right for all blots.
To monitor the downregulation of TRPV1 by PKCβII dynamically, we incorporated PKCβII into a pTRE2 vector containing a Tet-On promoter, the activity of which can be controlled by doxycycline (DOX). Under this Tet-On system, the expression of PKCβII can be induced dynamically by DOX. We then coexpressed TRPV1 with PKCβII-pTRE2 in Tet-On HEK293 cells. Fig. 3C shows that the expression of PKCβII is gradually increased after the addition of DOX and, correspondingly, the expression of TRPV1 begins to fall after 8 h. The downregulation of TRPV1 becomes prominent after 24 h of DOX induction. In all of following functional studies, we used 8 h of PKCβII induction to avoid excessive downregulation of TRPV1 protein.
We next examined the mechanisms causing the downregulation of TRPV1 by PKCβII. The downregulation of TRPV1 is not caused by the actions of known proteases such as caspase, calpain, and metalloprotease (Fig. 3D) nor by a nonspecific interference with TRPV1 transcription due to PKCβII coexpression (data not shown). It is also not a result of proteolysis in proteasomes or lysosomes, two principal proteolytic systems in cells, because inhibitors for these enzymes and organelles did not prevent the downregulation of TRPV1 caused by PKCβII (Fig. 3D,E, and data not shown). The downregulation of TRPV1 and PKCβII was, however, prevented by either the general TRP channel pore blocker ruthenium red (RR) or, even more potently, by the specific TRPV1 blocker N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC; Fig. 3F,G), suggesting that enhanced constitutive activity of the TRPV1 channel itself caused by PKCβII is the main cause of protein downregulation. Consistent with this idea, the expression of TRPV1 F641L, a channel pore mutant endowing channels with high constitutive basal activity (Myers et al., 2008; Myers et al., 2008), was barely detected, but the expression of this mutant was increased dramatically after blocking channels with RR (Fig. 3H). These highly active TRPV1 channels cause steady rises in intracellular calcium, leading to a cytotoxic effect (Myers et al., 2008), which may explain the downregulation of TRPV1.
We then investigated how the binding of PKCβII enhanced the basal activity of TRPV1. This effect could be achieved through either direct physical binding of PKCβII leading to conformational changes in TRPV1 or enhanced catalytic enzyme activity of PKCβII or both. To discriminate between these possibilities, we constructed two kinase-inactive PKCβII mutants by mutating two residues (K371 and T500) on PKCβII that are essential for catalytic activity. The downregulation of TRPV1 was abolished by both kinase-dead PKCβII mutations (Fig. 4A) despite the fact that both mutants still bound to TRPV1 (data not shown). Furthermore, coexpression of the kinase domain of PKCβII, an identified TRPV1-binding region, was not sufficient to cause the downregulation of TRPV1 (Fig. 4B). These results suggest that increased catalytic activity of PKCβII as a result of binding, not the direct physical binding per se, is mainly responsible for the enhanced function of TRPV1 and consequent downregulation of the channel, presumably through a phosphorylation-dependent mechanism.

Downregulation of TRPV1 by PKCβII depends on the phosphorylation of TRPV1 at Thr705. A, B, Catalytic activity of PKCβII is essential for the downregulation of TRPV1. Tet-On HEK-293 cells were transfected with TRPV1–V5 together with different PKCβII mutants or the kinase domain (KD) from PKCβII in pTRE2 vector as indicated. Twenty-four hours after transfection, PKCβII was induced with DOX for 24 h (+). Expression of TRPV1 was detected by anti-V5 (top blot). The blot was stripped and reprobed with anti-PKCβII (middle blot) and antitubulin (bottom blot). C, Two phosphorylation sites for PKCε are not involved in the downregulation of TRPV1by PKCβII. Tet-On HEK293 cells were transfected with wild-type (WT) TRPV1 or double mutant (DM)-TRPV1 (S502A/S801A) together with PKCβII-pTRE2. PKCβII induced 24 h after transfection. Expression of TRPV1and PKCβII was detected by anti-V5 (top blot) and anti-PKCβII (middle blot). The bottom blot shows equal expression of tubulin and sample loading for all groups. D, Downregulation of double mutants (DM)-TRPV1 was prevented by RR. Tet-On cells expressing DM-TRPV1–V5 and PKCβII-pTRE2 were treated with DOX in the presence of RR (10 μm) as indicated. TRPV1 was detected with anti-V5 (top). The blot was stripped and reprobed with antitubulin, showing equal loading (bottom blot). E, Downregulation of TRPV1 by PKCβII was prevented by mutating T705. Tet-On HEK293 cells were transfected with PKCβII-pTRE2, along with different TRPV1 mutants, as indicated. PKCβII expression was induced for 24 h with DOX. TRPV1 (top blot) and PKCβII (middle blot) were detected by anti-V5 and anti-PKCβII, respectively. Blots were stripped and reprobed with antitubulin (bottom blot). F, Quantification of downregulation of TRPV1 mutants by PKCβII in at least two experiments similar to those in E. All error bars indicate mean ± SEM. NS, Not significant. ***p < 0.001 compared with wild-type (WT) TRPV1. G, H, In vitro phosphorylation of purified GST-coupled TRPV1 C-terminal fragment and the T705A mutant (as indicated) by purified PKCβII (G) or immunoprecipitated PKCβII (H). Protein phosphorylation was detected by anti-phosphothreonine antibody (top blot). The bottom blot is a Coomassie blue stain of corresponding purified proteins. All blots were repeated at least three times, with similar results. Molecular weights are shown on the right for all blots.
Phosphorylation of TRPV1 by PKCε is a major mechanism underlying the sensitization of TRPV1 and consequent inflammatory hyperalgesia (Cesare et al., 1999; Zhang et al., 2007). Two PKCε phosphorylation sites, S502 and S801, have been identified as key residues for the sensitization of TRPV1 (Numazaki et al., 2002; Bhave et al., 2003; Zhang et al., 2008). However, mutating these two PKCε phosphorylation sites did not prevent the downregulation of TRPV1 elicited by PKCβII (Fig. 4C). Instead, the downregulation of the S502A/S801A TRPV1 mutant was prevented by RR, similar to wild-type TRPV1 (Figs. 3F, 4D). These results suggest that there are other, as yet unidentified PKCβII phosphorylation sites on TRPV1 that are essential for enhanced constitutive activity of TRPV1 channels.
Several serine and threonine residues on TRPV1, apart from S502 and S801, have been identified as PKC phosphorylation sites in vitro and most of these have no assigned functions (Bhave et al., 2003). We therefore mutated all other potential PKC phosphorylation sites and investigated whether the downregulation of TRPV1 caused by enhanced activity of channels evoked by PKCβII could be prevented. We found that the downregulation of TRPV1 was reduced dramatically by mutating Thr705, but not by mutating other potential PKC phosphorylation sites (Fig. 4E,F), suggesting that Thr705 on TRPV1 is a functionally relevant PKCβII phosphorylation site. To determine whether PKCβII phosphorylates Thr705 directly, we performed an in vitro PKC phosphorylation assay using the GST-coupled TRPV1 C-terminal fragment and the T705A C-terminal fusion protein. Figure 4G shows that PKCβII did not cause threonine phosphorylation of the control GST protein, but increased threonine phosphorylation of the GST-coupled TRPV1 C terminus dramatically. The increased threonine phosphorylation was substantially reduced by mutating Thr705, showing that Thr705 is indeed a major PKCβII phosphorylation site.
PKCβII enhances TRPV1 activity by phosphorylating Thr705. Our evidence also suggests that the enzyme activity of PKCβII is stimulated simply by a direct physical binding of TRPV1 without the need for additional signaling. To test this idea, PKCβII was immunoprecipitated from cell lysates containing PKCβII either alone or together with TRPV1 to perform an in vitro immunoprecipitation kinase assay. As expected, the presence of TRPV1 increased threonine phosphorylation of the TRPV1 C terminus significantly (Fig. 4H), demonstrating that TRPV1 binding facilitates the activation of PKCβII. Together, these results demonstrate that activated PKCβII, as a result of direct binding of TRPV1 to its kinase domain, downregulates TRPV1 channels by enhancing the basal constitutive activity of TRPV1 through phosphorylation of T705. This residue may therefore have a previously unrecognized role in tuning the activity of TRPV1 channels.
Sensitivity of TRPV1 channels is enhanced by PKCβII, but not by PKCε
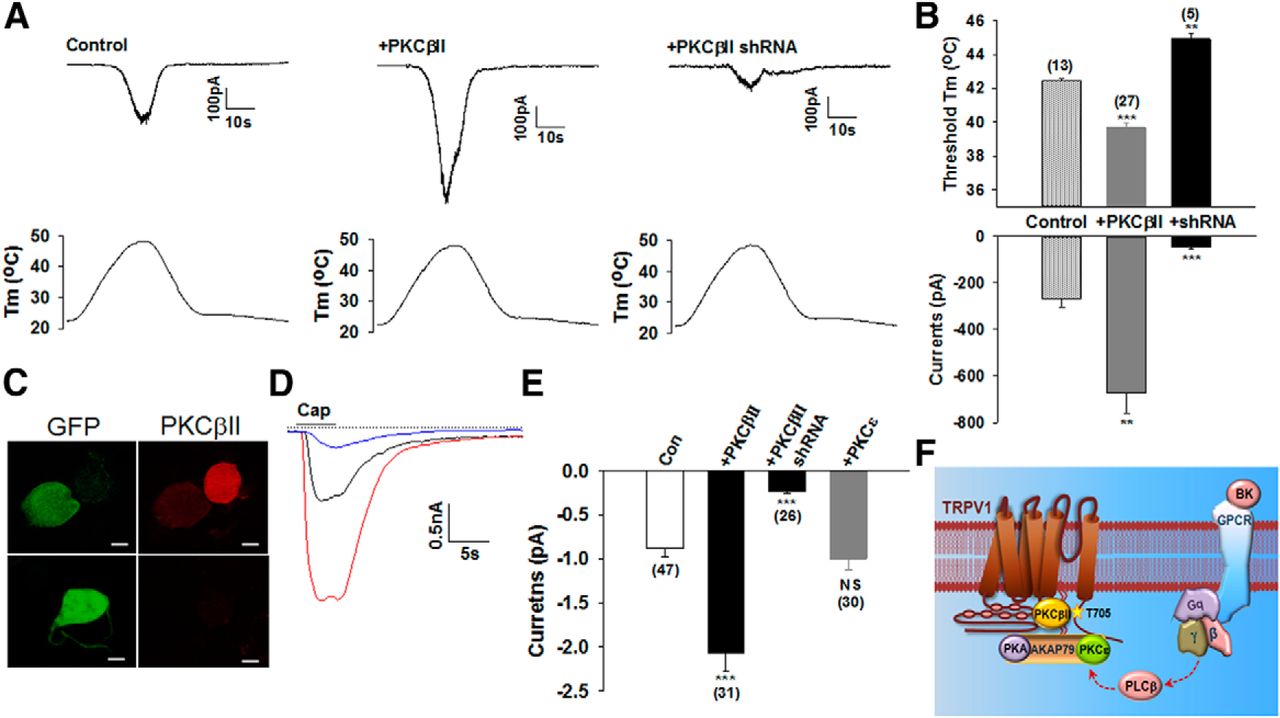
We designed shRNA constructs to knock down the expression of PKCβ and PKCε selectively. Both constructs potently reduced the expression of PKCβ and PKCε, respectively, in HEK293 cells, whereas corresponding scrambled shRNAs had no effect (Fig. 5A). To determine the effect of PKCβII on the function of TRPV1 directly, we measured inward currents carried by TRPV1 as a function of different concentrations of capsaicin. The sensitivity of TRPV1 to capsaicin was markedly increased after the induction of PKCβII for 8 h by DOX, resulting in a negative shift of the dose–response curve (Fig. 5B). This occurred despite the fact that a moderate downregulation of TRPV1 was induced at this time point, showing that functional enhancement elicited by PKCβII overrides PKCβII-induced TRPV1 downregulation. The increased sensitivity of TRPV1 was, however, abolished by inducing the catalytically inactive PKCβII mutant K371R (Fig. 5B), which also abolished the downregulation of TRPV1 (Fig. 4A). Conversely, knocking down endogenous PKCβII reduced the sensitivity of TRPV1 dramatically, yielding a positive shift of the curve (Fig. 5B). A similar reduction in the sensitivity of TRPV1 was also observed with the PKCβII phosphorylation site-ablated TRPV1 mutant T705A (Fig. 5C). Importantly, the T705A mutant abolished PKCβII-induced enhancement of the sensitivity of TRPV1 to capsaicin (Fig. 5C). These data show that the phosphorylation of TRPV1 by catalytically activated PKCβII underlies increases in the sensitivity of TRPV1. To determine the effect of PKCβII on the gating of TRPV1 by voltage, we monitored voltage-dependent currents mediated by TRPV1. The outward currents activated by depolarization were increased by induction of wild-type PKCβII, but not by induction of the enzyme-activity-deficient PKC mutant K371R (Fig. 5D). As expected, depolarization-induced currents were reduced robustly by either knocking down PKCβII (Fig. 5D) or mutating the PKCβII phosphorylation site T705 (Fig. 5F). These data show that phosphorylation of TRPV1 at T705 by PKCβII is critical for the gating of TRPV1 by both capsaicin and voltage.

PKCβII increases the sensitivity of TRPV1 through a phosphorylation-dependent mechanism. A, Knock down of PKCβII and PKCε by shRNAs. HEK-293 cells were transfected with scrambled or PKCβII shRNA (top two blots) or PKCε shRNA (bottom two blots), as indicated. PKCβII and PKCε were detected with anti-PKCβII and anti-PKCε, respectively. Blots were stripped and reprobed with antitubulin to show equal loading in all groups. B, Normalized whole-cell inward currents as a function of different capsaicin concentrations in Tet-On HEK293 cells. Shown are the control dose–response curve (♦), EC50 = 84.51 nm; PKCβII induction for 8 h with DOX (▴), EC50 = 38.15 nm; cotransfection with PKCβII shRNA (▾), EC50 = 155.01 nm; and cotransfection with PKCβII K371R with DOX induction (□), EC50 = 92.56 nm. PKCβII expression was induced by DOX for 8 h. All curves fitted with the Hill equation with a Hill coefficient of 1.5. C, Increased sensitivity of TRPV1 to capsaicin induced by PKCβII was abolished by mutating T705. Shown are the wild-type (WT) TRPV1 dose–response curve (♦), EC50 = 84.51 nm; the T705A TRPV1 dose–response curve (▾), EC50 = 201.2 nm; and the T705A TRPV1 dose–response curve induced with DOX for PKCβII expression (▴), EC50 = 171.3 nm. D, TRPV1 currents evoked by a voltage ramp (from −120 mV to +160 mV, 650 ms) were increased by PKCβII induction with DOX (blue curve), but inhibited by PKCβII shRNA (red curve). DOX-induced increases in TRPV1 currents were abolished by PKCβII K371R (pink curve). All I–V curves are averages of 16∼22 recordings. E, PKCε does not affect the sensitivity of TRPV1 to capsaicin. Normalized whole-cell inward currents plotted against different concentrations of capsaicin as in B. Control curve (○), EC50 = 86.94 nm; cotransfection with PKCε (▴), EC50 = 79.46 nm; and cotransfection with PKCε shRNA (▾), EC50 = 93.43 nm. The Hill coefficient is 1.5. F, TRPV1 currents evoked by a voltage ramp (from −120 mV to +160 mV, 650 ms) were reduced by mutating T705 (red curve), but not by mutating two PKCε phosphorylation sites S502A/S801A (DM TRPV1; pink curve) nor by coexpression with PKCε (blue curve). All I–V curves are averages of 16∼29 recordings.
PKCε is another PKC isoform involved in the potentiation of TRPV1 after activation by inflammatory mediators such as bradykinin (Cesare et al., 1999). Surprisingly, neither overexpression nor downregulation of PKCε had any effect on the sensitivity of TRPV1 (Fig. 5E). Furthermore, no effect on TRPV1 outward currents activated by depolarization was observed after overexpression of PKCε, and the PKCε phosphorylation-site-ablated TRPV1 mutant (S502A/S801A) showed similar voltage-dependent outward currents as wild-type TRPV1 (Fig. 5F). These findings suggest that the basal sensitivity of TRPV1 is not determined by PKCε and that PKCε is mainly in an inactive state under basal condition, so it cannot regulate TRPV1 until activation by additional signaling, such as from Gαq-protein-coupled receptors.
PKCβII determines the heat sensitivity of TRPV1
We next measured the heat activation of TRPV1 by exposing cells to a temperature ramp from 22°C up to 48°C in Tet-On HEK293 cells expressing TRPV1. TRPV1 was typically activated at ∼40.5°C in these cells (Fig. 6A,B,E). The induction of PKCβII with DOX significantly reduced the activation threshold of TRPV1 to 36.8°C (Fig. 6C,E), equivalent to the effect of a TRPV1 deletion mutant in which a proposed PIP2-binding region (777∼820) in the C terminus has been removed, thus rendering the channel hyper-heat sensitive (Prescott and Julius, 2003; Fig. 6F). The activation threshold for heat was further significantly reduced in the deletion mutant after PKCβII induction (Fig. 6F), suggesting a nonoverlapping mechanism used by PKCβII and the TRPV1 deletion mutant for enhancing the heat sensitivity of TRPV1. Concomitantly, the peak heat currents were also significantly larger after PKCβII induction (Fig. 6C,F). Remarkably, TRPV1 was almost heat insensitive after knocking down endogenous PKCβII and only a small, nonspecific membrane current was observed (Fig. 6D,E). The lack of heat-induced responses is not due to the absence of function of TRPV1, because a subsaturating concentration of capsaicin (1 μm) still induced a similar inward current as that in the control condition (Fig. 6D). In contrast, no effect was observed on either heat thresholds or peak heat currents by overexpression of PKCε or by knocking down endogenous PKCε (Fig. 6F). The N-terminal proximal domain of TRPV1 has been implicated as a thermal sensor of TRPV1 channels (Yao et al., 2011). Therefore, it was interesting to determine whether PKCβII may have caused changes in the thermal sensitivity of TRPV1 through a direct physical binding to the thermal sensor. Figure 6G shows that the heat activation threshold for TRPV1 was not significantly reduced after inducing the catalytically inactive PKCβII mutants (K371R and T500V), although there were small but significant increases in the peak heat currents. The small enhancement in peak heat currents may be caused by the direct physical binding of PKCβII to TRPV1. Collectively, these data suggest that the catalytic enzyme activity of PKCβII, but not PKCε, is the main cause of enhanced heat responses of TRPV1, presumably through a phosphorylation-dependent mechanism, and that the direct physical binding of PKCβII has only a minor role in regulating the heat sensitivity of TRPV1.

Heat sensitivity of TRPV1 is increased by PKCβII. A, Representative inward currents carried by TRPV1 (top left trace) induced by a temperature ramp shown underneath in a Tet-On HEK-293 cell expressing TRPV1 and PKCβII-pTRE2. Right, Inward currents evoked by capsaicin (1 μm, 5 s) applied at the end of temperature ramp. B, Arrhenius plot of inward current shown in A as a function of absolute temperature. The threshold heat temperature was estimated to be 40.1°C from the intersection of two different slopes (red lines). C, D, Example of TRPV1 inward current (top trace on the left) evoked by a temperature ramp shown underneath in a Tet-On HEK 293 cell when PKCβII was induced by DOX for 8 h (C) or coexpressing PKCβII shRNA (D). Capsaicin (1 μm, 5 s) applied after a temperature ramp (right). E, Current-temperature relationship from recorded cells in A, C, and D. F, Summary of heat threshold for TRPV1 and peak amplitudes of heat-induced currents in experiments similar to those in A–D. The number of experiments is given under each bar. All error bars indicate mean ± SEM. NS, Not significant. *p < 0.05; ***p < 0.001. Note that the heat threshold cannot be measured in cells treated with PKCβII shRNA due to small heat currents. The heat threshold was reduced by PKCβII to a similar level as in a hyper-heat-sensitive TRPV1 deletion mutant (CD777 TRPV1). G, Summary of heat thresholds and heat-induced peak currents in Tet-On HEK-293 cells expressing TRPV1 and PKCβII mutants (K371R, T500V)-pTRE2 (bars 1–4) or in cells expressing PKCβII-pTRE2 and double mutant (DM, S502A/S801A) TRPV1 (bars 5 and 6). The number of experiments is given above each bar. All error bars indicate mean ± SEM. NS, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
We have identified T705 on TRPV1 as a key PKCβII phosphorylation site. We next determined the heat sensitivity of the T705A TRPV1 mutant. Figure 7A shows that a similar heat ramp induced a much smaller current in T705A TRPV1 compared with wild-type TRPV1, with a significantly higher activation threshold. However, the peak current elicited by a submaximal concentration of capsaicin (1 μm) in the T705A mutant was similar to that in wild-type TRPV1 (Fig. 7A). The T705A mutant also abolished PKCβII-induced enhancement of the activation of TRPV1 by heat (Fig. 7B). In contrast, the PKCε phosphorylation-site-ablated mutant did not abolish the reduction in the heat threshold of TRPV1 induced by PKCβII (Fig. 6G), suggesting that T705 is a key residue in regulating the heat sensitivity of TRPV1.

Heat sensitivity of TRPV1 is determined by the phosphorylation of T705 induced by PKCβII. A, Example of inward current (top left trace) induced by a temperature ramp shown under Tet-On HEK-293 cells expressing T705A TRPV1. Right, current elicited by capsaicin (1 μm, 5 s) applied at the end of temperature ramp. B, Summary of heat thresholds and heat-induced peak currents in experiments similar to those in A. The number of experiments is given above each bar. All error bars indicate mean ± SEM. NS, Not significant. ***p < 0.001. C, Representative TRPV1 currents elicited by voltage steps from −140 mV to + 200 mV in 20 mV increments in HEK 293 cells expressing wild-type (WT) or T705A TRPV1, as indicated. D, Normalized conductance-voltage relationship for cells in C fitted by a Boltzmann function with values of V1/2 and steepness factors as follows: WT TRPV (Δ), 139.5 mV, 33.98 mV; T705A TRPV1 (▿), 198.9 mV, 33.05 mV. E, Summary of V1/2 values from experiments similar to those in C. The number of experiments is given above each bar. All error bars indicate mean ± SEM. ***p < 0.001.
We then examined the mechanisms causing T705A TRPV1 to be less responsive to heat. The activation of TRPV1 by heat was suggested to be caused by the shift in the transmembrane voltage toward a physiological membrane potential (Voets et al., 2004). As reported before (Voets et al., 2004; Zhang et al., 2008), wild-type TRPV1 showed strong voltage dependence after steps of depolarization, but similar voltage steps induced much smaller outward currents with T705A TRPV1 (Fig. 7C), causing a positive shift in voltage-dependent activation curve and a much higher V1/2 (Fig. 7D,E).
Together, these results suggest that the heat sensitivity of TRPV1 is critically dependent on PKCβII, which acts by phosphorylating T705. This residue thus has a pivotal role in determining the sensitivity of TRPV1 elicited by thermal and chemical stimuli.
PKCβII is not involved in the potentiation of TRPV1 evoked by the activation of a Gαq-protein-coupled receptor
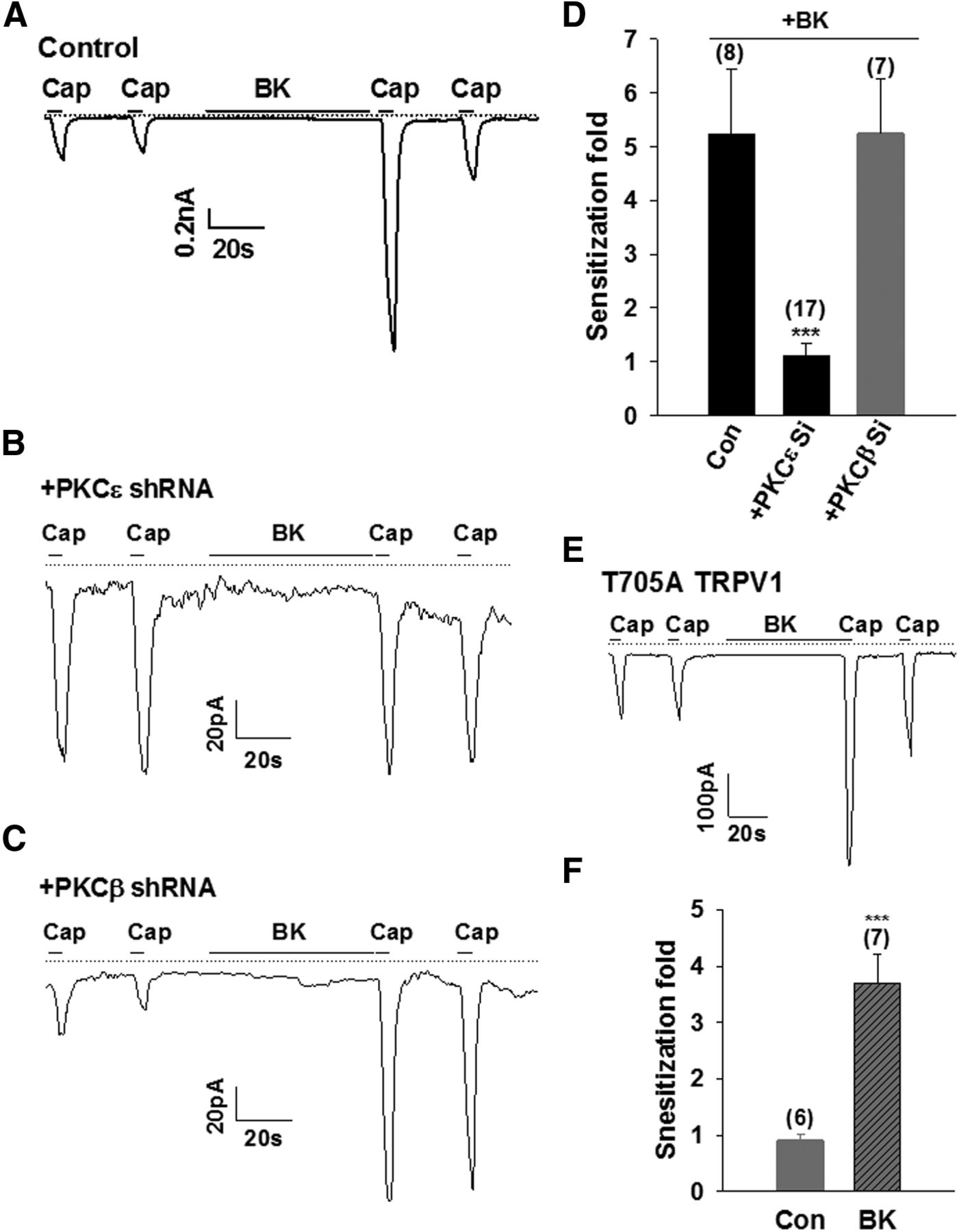
Because the basal sensitivity of TRPV1 is determined by PKCβII, we investigated whether PKCβII was also involved in the sensitization of TRPV1 induced by bradykinin (BK). BK binds to the BK B2 receptor (B2R), leading to the translocation and activation of PKCε, which then phosphorylates TRPV1 at two main residues, S502 and S801, leading to the sensitization of TRPV1 (Zhang et al., 2008). As shown previously (Zhang et al., 2008), BK induced a potent sensitization of TRPV1 in HEK293 cells expressing TRPV1 and B2R (Fig. 8A). The sensitization of TRPV1 evoked by BK was completely abolished by knocking down PKCε (Fig. 8B,D), confirming the critical role of PKCε in this process. However, downregulating PKCβII had no effect on the BK-induced sensitization of TRPV1 currents elicited by capsaicin (Fig. 8C,D). Moreover, the BK-induced sensitization of TRPV1 was not attenuated in the PKCβII phosphorylation-site-ablated TRPV1 mutant T705A (Fig. 8E,F), which, however, did abolish enhanced sensitivities of TRPV1 caused by PKCβII. These data indicate that PKCε, but not PKCβII, is critical for the potentiation of TRPV1 caused by the activation of Gαq protein-coupled receptor, but PKCε is not involved in regulating the basal sensitivity of the channel.

PKCε, but not PKCβII, is involved in the potentiation of TRPV1. A–C, Examples of whole-cell inward currents activated by capsaicin (50 nm, 5 s) from a HEK 293 cell expressing TRPV1 and the bradykinin B2 receptor (A; control) or together with PKCε shRNA (B) or PKCβII shRNA (C), as indicated. The dashed line is zero current. D, Collected results of experiments similar to those in A–C. The number of experiments is given above each bar. All error bars indicate mean ± SEM. ***p < 0.001. E, BK-induced sensitization of TRPV1 in PKCβII phosphorylation-site-ablated TRPV1 T705A mutant. Capsaicin (50 nm, 5 s) and BK (1 μm) were applied as indicated. The dashed line is zero current. F, Summary of experiments similar to those in E. The number of experiments is given above each bar. All error bars indicate mean ± SEM. ***p < 0.001.
PKCβII is required for the responsiveness of TRPV1 in DRG neurons
We next analyzed heat responses in cultured rat DRG neurons. A temperature ramp up to 48°C was used to activate low-threshold, heat-sensitive neurons, most of which are capsaicin sensitive and thus are mainly mediated by TRPV1. Conversely, another population of high-threshold heat-sensitive neurons (>50°C) are normally capsaicin insensitive and will not be activated by the temperature ramp used (Nagy and Rang, 1999a; Caterina et al., 2000). Of 38 recorded neurons, 13 neurons (34.2%) were heat sensitive and also activated by capsaicin, thus expressing TRPV1. These TRPV1+ neurons had a typical heat activation threshold of 42.4°C (Fig. 9A). The proportion of heat-sensitive neurons was significantly increased after expression of PKCβII in the DRG neurons (0.01 < p < 0001, χ2 test). In this case, 28 of 42 recorded neurons (66.7%) were heat sensitive and 27 of these heat-sensitive neurons were responsive to capsaicin, with a significantly lower thermal threshold of 39.7°C and much bigger peak heat currents (Fig. 9A,B). We then expressed PKCβ shRNA in the neurons, resulting in pronounced reductions in the expression of PKCβII (Fig. 9C). Interestingly, a significantly lower percentage of neurons (12.8%, 5 of 39, p < 0.05, χ2 test) were responsive to heat in neurons transfected with PKCβ shRNA. All five of these neurons were also capsaicin sensitive and had a significantly higher heat threshold (44.9°C) and much smaller peak heat currents (Fig. 9A,B). We further compared the peak current amplitudes elicited by capsaicin (1 μm). Transfection of neurons with PKCβII, but not PKCε, increased capsaicin-evoked currents robustly (Fig. 9D,E). Conversely, knocking down PKCβII reduced dramatically TRPV1 currents activated by capsaicin (Fig. 9D,E). It should be noted that native TRPV1 in DRG neurons displays a much lower sensitivity to capsaicin than that in transfected HEK293 cells (Kim et al., 2006). Therefore, 1 μm (i.e., submaximal) capsaicin can produce significant differences in TRPV1 peak currents in DRG neurons (Fig. 9D,E), but not in transfected HEK 293 cells (Fig. 6A,C,D). These results demonstrate that the heat sensitivity and responsiveness of TRPV1 depends critically on the coexpression of PKCβII in DRG neurons.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PKCβII is required for the responsiveness of DRG neurons to heat and capsaicin. A, Representative inward currents from a DRG neuron under control conditions (left), from a neuron transfected with GFP-PKCβII (middle), and from a neuron transfected with PKCβII shRNA (right) activated by a temperature ramp shown underneath for each condition. B, Summary of heat thresholds and peak heat currents in experiments similar to those in A. Only capsaicin-sensitive neurons, which express TRPV1, were included in the analysis. The number of neurons is given above each bar. All error bars indicate mean ± SEM. **p < 0.01; ***p < 0.001. C, DRG neurons were transfected with GFP-PKCβII shRNA. Neurons were then immunostained with anti-PKCβII. Left, GFP fluorescence indicating successfully transfected neurons. Right, expression of PKCβII. The expression of PKCβII was absent from all GFP+ neurons (n = 45). Scale bars, 10 μm. D, Example of inward currents activated by capsaicin (1 μm, 5 s) from a DRG neuron under control condition (in black), transfected with GFP-PKCβII (in red), and transfected with PKCβII shRNA (in blue). The dashed line is zero current. E, Summary of results in experiments similar to those in D. The last bar is from neurons transfected with GFP-PKCε. The number of experiments is given below each bar. All error bars indicate mean ± SEM. ***p < 0.001. F, Schematic summary of regulation of TRPV1 by PKCβII and PKCε. PKCβII binds directly to the N terminus of TRPV1, leading to the activation of PKCβII, which then phosphorylates T705 (shown by a star) in the C terminus of TRPV1. Phosphorylation at this residue enhances the excitability of TRPV1 channels. In contrast, PKCε interacts with TRPV1 indirectly through the scaffolding protein AKAP79, which binds to TRPV1 C terminus (Zhang et al., 2008). PKCε is in an inactive state within this complex and can only be activated by additional signaling such as via activation of the Gαq-coupled receptor B2R. Activated PKCε phosphorylates two residues at S502/S801, leading to the potentiation of TRPV1.
Discussion
The sensitivity of peripheral nociceptors is fundamental to the ability to elicit pain. The expression of TRPV1 channels is a molecular signature marking nociceptive neurons. TRPV1 itself can be targeted directly and excited by a variety of chemical and thermal stimuli producing pain and itch. Therefore, understanding the mechanisms for determining the sensitivity of TRPV1 is critical for understanding these TRPV1-channel-mediated conditions. In the present study, we report that the sensitivity of TRPV1 channels is determined by PKCβII. TRPV1 binds directly to PKCβII and subsequently activates PKCβII. Activated PKCβII then enhances the responsiveness of TRPV1 to thermal and chemical stimuli through a phosphorylation-dependent mechanism. Remarkably, TRPV1 is barely heat sensitive in the absence of PKCβII or after ablation of a PKCβII phosphorylation site on the channel. Furthermore, expression of PKCβII increased the proportion of heat-sensitive neurons and capsaicin-induced responses and knocking down PKCβII reduced the percentage of neurons responding to heat and capsaicin. PKCβII is thus an essential accessory protein for the gating of TRPV1, not only by chemical agonists, but also, importantly, by heat. This mechanism may underlie the varied thermal sensitivities of TRPV1 channels expressed in different populations of DRG neurons and in different systems in which there are different levels of PKCβII expression.
We have identified previously a TRPV1 signaling complex formed between TRPV1 and the scaffolding protein AKAP79/150, which then anchors PKA, PP2B, and PKC (Zhang et al., 2008). This signaling complex is normally inactive and becomes active only when anchored PKC becomes activated, for example, as a result of stimulation of G-protein-coupled receptors during inflammation and tissue injury. Therefore, the AKAP79/150-TRPV1 complex is mainly involved in the enhancement of heat pain (heat hyperalgesia). In the present study, TRPV1 was found to bind directly to PKCβII, but not to PKCε, forming a different local complex in a subpopulation of DRG neurons. This complex, however, was constitutively active and thus was mainly involved in regulating the basal sensitivity of TRPV1 (Fig. 9F). Increased sensitivity of TRPV1, determined by PKCβII, will therefore make nociceptive neurons more sensitive to pain and itch-producing chemicals and toxins.
PKCε is a primary PKC isoform in the TRPV1-AKAP79 complex involved in heat hyperalgesia. It acts by phosphorylating two functional sites, S502 and S801, on TRPV1. PKCβII enhances the sensitivity of TRPV1 by phosphorylating a distinct residue (T705) on TRPV1 (Fig. 9F). The sensitization of TRPV1 induced by BK was abolished by either knocking down PKCε or by mutating two PKCε phosphorylation sites; however, neither of these manipulations had any effect on the sensitivity of TRPV1 (i.e., no changes in the activation by capsaicin, voltage, and temperature). Conversely, the sensitivity of TRPV1 was reduced substantially by either downregulating PKCβII or ablating the PKCβII phosphorylation site, neither of which had any effect on the sensitization of TRPV1 induced by BK (Fig. 9F). These studies demonstrate clearly that the sensitivity of TRPV1, determined by the phosphorylation of T705, and the potentiation of TRPV1, determined by the phosphorylation of S502 and S801, are two separate processes involving different mechanisms. It is likely that the thermal threshold of TRPV1 will be reduced to an even lower level by activated PKCε after inflammation in a subset of PKCβII-expressing neurons in which the basal thermal threshold is already very low. Therefore, two different PKC isoforms act in concert to regulate different aspects of pain mediated by TRPV1.
In addition to enhancing the sensitivity of TRPV1, constitutively activated PKCβII, as a result of physical binding of TRPV1, may also target other ion channels and/or signaling messengers such as voltage-gated sodium and potassium channels, the activities of which are also regulated by PKC-mediated phosphorylation (Li et al., 1993; Enyedi and Czirjak, 2010). The regulation of these ion channels by PKC may further facilitate the transduction of pain and/or itch. Therefore, a subpopulation of DRG neurons expressing TRPV1-PKCβII complexes may represent a population of hypersensitive nociceptive neurons that have a more prominent role in thermal hyperalgesia and pain.
In many cases, ion channels are expressed as part of a specific protein complex containing auxiliary proteins, which strongly influence the activation, inactivation, and modulation of the channels (Isom et al., 1994). These ion channel complexes diversify the function of ion channels, allowing ion channels to perform appropriate functions in different cells and systems. In the present study, we discovered that the function of TRPV1 channels was contingent on coexpression of PKCβII, which binds constitutively to the channel. Therefore, PKCβII acts as an auxiliary subunit of TRPV1, controlling its sensitivity. The N terminus of TRPV1 contains several important structural domains, including the ankyrin repeat domain and the membrane proximal domain. In particular, the membrane proximal domain in the N terminus has been implicated as a thermal sensor of TRPV1 (Yao et al., 2011). It is thus tempting to assume that PKCβII might modulate TRPV1 through a physical interaction with these structural domains. However, the bound PKCβII modulates TRPV1 function mainly through induced catalytic enzyme activity as a result of binding; direct physical binding per se plays only a minor role, in contrast to many other ion channel complexes in which auxiliary subunits influence the function of channels through direct physical binding. This model of modulation is very special, because PKCβII and TRPV1 are mutually activated simply through a process of physical binding.
This study also raises the question of how the phosphorylation of TRPV1 by PKCβII affects the gating process of the channel. It is generally believed that temperature, voltage, and capsaicin open TRPV1 by acting on distinct structural modules (sensors), which then couple allosterically to a gating pathway on the channel (Latorre et al., 2007). In support of this idea, a tyrosine residue in the second transmembrane domain is specifically required for capsaicin gating, but does not affect either voltage or temperature gating (Jordt and Julius, 2002). The outer pore region of TRPV1 was suggested to act as a thermal sensor because mutations in this region specifically impair heat activation without grossly affecting either capsaicin- or voltage-induced activation of TRPV1 (Grandl et al., 2010; Yang et al., 2010). However, several other modular domains have also been implicated as thermal sensors, including the N-terminal proximal domain (Yao et al., 2011) and the C-terminal domain (Brauchi et al., 2007). It is unclear whether these domains serve as thermal sensors or function as allosteric couplers between temperature sensing and gate opening. In the present study, we observed that the phosphorylation of TRPV1 at T705 by PKCβII was critical for the activation of TRPV1 by heat, as well as by capsaicin and voltage. T705 is located in the TRP domain in the C terminus of TRPV1. The recently resolved structure of TRPV1 revealed significant contacts between the TRP domain and the pre-S1 helix, a region analogous to the N-terminal proximal domain, suggesting that these contacts are important for allosteric coupling between channel domains (Liao et al., 2013). It is likely that phosphorylation of TRPV1 by PKCβII at T705 in the TRP domain may influence these contacts, resulting in enhanced gating. Collectively, then, the Thr705 residue has a key role in the gating of TRPV1.
Our study demonstrates that the sensitivity of TRPV1, in particular to temperature, requires the presence of PKCβII and phosphorylation at T705. The phosphorylation by PKCβII is thus a required precondition for driving TRPV1 channels into an optimal state for activation, even though TRPV1 channels are intrinsically heat sensitive. We emphasize, then, that the thermosensor is not always a single molecule, but rather acts as a molecular complex. Consistent with this idea, our previous study also identified a cold complex consisting of the cold-sensitive TRPM8 channel and the Gαq protein, allowing efficient and rapid inactivation of TRPM8 channels during inflammation (Zhang et al., 2012; Li and Zhang, 2013).
Sensory neurons are highly heterogeneous in nature, enabling them to respond appropriately to a diversity of environmental and endogenous stimuli with different qualities and intensities. Ion channel–protein complexes have emerged as an elegant mechanism allowing for ion channels in different population of neurons to react differentially to even subtle changes in intensity. A subset of hyper-constitutively sensitive sensory DRG neurons expressing TRPV1-PKCβII complexes may be attractive targets for developing novel therapies against pain and itch.
Footnotes
- Received January 21, 2014.
- Revision received May 7, 2014.
- Accepted May 8, 2014.
This work was supported by the Medical Research Council (MRC New Investigator Research Grant G0801387 to X.Z.). R.H. was supported by a doctoral studentship from the Islamic Development Bank and the Cambridge Commonwealth Trust. We thank Jiehong Huang for work at the early stage of the project and Mike Edwards (University of Cambridge) for critical reading of this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Xuming Zhang, Rowett Institute of Nutrition and Health and Institute of Medical Sciences, University of Aberdeen, Foresterhill, Aberdeen AB25 2ZD, United Kingdom. x.zhang{at}abdn.ac.uk
- Copyright © 2014 the authors 0270-6474/14/348246-13$15.00/0