
Abstract
Ubiquilin-1 (Ubqln1 or Ubqln), a ubiquitin-like protein, mediates degradation of misfolded proteins and has been implicated in a number of pathological and physiological conditions. To better understand its function in vivo, we recently generated transgenic (Tg) mice that globally overexpress mouse Ubqln in a variety of tissues and ubqln conditional knock-out mice. The Tg mice were viable and did not show any developmental or behavioral abnormalities compared with their wild-type (WT) littermates. When subjected to oxidative stress or ischemia/reperfusion, however, ubqln Tg mice but not the WT littermates showed increased tolerance to these insults. Following ischemic stroke, ubqln Tg mice recovered motor function more rapidly than did the WT mice. In contrast, KO of ubqln exacerbated neuronal damage after stroke. In addition, KO of ubqln also caused accumulation of ubiquitinated proteins. When ubqln KO mice were crossed with a ubiquitin-proteasome system function reporter mouse, the accumulation of a proteasome surrogate substrate was observed. These results suggest that Ubqln protects mice from oxidative stress and ischemic stroke-caused neuronal injury through facilitating removal of damaged proteins. Thus, enhanced removal of unwanted proteins is a potential therapeutic strategy for treating stroke-caused neuronal injury.
Introduction
Stroke is a major health problem in both developed and developing countries, and is associated with a high mortality rate, long-term disability, and economic consequences. Oxidative stress has been established as one of the major causative mechanisms underlying neuronal injury following stroke due to the overproduction and accumulation of reactive oxygen species (ROS) and reactive nitrogen species (RNS; Facchinetti et al., 1998; Slemmer et al., 2008). Both ROS and RNS oxidize a variety of proteins, resulting in protein misfolding and changes of their proper three-dimensional structures (Zhang and Saunders, 2009). When these events occur, misfolded proteins become prone to aggregation (Gregersen and Bross, 2010). Inefficient clearance of these misfolded proteins not only causes cell dysfunction but ultimately leads to cell death (Ge et al., 2007).
The ubiquitin-proteasome system (UPS) plays an essential role in maintaining neuronal homeostasis through removal of misfolded/aggregating proteins (Ge et al., 2007). Despite the great interest in regulation of protein degradation in developing therapies against neuronal injury following stroke, the role of aberrant protein clearance in the outcome of ischemic stroke remains far from being understood. Although it is speculated that appropriate removal of aberrant proteins following brief ischemia is neuroprotective, direct evidence for this role is lacking. To develop effective therapies for stroke, it is important to determine whether augmentation of unwanted protein clearance is neuroprotective or deleterious.
Ubiquilin-1 (Ubqln1), also commonly referred to as Ubqln, is a ubiquitin (Ub)-like (UbL) protein containing an N-terminal UbL domain and a C-terminal Ub associated (UbA) domain (Mah et al., 2000). Previous studies have linked Ubqln to UPS function by enhancing degradation of polyubiquitinated proteins (Ko et al., 2004). The UbA domain of Ubqln binds to polyUb chains, whereas the UbL domain of Ubqln interacts with the proteasome. Thus, Ubqln is regarded as a shuttle factor facilitating degradation of unwanted proteins through augmented delivery of polyUb-proteins to the proteasome (Ko et al., 2004). Indeed, our previous observations using cell culture models have demonstrated that overexpression of Ubqln enhances the degradation of misfolded proteins and reduces misfolded protein-caused cytotoxicity, whereas knockdown of Ubqln exacerbates misfolded protein-caused toxicity (Wang et al., 2006; Wang and Monteiro, 2007a,b). Thus far, however, the biological function of Ubqln has not been investigated in vertebrates. Here, we created ubqln transgenic (Tg) and conditional knock-out (cKO) mouse models and studied the role of Ubqln in oxidative stress and ischemic stroke-caused cell death.
Materials and Methods
Animals.
All animal maintenance and experimental procedures were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee of the University of South Dakota. Mice were maintained in a temperature and humidity controlled environment with a 12 h light/dark cycle and with ad libitum access to food and water.
Generation of ubqln Tg mice.
The open reading frame of the full-length mouse ubqln cDNA sequence (OriGene) was inserted into the EcoRI site of the pCAGGS vector (Kumarapeli et al., 2005) such that expression of the ubqln cDNA was driven by a hybrid promoter composed of the human cytomegalovirus (CMV) immediate-early enhancer and the chicken β-actin promoter along with its first intron, in front of the rabbit β-globin polyadenylation site (Marber et al., 1995; Kumarapeli et al., 2005). The sequence was confirmed by both restriction enzyme digestion and DNA sequencing. The overexpression construct was liberated from the vector with SalI and StuI and then used for pronuclear microinjection to create ubqln Tg mice in a C57BL/6/SJL background. Tg-positive mice were identified by PCR using the following primer pair: 5′-GCCTGG GGTGCCTAATGAGTGA-3′ and 5′-CGGCCTCGGCCTCTGCATAAAT-3′. All ubqln mice used for experiments in the present studies were heterozygous Tg mice.
Generation of floxed ubqln mice.
The floxed ubqln mouse (on a C57BL/6 genetic background) was generated by Taconic. Briefly, the mouse RP23-41J1 BAC DNA was used as template for generating the 5′ and 3′ homology arms, the conditional KO region for the gene targeting vector, as well as the probes for screening for the recombination. The 5′ homology arm (∼5.4 kb), cKO region (∼3.4 kb, containing exons 2 and 3), the 3′ homology arm (∼3.2 kb), the 5′ probe and 3′ probe were generated by PCR. The 5′ homology arm, cKO region containing exons 2 and 3, and the 3′ homology arm were cloned into the LoxFtNwCD or pCR4.0 vector and the resultant constructs were confirmed by restriction digestion and end-sequencing. Aside from the homology arms, the final targeting vector also contains LoxP sequences flanking the conditional KO region, Frt sequences flanking the Neo expression cassette that was used for positive selection of the electroporated embryonic stem (ES) cells, and a DTA expression cassette (used for negative selection of the ES cells). NotI was used for linearizing the final targeting vector before electroporation. The liberated targeting construct was introduced into C57BL/6 ES cells by electroporation and G418 resistant clones were screened for homologous recombination by Southern blotting using both 5′ and 3′ probes. The positively targeted ES cell colonies were identified and confirmed by Southern blotting and PCR. Following removal of the neo cassette from the positively targeted ES cells, they were injected into blastocysts, which were then transferred to pseudopregnant females. The chimeric offspring were identified by PCR. Thirteen floxed Ublqn mice belonging to three different lines were generated. Germline transmission of the targeted allele was confirmed by both Southern blot using both the 5′ and 3′ probes as well as PCR using the primer pairs: 5′-GAGTCTCTGGAAAAGATTTCCCTGTTTC-3′ and 5′-GACCCAATGGAGTAGGGGTCACAAA-3′. To generate ubqln conditional KO mice, the floxed mice were crossed with neuron-specific synapsin-I promoter-driven Cre Tg mice (Zhu et al., 2001; The Jackson Laboratory). The floxed mice carrying a Cre transgene were then crossed with the floxed mice will produce cKO mice.
Treatments of mice with oxidative stress.
Acute oxidative stress in liver was induced by intraperitoneal injection of menadione (150 mg/kg) as previously described (Hong et al., 2009). Mice (at 2 months) were killed 6 h after the injection. The liver was immediately removed for cell viability and caspase-3 activation assays. Alternatively, livers were removed, fixed with 4% paraformaldehyde, and sectioned for analysis of apoptotic cells. To induce oxidative stress in the brain, mice were intraperitoneally injected with menadione (100 mg/kg) daily and previous data have shown that menadione can cross the lipid bilayer and blood–brain barrier (Voigt et al., 2004). Three days after the injection, mice were killed and mouse brains were removed for assessing caspase-3 activity or fixed and sectioned for examining apoptotic cells.
Assessment of intracellular ROS.
Following 6 h or 3 d of menadione injection, mice were transcardially perfused with 5 μm CellROX Green (Life Technologies) oxidative stress reagent (in PBS, pH 7.4, containing heparin, Sigma-Aldrich; 10 U/ml), subsequently with PBS, and finally with 4% paraformaldehyde to fix tissues. Mouse livers and brains were then removed, postfixed in the same fixative, and cryoprotected by incubation with 30% sucrose. Tissues were cut in 7–15 μm thick using a cryostat (Leica Biosystems) at −20°C. Replicate liver or brain sections were then subjected to fluorescence microscopy analysis. Fluorescence intensity in each field was measured using the ImageJ software.
Liver H&E staining.
Fixed liver tissues were cryoprotected by incubate with 30% sucrose and then cryosectioned at 7–15 μm thickness. Replicate liver sections were stained with hematoxylin and eosin (H&E) for evaluation of necrosis as previously described (Gujral et al., 2001). Images of H&E stained liver sections were captured under 10× and 40× objective lenses using an upright microscope (iScope) equipped with a color digital camera.
Transient focal cerebral ischemia/reperfusion.
Transient focal cerebral ischemia/reperfusion of adult male mice (at 2–3 months) was induces by middle cerebral artery occlusion (MCAO) as previously described (Lü and Wang, 2012). After 1 h of occlusion of the MCAO, the mouse brains were allowed to reperfuse for 24 h before being killed for 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) staining as previously described (Lü and Wang, 2012) or to reperfuse for different periods of time to measure animals' body weight and motor function recovery. After surgery, body temperature of the mice was maintained at 37°C using a heating pad. When the mice regained complete consciousness, their neurological deficit was evaluated using a 5-point scale as previously described (Atochin et al., 2003). The animals that scored between 2 and 3 were included in the experiments.
Rotarod test.
Mouse motor behavior was measured using an accelerating rotarod machine (Med Associates). The cylinder rotated at an initial rate of 3.5 rpm and accelerated gradually at 20 rpm/min to a maximum of 40 rpm. When a mouse could no longer stay on the rod, it fell and the amount of time that the mouse remained on the accelerating rod was recorded.
Tissue cell viability and caspase-3 activity assays.
Tissue cell viability was assessed either by measuring tissue ATP levels using an ATP Determination Kit (Life Technologies) or by assessing caspase-3-like activity using our previously described methods (Dong et al., 2012). Briefly, a fluorogenic synthetic caspase-3 substrate, acetyl-Asp-Glu-Val-Asp-7-amido-trifluoromethyl-coumarin (Ac-DEVD-AFC) was incubated with cell lysates. The cleaved free AFC was measured using a fluorescence multiwell plate reader (Biotek) with an excitation at 400 nm and emission at 505 nm.
Detection of apoptotic cells and confocal microscopy.
Mice were immediately perfused with ice-cold PBS, followed by 4% paraformaldehyde in PBS. The mouse organs were then isolated and incubated with the same fixative for 3 d with rotation at 4°C. The samples were then transferred to 20% sucrose and incubated overnight before being frozen and subjected to cryostat sectioning to make 15-μm-thick sections. Apoptotic cell death was examined using the ApopTag Plus Fluorescein In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer's instructions. Stained cells were observed with a laser scanning confocal (Olympus) equipped with an argon laser and two HeNe lasers and FluoView 1000 software. Apoptotic rate was calculated as follows: apoptotic (or TUNEL positively stained cell) rate (%) = number of TUNEL-positive staining cells/number of total cells × 100% (at least 600 cells were counted in each group).
FJB staining of degeneration neurons.
Staining of degeneration neurons with Fluoro-Jade B (FJB) in brain sections was according to a previously described method (Schmued and Hopkins, 2000). Images were captured from the cortex with a Zeiss Axiovert fluorescence microscope and quantification of FJB staining in each field was performed using the ImageJ software.
Western blot analysis.
We used previously described methods for Western blot analysis (Dong et al., 2012; Lü and Wang, 2012). Primary antibodies used in the studies include anti-Ubqln antibody (1:1000, Abcam), anti-β-tubulin (1:2000, Applied Biological Materials), anti-ubiquitin (against both polyubiquitin and monoubiquitin, 1:1000, Cell Signaling Technology), anti-GFP (1:1000, Santa Cruz Biotechnology), and anti-actin (1:1000, Santa Cruz Biotechnology). Detection was performed using HRP-conjugated anti-rabbit, -mouse, or -goat antibody (1:3000, Santa Cruz Biotechnology) and Super-Signals West Pico substrate (Pierce).
Statistical analysis.
Statistical comparisons of biochemical and histology staining data between two groups were evaluated using two-tailed Student's t test. p < 0.05 was regarded as statistically significant. Two-way ANOVA was conducted on the experimental data using two genotypes of mice and time as grouping factors.
Results
Generation of ubqln Tg mice
To generate ubqln Tg mice, we cloned the entire mouse ubqln cDNA into an expression vector under the regulation of a combination cytomegalovirus early enhancer element and chicken β-actin globin promoter (Kumarapeli et al., 2005). Correct clones were identified by restriction enzyme digestion and confirmed by direct sequencing of the DNAs in both forward and reverse directions. Additionally, cultured cells were transfected with the construct to verify that the codons are in-frame as determined by Western blot (data not shown). After linearization, the transgenic vector was injected into fertilized eggs to generate Tg mice. The Tg mice were originally generated in C57BL/6/SJL hybrid background and then repeatedly backcrossed with the C57BL/6 mouse for at least six generations before being used for experiments. We have established and maintained two independent ubqln Tg mouse lines, Tg-2 and Tg-4, and immunoblotting indicates that ubqln transgene exhibited striking overexpression (OE) in the brain, heart, liver, and skeletal muscle, although most of other tissues also showed evident OE of Ubqln in the two lines of the Tg mice (Fig. 1A,B). As OE of Ubqln was observed in two independent stable Tg mouse lines, it is unlikely that the effect of OE of the Tg gene was caused by an insertional mutation. Because we did not find any difference between the two lines of Tg mice, the experiments performed in this research were mainly used the Tg-2 line mice. The two lines of Tg mice did not show any developmental or behavioral abnormalities (data not shown). The litter size and morphology of the Tg mice did not differ from those of their wild-type (WT) littermates (data not shown). These data suggest that OE of Ubqln does not cause deleterious effect on the animal.

OE of Ubqln in the Tg mice reduces oxidative stress-induced cell death in the liver. A, B, Western blot analysis showing Ubqln OE in the indicated tissues in the Tg-2 (A) and Tg-4 line (B) mice at 2 months. Immunoblotting of β-tubulin was used as a loading control. C, Fluorescence microscopy analysis of intracellular ROS levels in hepatocytes. WT or Tg mice were intraperitoneally injected with 150 mg/kg menadione or vehicle (control). After 6 h following the injection, animals were killed and subjected to ROS analysis. D, Graph showing the measurement of ROS levels in WT and Tg livers in presence or absence of menadione treatment. Data are shown as mean ± SEM; n = 3; *p < 0.001, **p < 0.05. E, Representative liver sections were stained with H&E. Note that compared with the Tg mouse liver, menadione treatment in the WT mouse liver caused increased vacuolization, cell disruption, and karyolysis (pointed by arrows). F, Measurement of ATP levels of liver cells of Tg and WT mice at 2 months after 6 h of menadione treatment. Data are shown as mean ± SD; n = 3 for each group; *p < 0.01. G, Measurement of caspase-3 activity of liver cells of Tg and WT mice (at 2 months) following 6 h of menadione treatment. Data are shown as mean ± SD; n = 3 for each group; *p < 0.05. H, Confocal microscopy showing TUNEL positively stained cells (green) in the liver of the WT and Tg mice (at 2 months) after menadione treatment. Scale bar, 50 μm. I, Graph showing the percentage of TUNEL positively stained cells in livers. Data are shown as mean ± SD; n = 4 for each group; *p < 0.05.
Liver cells of the ubqln Tg mice showed increased tolerance to oxidative stress
With the Tg mice, we next determined whether OE of Ubqln in mice is protective or deleterious in oxidative stress condition. Accordingly, we examined cell viability by assessing intracellular ATP levels and caspase-3 activity in both the Tg mice and their WT littermates following oxidative stress. Menadione can cross the lipid bilayer and is therefore considered an intracellular oxyradical producing agent (Thor et al., 1982; Voigt et al., 2004; Hong et al., 2009). We injected menadione intraperitoneally into the mice and assessed the animals for ROS levels and tissue cell injury 6 h after the injection. As shown in Figure 1C,D, liver cells showed distinct responses to the acute treatment of menadione in the two types of mice. Although treatment of mice with menadione enhanced the generation of ROS in both WT and Tg mouse livers, compared with the WT animals, OE of Ubqln strikingly suppressed ROS production in the Tg mouse liver (Figs. 1C,D). Liver cells derived from the Tg mice showed significantly higher viability than the cells derived from WT mice as indicated by their reduced vacuolization, cell disruption, and karyolysis in H&E-stained tissue sections (Fig. 1E) and higher ATP levels (Fig. 1F). In addition, the hepatocytes of the Tg mice showed lower caspase-3 activity than those of WT mice (Fig. 1G). Consistent with these results, the increased tolerance of the Tg mice-derived liver cells to oxidative stress is further supported by a reduction in the percentage of apoptotic cells demonstrated by TUNEL assay in the livers of Tg mice (Fig. 1H,I). These data indicate that OE of Ubqln in mice protects liver cells from oxidative stress-caused tissue injury.
Brain cells of the ubqln Tg mice showed increased tolerance to oxidative stress
To further test whether the increased tolerance to oxidative stress also occurs in the CNS of the Tg mice, we examined the response of brain cells to oxidative stress in both the Tg and WT mice. Although it has been reported that menadione can cross the lipid bilayer and enter cells (Voigt et al., 2004), we did not detect differences of brain cell viability and caspase-3 activity in the two types of mice after 6 h of menadione treatment. This may be due to the blood–brain barrier that delays transport of the free radical producer. Thus, we tested whether repeated intraperitoneal injections of menadione into the two types of mice cause different responses. Although ROS levels were significantly elevated in both WT and Tg mouse brains following 3 d of oxidative stress induced by daily menadione treatment (Fig. 2A,B), caspase-3 activity in brain cells of the Tg mice was significantly lower than that of the WT animals (Fig. 2C). To further determine whether menadione-induced oxidative stress causes differential cell death in the two types of animal brains, we stained brain sections with FJB, a high affinity fluorescent marker for selectively localizing degeneration neurons (Schmued and Hopkins, 2000). As shown in Figure 2D,E, the WT mouse brain cells showed more frequent neurodegeneration than did the Tg mouse brain cells following menadione treatment. In agreement with this, apoptotic cells determined by the TUNEL assay in both the cortex (Figs. 2F,G) and striatum (Figs. 2H,I) of the Tg mice were remarkably fewer than those in WT mice. These results suggest that OE of Ubqln in mice also reduces oxidative stress-caused cell death in the brain.

OE of Ubqln in the Tg mice reduces oxidative stress-induced cell death in the brain. A, Fluorescence microscopy analysis of intracellular ROS levels in the brain cortex. WT or Tg mice were intraperitoneally injected with 100 mg/kg menadione or vehicle (control) daily. After 3 d injections, animals were killed and subjected to ROS analysis. B, Graph showing the measurement of ROS levels in WT and Tg brain cortex in presence or absence of menadione treatment. Data are shown as mean ± SEM; n = 3; *p < 0.05. C, Caspase-3 activity of brain cortex of Tg and WT mice at 2 months after 3 d menadione treatment. Data are shown as mean ± SD; n = 3 for each group; *p < 0.01. D, Brain sections were stained with FJB to identify degeneration neurons. E, Graph showing the quantification of neurodegeneration in the WT and Tg mouse brain cortex after 3-d menadione injection. Data are shown as mean ± SEM; n = 3; *p < 0.05. F, Confocal microscopy showing apoptotic cells (green) in the brain cortex after 3 d menadione treatment in the mice at 2 months. Scale bar, 50 μm. G, Graph showing the percentage of TUNEL positively stained cells in brain cortex after 3 d menadione treatment. Data are shown as mean ± SD; n = 4 for each group; *p < 0.001. H, Confocal microscopy showing TUNEL positively stained cells (green) in the brain striatum after 3 d menadione treatment in the mice at 2 months. Scale bar, 50 μm. I, Graph showing the percentage of TUNEL positively stained cells in brain striatum after 3 d menadione treatment. Data are shown as mean ± SD; n = 4 for each group; *p < 0.001.
OE of Ubqln reduces ischemia/reperfusion-caused brain injury in mice
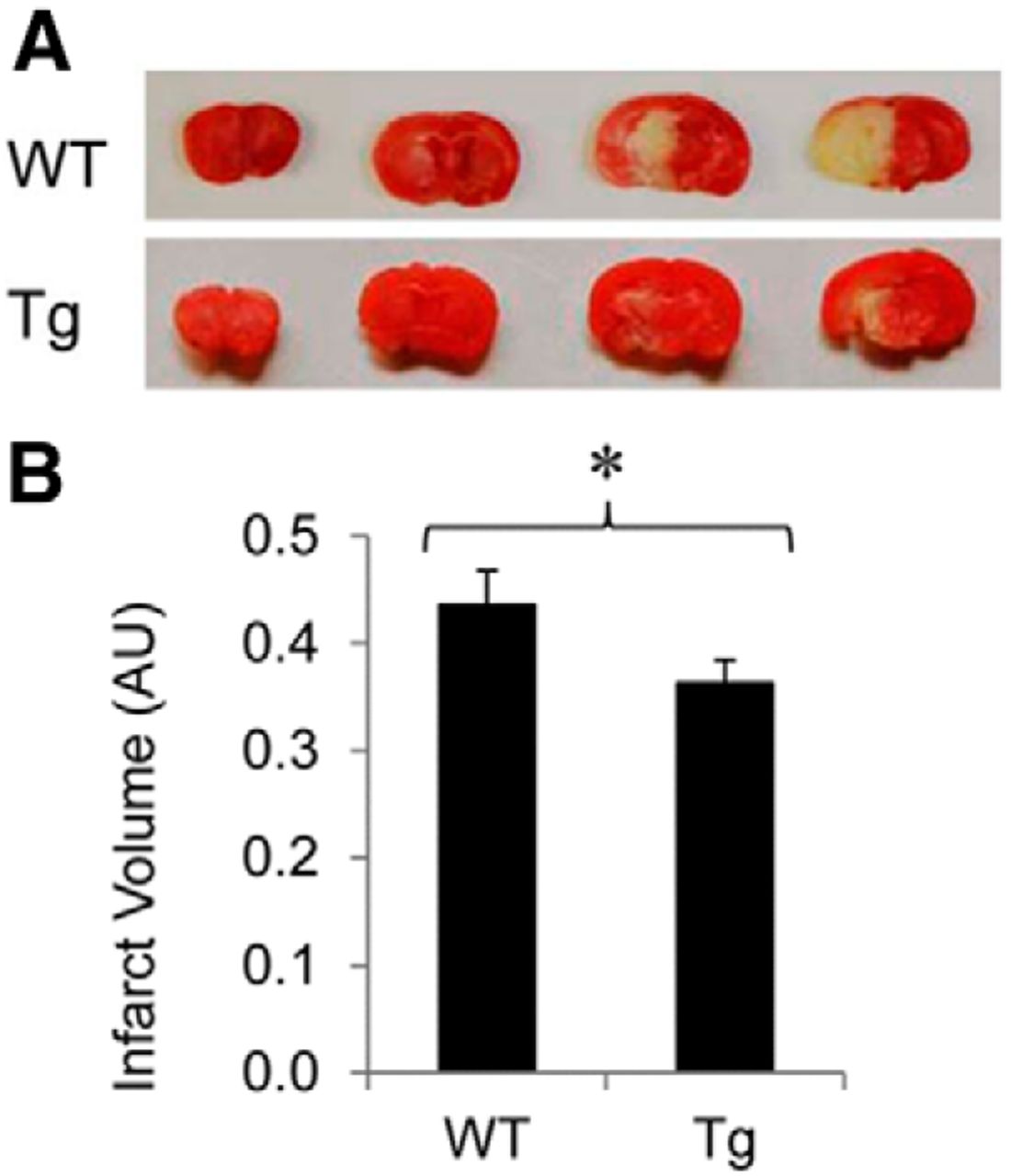
Oxidative stress has been established as one of the major causative mechanisms underlying neuronal injury following ischemia/reperfusion in the brain (Facchinetti et al., 1998; Slemmer et al., 2008; Gregersen and Bross, 2010; Hochrainer et al., 2012). To further examine whether OE of Ubqln reduces ischemia/reperfusion-caused brain injury in mice, we subjected the mice to ischemia by transient MCAO (Lü and Wang, 2012). As shown in Figure 3A,B, compared with the WT animals, the Tg mouse brains showed a significant reduction of infarct volume following 1 h MCAO and 24 h reperfusion, indicating that OE of Ubqln in the Tg mice attenuates ischemia/reperfusion-caused brain injury.

ubqln Tg mice showed reduced neuronal damage after stroke. A, TTC staining of coronal sections from ischemic mouse brains. The white indicates infarction. B, Measurement of infarct volume. Data are shown as mean ± SD; n = 7 for the WT mice; n = 5 for the Tg mice; *p < 0.05.
OE of Ubqln enhances functional recovery after ischemia/reperfusion
To determine whether OE of Ubqln promotes functional recovery we closely monitored mouse behaviors after the ischemia/reperfusion procedure. We found that following the surgery, the Tg mice started to walk and search for food sooner than the WT mice (data not shown). We measured the animal body weight daily from 0 to 10 d after the surgery. Both the Tg and WT mice rapidly lost weight after the surgery (Fig. 4A), due to the impairment of their motor function. However, the average body weight of the Tg mice showed a trend of recovering faster than the WT animals, although there was no statistically significant difference between the two types of animals (Fig. 4A). We used the rotarod test to assess mouse motor function following ischemia/reperfusion, and found that the Tg mice showed a relatively more rapid recovery of motor function compared with the WT mice (Fig. 4B). Only 3 d after the surgery, the motor function of the Tg mice had recovered to the presurgery level, whereas it took 10 d for the WT mice to completely recover to their presurgery level. These data reveal that OE of Ubqln not only reduces neuronal injury but also enhances functional recovery in mice following ischemia/reperfusion-induced brain injury.

ubqln Tg mice show enhanced motor function recovery after transient focal cerebral ischemia. A, Body weight of Tg and WT mice measured each d after surgery. n = 9 for the WT mice; n = 11 for the Tg mice. B, Rotarod test results following the surgery. The cutoff time for mice staying on the rotarod was set at 250 s. A two-way ANOVA (time by genotype) revealed a significant effect of genotype (F(10,241) = 53.83, p < 0.0001), and a significant effect of time (F(10,241) = 5.79, p < 0.001). Data are shown as mean ± SD; n = 9 for the wild-type mice; n = 11 for the Tg mice.
Generation of ubqln cKO mice
In addition to using gain-of-function approaches we also generated ubqln cKO mice to perform loss-of-function studies to the mice. Mouse Ubqln shares 88% amino acid sequence identity with the human Ubqln. Exons 2 and 3 of the mouse ubqln gene were selected as the cKO region (Fig. 5A). The deletion of exons 2 and 3 causes a frameshift of the downstream exons and results in a stop codon downstream of and close to exon 1. The first loxP sequence was inserted upstream of the 5′ end of exon 2 and the second loxP sequence along with a Neo cassette, was placed downstream of the 3′end of exon 3. The targeting vector was confirmed by both restriction digestion and end sequencing analysis. The targeting construct was introduced into C57BL/6 ES cells by electroporation and G418 resistant clones were screened for homologous recombination by Southern blotting using both 5′ and 3′ probes. The Neo cassette in the targeting vector was subsequently removed from the ES cells to avoid its potential influence on the floxed mice. Targeted ES cell colonies were identified and confirmed by Southern blotting and PCR before being injected into blastocysts. The blastocysts were then transferred to pseudopregnant females and chimeric offspring were identified by PCR. Thirteen floxed ubqln mice were generated and stable germline transmission of the targeted allele from one generation to the next was confirmed by both Southern blot using the 5′ probe (Fig. 5B) and the 3′ probe (Fig. 5C) and PCR (Fig. 5D). The litter size, morphology, and body weight of the floxed mice did not differ from the WT mice (data not shown). To verify the functionality of the loxP/Cre system, we crossed ubqln floxed mice with synapsin-I promoter-driven Cre Tg mice to generate neuron-specific ubqln KO mice (Zhu et al., 2001). Western blot analysis indicated that neuron-specific Cre led to apparent KO of ubqln in the brain cortex (Fig. 5E). Neuron-specific KO of ubqln did not affect animal growth, as both the WT and cKO adult mice had the similar body weights at 2 months of age (data not shown).

Generation of ubqln cKO mice. A, An illustration of ubqln WT allele, targeting vector, and recombinant ubqln alleles. Red bars, probes for Southern blot. B, C, Genotyping of ubqln-floxed mice by Southern blot analysis of BamHI digested genomic DNA with a 5′ probe (B) or with a 3′ probe (C) from the WT mice (lanes 1, 2), targeted embryonic stem (ES) cells (lanes 3, 4), and heterozygous floxed F2 generation mice (lanes 5–7). D, PCR analysis of the floxed F2 generation mice following chimera breeding. WT: lanes 2 and 9; targeted ES cells: lanes 3 and 4; buffer control: lane 5; heterozygous floxed mice: lanes 6–8. Lane 1, DNA ladder. E, Examination of Ubqln protein levels in the brain in the synapsin-I promoter-regulated Cre recombinase mediated neuron-specific KO and WT control mice by Western blotting.
Neuron-specific KO of ubqln exacerbates ischemia/reperfusion-caused brain injury and delays functional recovery
With the cKO mice, we next determined whether KO of ubqln affected brain cell injury following ischemic stroke. The cKO mice were subjected to 1 h MCAO followed by 24 h reperfusion. The KO mice showed exacerbated brain injury compared with the WT mice as indicated by the increased infarct volume (Figs. 6A,B). Moreover, rotarod test results showed that the neuron-specific KO mice demonstrated remarkably slower recovery of motor function than the WT mice after the surgery (Fig. 6C). Thus, these results further confirm that Ubqln is important for neuroprotection and that loss of Ubqln exacerbates neuronal injury and delays functional recovery following ischemia/reperfusion.

Synapsin-I neuron-specific KO of ubqln exacerbates ischemia/reperfusion-caused brain injury and delays functional recovery. A, KO of ubqln in neurons exacerbates neuronal injury as indicated by TTC staining 24 h after ischemic stroke. B, Measurement of infarct volume. Data are shown as mean ± SD; n = 7 for each group; *p < 0.05. C, Rotarod test results suggesting that KO of ubqln in neurons delays functional recovery after stroke. The cutoff time for mice staying on the rotarod was set at 250 s. A two-way ANOVA (time by genotype) revealed a significant effect of genotype (F(10,248) = 6.30, p = 0.0127), and a significant effect of time (F(10,248) = 7.04, p < 0.001). Data are shown as mean ± SD; n = 6 for the WT mice; n = 7 for the KO mice.
KO of ubqln causes accumulation of ubiquitinated proteins in the brain
To shed light on how Ubqln protects neurons from oxidative stress and ischemic stroke-caused injury, we examined whether neuron-specific KO of ubqln alters protein degradation in the brain by assessing the total ubiquitinated-protein levels in the KO mouse brain. Compared with the control animals, KO of ubqln resulted in a significant accumulation of ubiquitinated-proteins in the brain (Figs. 7A,B). In addition, we also examined the functionality of UPS using a UPS function reporter mouse in the absence of the ubqln gene. This UPS function reporter mouse expresses a CAG (chicken β-actin promoter with CMV enhancer) promoter-driven green fluorescent protein (GFP) transgene fused with the degradation signal (also referred to as degron) CL1 at its C-terminus which targets it for ubiquitination and degradation by the UPS (Su et al., 2011). The modified GFP is referred to as GFPu or GFPdgn. Because GFPu serves as a surrogate substrate for the UPS, its level inversely reflects UPS function. The GFPu Tg mice were crossed with ubqln neuron-specific KO mice to generate ubqln-cKO/ubqln mice. As shown in Figure 7C,D, GFPu accumulated ∼1.7-fold when ubqln was knocked-out in neurons in the mice. Together, these data indicate that Ubqln is important for degradation of ubiquitinated proteins and disruption of the gene leads to accumulation of UPS substrates.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Accumulation of ubiquitinated proteins and a UPS function reporter protein, GFPu, in the brain cortex of the synapsin-I neuron-specific ubqln cKO mice. A, Western blot analysis of ubiquitinated proteins in WT and ubqln KO brain cortex at 2 months. B, Quantification of ubiquitinated protein levels. Data are shown as mean ± SD; n = 3; *p < 0.05. C, Western blot analysis of GFPu expression in brain cortex derived from GFPu, WT control, and ubqln-KO/ubqln mice at 2 months. D, GFP expression was quantified and normalized against tubulin levels. Data are shown as mean ± SD; n = 3; *p < 0.05.
Discussion
To perform both gain-of-function and loss-of-function analysis of Ubqln, we developed both ubqln Tg and cKO mouse models. The Tg mice overexpressing Ubqln in all tissues examined were viable and did not show any developmental or behavioral abnormalities. When subjected to oxidative stress or ischemia/reperfusion the Tg mice showed increased tolerance to these insults compared with the WT mice. Following ischemic stroke, the ubqln Tg mice recovered motor function more rapidly than did the WT mice. In contrast, KO of ubqln not only exacerbated ischemia-induced neuronal damage, but also delayed brain functional recovery compared with the WT mice. Interestingly, the ubqln mice deficient in ubqln gene showed accumulation of the surrogate GFPu substrate. Together, these results indicate that Ubqln is required for UPS function and for neuroprotection against oxidative stress/ischemia caused injury.
The results obtained here are consistent with our previous studies that OE of Ubqln in cell cultures protects cells from misfolded protein-caused cytotoxicity (Wang et al., 2006; Wang and Monteiro, 2007a). The protective effect exerted by Ubqln is likely due to the enhanced removal of unwanted proteins through the UPS pathway (Ko et al., 2004; Wang and Monteiro, 2007b). However, our results cannot exclude other mechanisms because Ubqln is also involved in autophagy (N′Diaye and Brown, 2003; Rothenberg et al., 2010). Additionally, recent studies have shown that Ubqln itself may be an authentic chaperone protein that prevents the aggregation of amyloid precursor protein and ameliorates neuronal death (Stieren et al., 2011). It is possible that after binding to misfolded proteins, Ubqln will mediate them to UPS and/or autophagy for removal, resulting in reduced toxicity and neuroprotection. Indeed, this has been supported by previous observations that OE of Ubqln results in enhanced degradation of polyubiquitinated/misfolded proteins (Ko et al., 2004; Wang and Monteiro, 2007b). However, some studies have indicated that Ubqln interacts with and stabilizes some specific proteins, such as an anti-apoptotic BCL2-like protein, BCL2L/BCLb, and interestingly, the BCLb protein stabilized by Ubqln is monoubiquitinated (Beverly et al., 2012). It remains unclear how Ubqln increases the half-life of this protein while it enhances the degradation of polyubiquitinated proteins. In this research, we observed an association of OE of Ubqln with increased tolerance to oxidative stress and ischemic stroke in mice. The Tg mice showed less cell damage and enhanced brain recovery following oxidative stress or ischemia insults. Thus, our data are in accordance with several lines of previous in vitro studies, supporting the idea that Ubqln improves cell survival under oxidative stress in vivo through promoting removal of unwanted proteins.
The protective role of Ubqln observed in the Tg mice is also supported by our ubqln cKO data. In contrast to the Tg mice, neuron-specific KO of ubqln in mice makes brain cells more vulnerable to ischemia, as indicated not only by increased neuronal loss but also by delayed functional recovery. In addition, KO of ubqln increases the accumulation of ubiquitinated proteins. When ubqln KO mice were crossed with a UPS function reporter mouse, the accumulation of a proteasome surrogate substrate was observed. Although mouse Ubqln subfamily proteins, similar to the corresponding human orthologs, contain multiple Ubqln members (Kleijnen et al., 2000; Mah et al., 2000), our results indicate that the effect of loss-of-function caused by cKO of the ubqln gene cannot be completely compensated for by other members of the subfamily. This is consistent with another report showing that loss of only Ubqln2 function caused by a mutation in humans leads to impaired protein degradation and neurodegeneration (Deng et al., 2011). Therefore, enhancement of unwanted protein degradation is potential therapeutic strategy not only for stroke but also for other neurodegenerative disorders.
In summary, despite a number of studies regarding the role of Ubqln, no ubqln Tg or KO mouse has been reported. Here we report the generation of two novel mouse models and the initial characterization of the response to challenging them with oxidative stress and cerebral ischemia. Our data indicate that OE of Ubqln in the Tg mice provides the animals with increased tolerance to oxidative stress and ischemia, whereas KO of the gene makes cells more vulnerable to these insults. These results reveal that enhanced removal of unwanted proteins is a potential therapeutic strategy for treating stroke-caused neuronal injury. In addition, Ubqln has been implicated in a number of physiological and pathological conditions, including viral infection (Feng et al., 2004), neural stem cell differentiation (Hoffrogge et al., 2006), tumorigenesis (Chen et al., 2007), endoplasmic reticulum stress (Ko et al., 2002; Lim et al., 2009), Alzheimer's disease (Bertram et al., 2005; Haapasalo et al., 2010; El Ayadi et al., 2012), Huntington's disease (Doi et al., 2004; Wang et al., 2006; Wang and Monteiro, 2007b), and amyotrophic lateral sclerosis (Kim et al., 2009; Hanson et al., 2010). Thus, the mouse models will provide useful tools for defining the (patho)physiological significance of Ubqln in different tissues/organs as well as for exploring its role in a number of neurologic diseases.
Footnotes
- Received August 16, 2013.
- Revision received January 5, 2014.
- Accepted January 9, 2014.
This work was supported by a Competitive Research Grant Award of the South Dakota Board of Regents (H.W.) and Start-up Funds from the University of South Dakota (H.W.). We thank Dr Robin Miskimins for critical reading of the paper, Drs Michael Watt and Gina Forster at the University of South Dakota (USD) Animal Behavioral Core Facility for assistance in mouse rotarod test, Drs Evelyn Schlenker and J. Scott Pattison for help in statistical analysis, Fangfang Qiao for preparation of mouse tissues, Suleman Said at the USD Histopathology Core Facility for assistance in the preparation of brain sections and histology staining, and Dr Fran Day at the USD Imaging Core Facility for help in confocal microscopy. X.W. was partially supported by NIH Grants R01 HL072166 and R01 HL085629 and H.W. was supported in part by an NIH Grant R15NS071459.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr Hongmin Wang, Division of Basic Biomedical Sciences, Sanford School of Medicine, University of South Dakota, Vermillion, SD 57069. Hongmin.Wang{at}usd.edu
- Copyright © 2014 the authors 0270-6474/14/342813-09$15.00/0